
Laguncularia racemosa Phenolics Profiling by Three-Phase Solvent System Step-Gradient Using High-Performance Countercurrent Chromatography with Off-Line Electrospray Mass-Spectrometry Detection

 ,
,
Abstract
1. Introduction
2. Results and Discussion
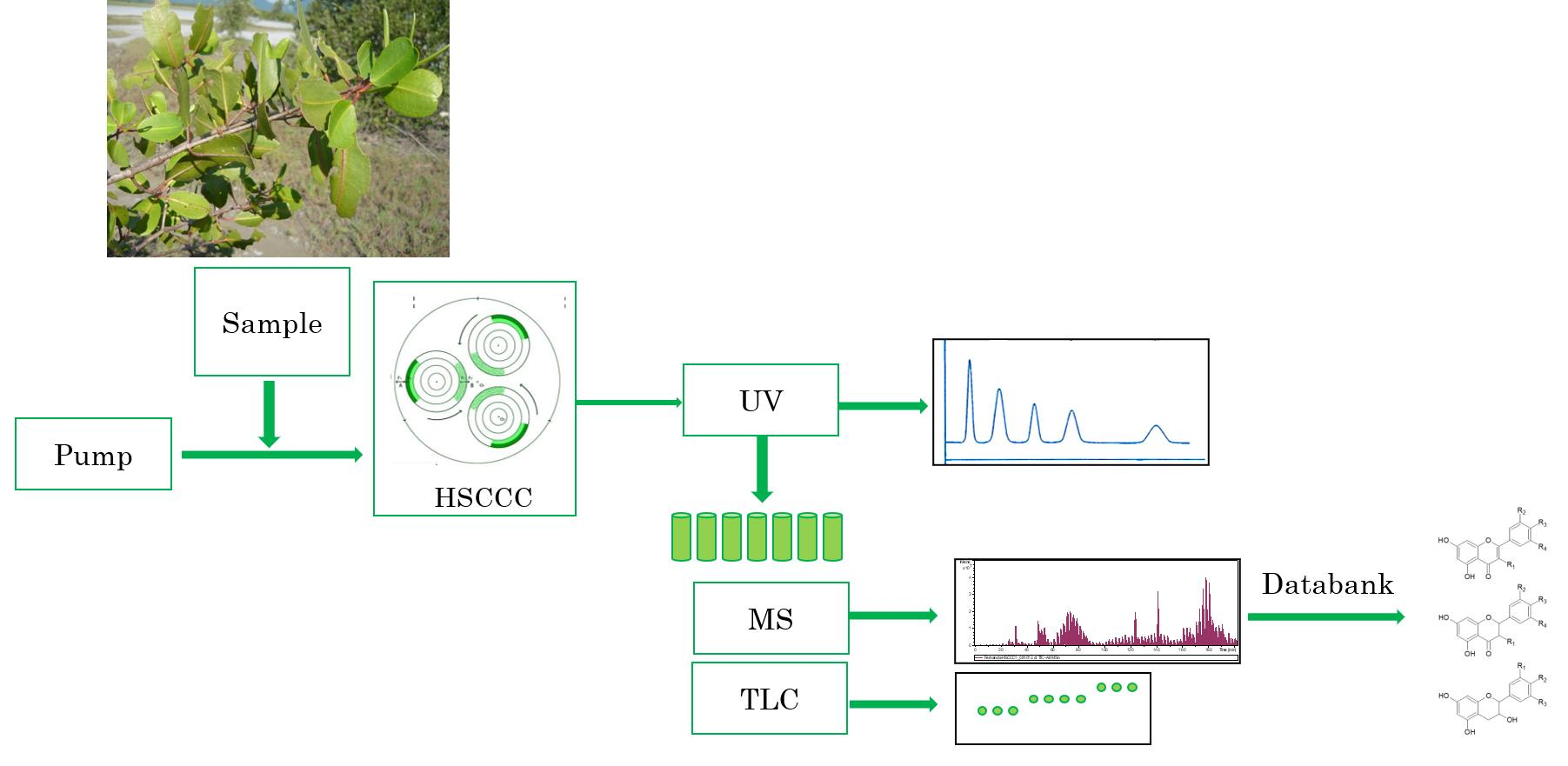
2.1. HPCCC of L. racemosa Metabolites by Off-Line ESI-MS/MS Profile Detection in the Sequential Order of Recovery
2.1.1. Flavonoids and Derivatives
2.1.2. Hydrolysable Tannins
2.1.3. Condensed Tannins
2.1.4. Low Molecular Weight Polyphenols
3. Materials and Methods
3.1. Chemical Reagents and Solvents
3.2. Preparation of the Extract
3.3. Thin Layer Chromatography
3.4. LC-ESI/TOF MS Preliminary Analysis
3.5. High Performance Countercurrent Chromatography
3.5.1. Equipment
3.5.2. Three-Phase Solvent System Test Evaluation
3.5.3. Solvent System and Sample Preparation
3.5.4. HPCCC Separation Procedure
3.6. Metabolite Profiling by Offline Injections to ESI-MS/MS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef]
- Marston, A.; Hostettmann, K. Developments in the application of counter-current chromatography to plant analysis. J. Chromatogr. A 2006, 1112, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.A. Recent progress on the industrial scale-up of counter-current chromatography. J. Chromatogr. A 2007, 1151, 6–13. [Google Scholar] [CrossRef]
- Berthod, A.; Maryutina, T.; Spivakov, B.; Shpigun, O.; Sutherland, I.A. Countercurrent chromatography in analytical chemistry (IUPAC Technical Report). Pure Appl. Chem. 2009, 81, 355–387. [Google Scholar] [CrossRef]
- Gutzeit, D.; Winterhalter, P.; Jerz, G. Application of preparative high-speed counter-current chromatography/electrospray ionization mass spectrometry for a fast screening and fractionation of polyphenols. J. Chromatogr. A 2007, 1172, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rivera, M.P.; Lugo-Cervantes, E.; Winterhalter, P.; Jerz, G. Metabolite profiling of polyphenols in peels of Citrus limetta Risso by combination of preparative high-speed countercurrent chromatography and LC–ESI–MS/MS. Food Chem. 2014, 158, 139–152. [Google Scholar] [CrossRef]
- Vieira, M.N.; Costa, F.N.; Leitão, G.G.; Garrard, I.; Hewitson, P.; Ignatova, S.; Winterhalter, P.; Jerz, G. Schinus tere-binthifolius scale-up countercurrent chromatography (Part I): High performance countercurrent chromatography fractionation of triterpene acids with off-line detection using atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A. 2015, 1389, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.D.N.; Borges, R.M.; Leitão, G.G.; Jerz, G. Preparative mass-spectrometry profiling of minor concentrated metabolites in Salicornia gaudichaudiana Moq by high-speed countercurrent chromatography and off-line electrospray mass-spectrometry injection. J. Sep. Sci. 2019, 42, 1528–1541. [Google Scholar] [CrossRef] [PubMed]
