
Complexes of Dichlorogermylene with Phosphine/Sulfoxide-Supported Carbone as Ligand †

 ,
,
Abstract
1. Introduction
2. Results and Discussion
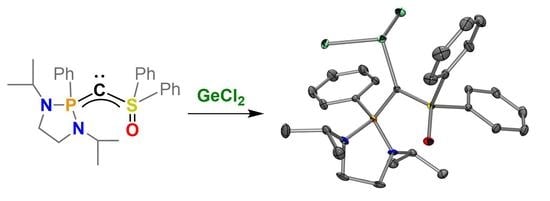
2.1. Synthesis
2.2. Dichlorogermylene Coordination
2.3. Conclusions
3. Materials and Methods
3.1. General Comments
3.2. Synthesis
3.3. X-ray Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mortreux, A.; Petit, F. Industrial Applications of Homogeneous Catalysis; Springer: Dordrecht/Holland, The Netherlands, 1988. [Google Scholar]
- Igau, A.; Grützmacher, H.; Baceiredo, A.; Bertrand, G. Analogous α,α’-Bis-Carbenoid Triply Bonded Species: Synthesis of a Stable λ3-Phosphinocarbene-λ5-Phosphaacetylene. J. Am. Chem. Soc. 1988, 110, 6463–6466. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Glorius, F. N-Heterocyclic Carbenes in Transition Metal Catalysis; Springer: Berlin/Heidelberg, Germany, 2007; Volume 21. [Google Scholar]
- Nolan, S.P. N-Heterocyclic Carbenes in Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Herrmann, W.A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- César, V.; Bellemin-Laponnaz, S.; Gade, L.H. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. Chem. Soc. Rev. 2004, 33, 619–636. [Google Scholar] [CrossRef]
- Ramirez, F.; Desai, N.B.; Hansen, B.; McKelvie, N. Hexaphenylcarbodiphosphorane, (C6H5)3PCP(C6H5)3. J. Am. Chem. Soc. 1961, 83, 3539–3540. [Google Scholar] [CrossRef]
- Tonner, R.; Öxler, F.; Neumüller, B.; Petz, W.; Frenking, G. Carbodiphosphoranes: The Chemistry of Divalent Carbon(0). Angew. Chem. Int. Ed. 2006, 45, 8038–8042. [Google Scholar] [CrossRef] [PubMed]
- Tonner, R.; Frenking, G. C(NHC)2: Divalent Carbon(0) Compounds with N-Heterocyclic Carbene Ligands—Theoretical Evidence for a Class of Molecules with Promising Chemical Properties. Angew. Chem. Int. Ed. 2007, 46, 8695–8698. [Google Scholar] [CrossRef]
- Tonner, R.; Frenking, G. Divalent Carbon(0) Chemistry, Part 1: Parent Compounds. Chem. Eur. J. 2008, 14, 3260–3272. [Google Scholar] [CrossRef]
- Tonner, R.; Frenking, G. Divalent Carbon(0) Chemistry, Part 2: Protonation and Complexes with Main Group and Transition Metal Lewis Acids. Chem. Eur. J. 2008, 14, 3273–3289. [Google Scholar] [CrossRef]
- Kaska, W.C.; Mitchell, D.K.; Reichelderfer, R.F. Transition metal complexes of hexaphenylcarbodiphosphorane. J. Organomet. Chem. 1973, 47, 391–402. [Google Scholar] [CrossRef]
- Fujii, T.; Ikeda, T.; Mikami, T.; Suzuki, T.; Yoshimura, T. Synthesis and structure of (MeN)Ph2S=C=SPh2(NMe). Angew. Chem. Int. Ed. 2002, 41, 2576–2578. [Google Scholar] [CrossRef]
- Alcarazo, M.; Lehmann, C.W.; Anoop, A.; Thiel, W.; Fürstner, A. Coordination chemistry at carbon. Nat. Chem. 2009, 1, 295–301. [Google Scholar] [CrossRef]
- Dellus, N.; Kato, T.; Bagan, X.; Saffon, N.; Branchadell, V.; Baceiredo, A. An isolable mixed P,S-bis(ylide) as an asymmetric carbon atom source. Angew. Chem. Int. Ed. 2010, 49, 6798–6801. [Google Scholar] [CrossRef]
- Alcarazo, M.; Radkowski, K.; Mehler, G.; Goddard, R.; Fürstner, A. Chiral heterobimetallic complexes of carbodiphosphoranes and phosphinidene–carbene adducts. Chem. Commun. 2013, 49, 3140–3142. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Schier, A. Coordination Chemistry at Carbon: The Patchwork Family Comprising (Ph3P)2C, (Ph3P)C(C2H4), and (C2H4)2C. Angew. Chem. Int. Ed. 2013, 52, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Morosaki, T.; Suzuki, T.; Wang, W.W.; Nagase, S.; Fujii, T. Syntheses, Structures, and Reactivities of Two Chalcogen-Stabilized Carbones. Angew. Chem. Int. Ed. 2014, 53, 9569–9571. [Google Scholar] [CrossRef]