- Berthod, A.; Ruiz-Angel, M.-J.; Carda-Broch, S. Countercurrent chromatography: People and applications. J. Chromatogr. A 2009, 1216, 4206–4217. [Google Scholar] [CrossRef]
- Huang, X.-Y.; Ignatova, S.; Hewitson, P.; Di, D.-L. An overview of recent progress in elution mode of counter current chro-matography. Trends Anal. Chem. 2016, 77, 214–225. [Google Scholar] [CrossRef]
- Shibusawa, Y.; Yamakawa, Y.; Noji, R.; Yanagida, A.; Shindo, H.; Ito, Y. Three-phase solvent systems for comprehensive separation of a wide variety of compounds by high-speed counter-current chromatography. J. Chromatogr. A 2006, 1133, 119–125. [Google Scholar] [CrossRef]
- Yanagida, A.; Yamakawa, Y.; Noji, R.; Oda, A.; Shindo, H.; Ito, Y.; Shibusawa, Y. Comprehensive separation of secondary metabolites in natural products by high-speed counter-current chromatography using a three-phase solvent system. J. Chromatogr. A 2007, 1151, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, S.; Long, L.; Yin, H.; Tian, X.; Luo, X.; Nan, H.; He, S. The separation of flavonoids from Pongamia pinnata using combination columns in high-speed counter-current chromatography with a three-phase solvent system. J. Chromatogr. A 2013, 1315, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Shinomiya, K.; Ito, Y. Countercurrent Chromatographic Separation of Biotic Compounds with Extremely Hydrophilic Or-ganic-Aqueous Two-Phase Solvent Systems and Organic-Aqueous Three-Phase Solvent Systems. J. Liq. Chromatogr. Rel. Technol. 2006, 29, 733–750. [Google Scholar] [CrossRef]
- Hamzaoui, M.; Renault, J.-H.; Nuzillard, J.-M.; Reynaud, R.; Hubert, J. Stepwise Elution of a Three-phase Solvent System in Centrifugal Partition Extraction: A New Strategy for the Fractionation and Phytochemical Screening of a Crude Bark Extract. Phytochem. Anal. 2013, 24, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Leitão, G.G.; Costa, F.D.N. Gradient Elution in Countercurrent Chromatography. Planta Medica 2015, 81, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.D.N.; Garrard, I.; Da Silva, A.J.R.; Leitão, G.G. Changes in the mobile phase composition on a stepwise counter-current chromatography elution for the isolation of flavonoids from Siparuna glycycarpa. J. Sep. Sci. 2013, 36, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- The Plant List. Available online: http://www.theplantlist.org/tpl1.1/search?q=laguncularia+racemosa (accessed on 6 December 2019).
- Dodd, R.S.; Rafii, Z.A.; Fromard, F.; Blasco, F. Evolutionary diversity among Atlantic coast mangroves. Acta Oecol. 1998, 19, 323–330. [Google Scholar] [CrossRef]
- Lallier-Vergès, E.; Marchand, C.; Disnar, J.-R.; Lottier, N. Origin and diagenesis of lignin and carbohydrates in mangrove sediments of Guadeloupe (French West Indies): Evidence for a two-step evolution of organic deposits. Chem. Geol. 2008, 255, 388–398. [Google Scholar] [CrossRef]
- Nyananyo, B.; Briyai, F.; Kiesekime, S. Laguncularia recemosa (L.) Gaertner f. (Family Combretaceae): Gross morphology, phytochemistry, ecology and distribution in the Niger Delta. J. Appl. Sci. Environ. Manag. 2010, 13, 47–49. [Google Scholar] [CrossRef][Green Version]
- Frerichs, G.; Arends, G.; Zörnig, H. Hager’s Handbuch der Pharmazeutischen Praxis; Springer: Berlin/Heidelberg, Germany, 1994. [Google Scholar]
- Xue, D.-Q.; Wang, J.-D.; Guo, Y.-W. A new sulphated nor-sesquiterpene from mangrove Laguncularia racemosa (L.) Gaertn. F. J. Asian Nat. Prod. Res. 2008, 10, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.F.B.; Gaeta, H.H.; Belchor, M.N.; Ferreira, M.J.F.; Pinho, M.V.T.; Toyama, D.O.; Toyama, M.H. Evaluation of Potential Thrombin Inhibitors from the White Mangrove (Laguncularia racemosa (L.) C.F. Gaertn.). Mar. Drugs 2015, 13, 4505–4519. [Google Scholar] [CrossRef] [PubMed]
- Baxter, H.; Harborne, J.B.; Moss, G.P. Phytochemical Dictionary: A Handbook of Bioactive Compounds from Plants; Taylor & Francis: London, UK, 1999; p. 598. [Google Scholar]
- Shi, C.; Xu, M.-J.; Bayer, M.; Deng, Z.-W.; Kubbutat, M.H.; Waejen, W.; Proksch, P.; Lin, W.-H. Phenolic compounds and their anti-oxidative properties and protein kinase inhibition from the Chinese mangrove plant Laguncularia racemosa. Phytochemistry 2010, 71, 435–442. [Google Scholar] [CrossRef]
- Koch, B.P.; Rullkötter, J.; Lara, R.J. Evaluation of triterpenols and sterols as organic matter biomarkers in a mangrove eco-system in northern Brazil. Wetl. Ecol. Manag. 2003, 11, 257–263. [Google Scholar] [CrossRef]
- Costa, F.N.; Leitão, G.G. Strategies of solvent system selection for the isolation of flavonoids by countercurrent chroma-tography. J. Sep. Sci. 2010, 33, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, D.; Liang, J.; Berthod, A. Modeling gradient elution in countercurrent chromatography: Efficient separation of tanshinones from Salvia miltiorrhiza Bunge. J. Sep. Sci. 2012, 35, 964–976. [Google Scholar] [CrossRef]