- Morosaki, T.; Wang, W.-W.; Nagase, S.; Fujii, T. Synthesis, Structure, and Reactivities of Iminosulfane- and Phosphane-Stabilized Carbones Exhibiting Four-Electron Donor Ability. Chem. Eur. J. 2015, 21, 15405–15411. [Google Scholar] [CrossRef]
- Chen, W.-C.; Shen, J.-S.; Juca, T.; Peng, C.-J.; Lin, Y.-H.; Wang, Y.-P.; Shih, W.-C.; Yap, G.P.A.; Ong, T.-G. Expanding the Ligand Framework Diversity of Carbodicarbenes and Direct Detection of Boron Activation in the Methylation of Amines with CO2. Angew. Chem. Int. Ed. 2015, 54, 15207–15212. [Google Scholar] [CrossRef] [PubMed]
- Troadec, T.; Wasano, T.; Lenk, R.; Baceiredo, A.; Saffon-Merceron, N.; Hashizume, D.; Saito, Y.; Nakata, N.; Branchadell, V.; Kato, T. Donor-Stabilized Silylene/Phosphine-Supported Carbon(0) Center with High Electron Density. Angew. Chem. Int. Ed. 2017, 56, 6891–6895. [Google Scholar] [CrossRef]
- Corberan, R.; Marrot, S.; Dellus, N.; Merceron-Saffon, N.; Kato, T.; Peris, E.; Baceiredo, A. First Cyclic Carbodiphosphoranes of Copper(I) and Gold(I) and Their Application in The Catalytic Cleavage of X-H bonds (X = N and O). Organometallics 2009, 28, 326–330. [Google Scholar] [CrossRef]
- Goldfogel, M.J.; Roberts, C.C.; Meek, S.J. Intermolecular hydroamination of 1,3-dienes catalyzed by bis(phosphine)carbodicarbene-rhodium complexes. J. Am. Chem. Soc. 2014, 136, 6227–6230. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-C.; Shen, J.-S.; Lin, B.-C.; Chen, W.-C.; Chan, Y.-T.; Ching, W.-M.; Yap, G.P.A.; Hsu, C.-P.; Ong, T.-G. Synthesis and isolation of an acyclic tridentate bis(pyridine)carbodicarbene and studies on its structural implications and reactivities. Angew. Chem. Int. Ed. 2015, 54, 2420–2424. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.C.; Matias, D.M.; Goldfogel, M.J.; Meek, S.J. Lewis Acid Activation of Carbodicarbene Catalysts for Rh-Catalyzed Hydroarylation of Dienes. J. Am. Chem. Soc. 2015, 137, 6488–6491. [Google Scholar] [CrossRef]
- Pranckevicius, C.; Fan, L.; Stephan, D.W. Cyclic Bent Allene Hydrido-Carbonyl Complexes of Ruthenium: Highly Active Catalysts for Hydrogenation of Olefins. J. Am. Chem. Soc. 2015, 137, 5582–5589. [Google Scholar] [CrossRef]
- Marcum, J.S.; Roberts, C.C.; Manan, R.S.; Cervarich, T.N.; Meek, S.J. Chiral Pincer Carbodicarbene Ligands for Enantioselective Rhodium-Catalyzed Hydroarylation of Terminal and Internal 1,3-Dienes with Indoles. J. Am. Chem. Soc. 2017, 139, 15580–15583. [Google Scholar] [CrossRef] [PubMed]
- Liberman-Martin, A.L.; Grubss, R.H. Ruthenium Olefin Metathesis Catalysts Featuring a Labile Carbodicarbene Ligand. Organometallics 2017, 36, 4091–4094. [Google Scholar] [CrossRef]
- Inés, B.; Patil, M.; Carreras, J.; Goddard, R.; Thiel, W.; Alcarazo, M. Synthesis, Structure, and Reactivity of a Dihydrido Borenium Cation. Angew. Chem. Int. Ed. 2011, 50, 8400–8403. [Google Scholar] [CrossRef]
- Xiong, Y.; Yao, S.; Tan, G.; Inoue, S.; Driess, M. A cyclic Germadicarbene (“Germylone”) from Germyliumylidene. J. Am. Chem. Soc. 2013, 135, 5004–5007. [Google Scholar] [CrossRef]
- Li, Y.; Mondal, C.; Roesky, H.W.; Zhu, H.; Stollberg, P.; Herbst-Irmer, R.; Stalke, D.; Andrada, D. Acyclic Germylones: Congeners of Allenes with a CentralGermanium. J. Am. Chem. Soc. 2013, 135, 12422–12428. [Google Scholar] [CrossRef]
- Khan, S.; Gopakumar, G.; Thiel, W.; Alcarazo, M. Stabilization of a two-Coordinate [GeCl]+ Cation by Simultaneous s and p Donation from a Monodentate Carbodiphosphorane. Angew. Chem. Int. Ed. 2013, 52, 5644–5647. [Google Scholar] [CrossRef]