- Ignatova, S.; Sumner, N.; Colclough, N.; Sutherland, I. Gradient elution in counter-current chromatography: A new layout for an old path. J. Chromatogr. A 2011, 1218, 6053–6060. [Google Scholar] [CrossRef]
- McNab, H.; Ferreira, E.S.B.; Hulme, A.N.; Quye, A. Negative ion ESI–MS analysis of natural yellow dye flavonoids—An isotopic labelling study. Int. J. Mass. Spectrom. 2009, 284, 5757–5765. [Google Scholar] [CrossRef]
- Fabre, N.; Rustan, I.; Hoffmann, E.; Quetin-Leclercq, J. Determination of flavone, flavonol, and flavanone aglycones by neg-ative ion liquid chromatography electrospray ion trap mass spectrometry. J. Am. Soc. Mass. Spectrom. 2001, 12, 707–715. [Google Scholar] [CrossRef]
- Cuyckens, F.; Claeys, M. Mass spectrometry in the structural analysis of flavonoids. J. Mass Spectrom. 2004, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.F.; Justino, G.C. Structural Analysis of Flavonoids and Related Compounds—A Review of Spectroscopic Applications. In Phytochemicals—A Global Perspective of Their Role in Nutrition and Health; IntechOpen: London, UK, 2012. [Google Scholar]
- Koolen, H.H.F.; Silva, F.M.A.; Gozzo, F.C.; Souza, A.Q.L.; Souza, A.D.L. Antioxidant, antimicrobial activities and characteri-zation of phenolic compounds from buriti (Mauritia flexuosa L. f.) by UPLC–ESI-MS/MS. Food Res. Int. 2013, 51, 467–473. [Google Scholar] [CrossRef]
- Singh, A.P.; Wilson, T.; Luthria, D.; Freeman, M.R.; Scott, R.M.; Bilenker, D.; Shah, S.; Somasundaram, S.; Vorsa, N. LC-MS–MS characterisation of curry leaf flavonols and antioxidant activity. Food Chem. 2011, 127, 80–85. [Google Scholar] [CrossRef]
- Chen, L.; Qi, J.; Chang, Y.-X.; Zhu, D.; Yu, B. Identification and determination of the major constituents in Traditional Chinese Medicinal formula Danggui-Shaoyao-San by HPLC–DAD–ESI-MS/MS. J. Pharm. Biomed. Anal. 2009, 50, 127–137. [Google Scholar] [CrossRef]
- Lee, J.-H.; Johnson, J.V.; Talcott, S.T. Identification of Ellagic Acid Conjugates and Other Polyphenolics in Muscadine Grapes by HPLC-ESI-MS. J. Agric. Food Chem. 2005, 53, 6003–6010. [Google Scholar] [CrossRef] [PubMed]
- Zehl, M.; Braunberger, C.; Conrad, J.; Crnogorac, M.; Krasteva, S.; Vogler, B.; Beifuss, U.; Krenn, L. Identification and quan-tification of flavonoids and ellagic acid derivatives in therapeutically important Drosera species by LC–DAD, LC–NMR, NMR, and LC–MS. Anal. Bioanal. Chem. 2011, 400, 2565–2576. [Google Scholar] [CrossRef]