- Roy, M.M.D.; Lummis, P.A.; Ferguson, M.J.; McDonald, R.; Rivard, E. Accessing Low-Valent Inorganic Cations by Using an Extremely Bulky N-Heterocyclic Carbene. Chem. Eur. J. 2017, 23, 11249–11252. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.; Belding, L.; Van der Est, A.; Dudding, T.; Korobkov, I.; Nikonov, G.I. A coordination Compound of Ge0 Stabilized by a Diiminopyridine Ligand. Angew. Chem. Int. Ed. 2014, 53, 2711–2715. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.-L.; Yim, W.-L.; So, C.-W. An N-Heterocyclic Silylene-Stabilized Digermanium(0) Complex. Angew. Chem. Int. Ed. 2014, 53, 13155–13158. [Google Scholar] [CrossRef]
- Zhou, Y.-P.; Karni, M.; Yao, S.; Apeloig, Y.; Driess, M. A Bis(silylenyl)pyridine Zero-Valent Germanium Complex and Its Remarkable Reactivity. Angew. Chem. Int. Ed. 2016, 55, 15096–15099. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Gusev, D.; Dmitrienko, A.; Gabidullin, B.M.; Spasyuk, D.; Pilkington, M.; Nikonov, G.I. Ge(0) Compound Stabilized by a Diimino-Carbene Ligand: Synthesis and Ambiphilic Reactivity. J. Am. Chem. Soc. 2020, 142, 5852–5861. [Google Scholar] [CrossRef]
- Gonzalez, M.L.; Bousquet, L.; Hameury, S.; Alvarez Toledano, C.; Saffon-Merceron, N.; Branchadell, V.; Maerten, E.; Baceiredo, A. Phosphine/Sulfoxide-Supported Carbon(0) Complex. Chem. Eur. J. 2018, 24, 2570–2574. [Google Scholar] [CrossRef]
- Meiners, F.; Saak, W.; Weidenbruch, M. Reaction of a diarylgermylene with a phosphaalkyne: Formation of a germadiphosphacyclobutene with an exocyclic C=Ge double bond. Chem. Commun. 2001, 215–216. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Bresch, S.; Manoharan, M. Hybridization Trends for Main Group Elements and Expanding the Bent’s Rule beyond Carbon: More than Electronegativity. J. Phys. Chem. A 2014, 118, 3663–3677. [Google Scholar] [CrossRef]
- Amadoruge, M.L.; Weinert, C.S. Singly Bonded Catenated Germanes: Eighty Years of Progress. Chem. Rev. 2008, 108, 4253–4294. [Google Scholar] [CrossRef]
- Ibrahim Al-Rafia, S.M.; Momeni, M.R.; McDonald, R.; Ferguson, M.J.; Brown, A.; Rivard, E. Controlled Growth of Dichlorogermanium Oligomers from Lewis Basic Hosts. Angew. Chem. Int. Ed. 2013, 52, 6390–6395. [Google Scholar] [CrossRef]
- Fustier-Boutignon, M.; Nebra, N.; Mézailles, N. Geminal Dianions Stabilized by Main Group Elements. Chem. Rev. 2019, 119, 8555–8700. [Google Scholar] [CrossRef] [PubMed]
- SADABS. Program for Data Correction, Version 2016/2; Bruker−AXS: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 4 | 5 | 6 | |
|---|---|---|---|---|
| P1-C1 | 1.719(15) | 1.800(1) | 1.725(2) | 1.748(2) |
| S1-C1 | 1.653(13) | 1.593(1) | 1.650(2) | 1.665(2) |
| P1-C1-S1 | 120.98(9) | 120.74(8) | 113.73(14) | 112.40(10) |
| C1-Ge1 | - | - | 2.071(2) | 1.980(2) |
| Ge1-Ge2 | - | - | - | 2.582(1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Authesserre, U.; Hameury, S.; Dajnak, A.; Saffon-Merceron, N.; Baceiredo, A.; Madec, D.; Maerten, E. Complexes of Dichlorogermylene with Phosphine/Sulfoxide-Supported Carbone as Ligand. Molecules 2021, 26, 2005. https://doi.org/10.3390/molecules26072005
Authesserre U, Hameury S, Dajnak A, Saffon-Merceron N, Baceiredo A, Madec D, Maerten E. Complexes of Dichlorogermylene with Phosphine/Sulfoxide-Supported Carbone as Ligand. Molecules. 2021; 26(7):2005. https://doi.org/10.3390/molecules26072005
Chicago/Turabian StyleAuthesserre, Ugo, Sophie Hameury, Aymeric Dajnak, Nathalie Saffon-Merceron, Antoine Baceiredo, David Madec, and Eddy Maerten. 2021. "Complexes of Dichlorogermylene with Phosphine/Sulfoxide-Supported Carbone as Ligand" Molecules 26, no. 7: 2005. https://doi.org/10.3390/molecules26072005
APA StyleAuthesserre, U., Hameury, S., Dajnak, A., Saffon-Merceron, N., Baceiredo, A., Madec, D., & Maerten, E. (2021). Complexes of Dichlorogermylene with Phosphine/Sulfoxide-Supported Carbone as Ligand. Molecules, 26(7), 2005. https://doi.org/10.3390/molecules26072005