- Fracassetti, D.; Costa, C.; Moulay, L.; Tomás-Barberán, F.A. Ellagic acid derivatives, ellagitannins, proanthocyanidins and other phenolics, vitamin C and antioxidant capacity of two powder products from camu-camu fruit (Myrciaria dubia). Food Chem. 2013, 139, 578–588. [Google Scholar] [CrossRef]
- Negri, G.; Tabach, R. Saponins, tannins and flavonols found in hydroethanolic extract from Periandra dulcis roots. Rev. Bras. de Farm. 2013, 23, 851–860. [Google Scholar] [CrossRef]
- Wyrepkowski, C.C.; Da Costa, D.L.M.G.; Sinhorin, A.P.; Vilegas, W.; De Grandis, R.A.; Resende, F.A.; Varanda, E.A.; Dos Santos, L.C. Characterization and Quantification of the Compounds of the Ethanolic Extract from Caesalpinia ferrea Stem Bark and Evaluation of Their Mutagenic Activity. Molecules 2014, 19, 16039–16057. [Google Scholar] [CrossRef]
- Ferreres, F.; Grosso, C.; Gil-Izquierdo, A.; Valentão, P.; Andrade, P.B. Ellagic acid and derivatives from Cochlospermum ango-lensis Welw. Extracts: HPLC–DAD–ESI/MSn profiling, quantification and in vitro anti-depressant, anti-cholinesterase and anti-oxidant activities. Phytochem. Anal. 2013, 24, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.C.; Chien, S.C.; Chen, H.F.; Hsu, F.L. Tannins and related compounds from Combretaceae plants. J. Chin. Pharm. Sci. 2000, 52, 1–26. [Google Scholar]
- Okuda, T.; Yoshida, T.; Hatano, T. Classification of oligomeric hydrolysable tannins and specificity of their occurrence in plants. Phytochemistry 1993, 32, 507–521. [Google Scholar] [CrossRef]
- Chaabi, M.; Benayache, S.; Benayache, F.; N’Gom, S.; Koné, M.; Anton, R.; Weniger, B.; Lobstein, A. Triterpenes and polyphenols from Anogeissus leiocarpus (Combretaceae). Biochem. Syst. Ecol. 2008, 36, 59–62. [Google Scholar] [CrossRef]
- Barry, K.M.; Davies, N.W.; Mohammed, C.L. Identification of hydrolysable tannins in the reaction zone of Eucalyptus nitens wood by high performance liquid chromatography-electrospray ionisation mass spectrometry. Phytochem. Anal. 2001, 12, 120–127. [Google Scholar] [CrossRef]
- McKee, K.L. Interspecific variation in growth, biomass partitioning, and defensive characteristics of neotropical mangrove seedlings: Response to light and nutrient availability. Am. J. Bot. 1995, 82, 299–307. [Google Scholar] [CrossRef]
- Vargas-Magaña, J.J.; Torres-Acosta, J.F.J.; Aguilar-Caballero, A.J.; Sandoval-Castro, C.A.; Hoste, H.; Chan-Péreza, J.I. An-thelmintic activity of acetone–water extracts against Haemonchus contortus eggs: Interactions between tannins and other plant secondary compounds. Vet. Parasitol. 2014, 206, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Maie, N.; Pisani, O.; Jaffé, R. Mangrove tannins in aquatic ecosystems: Their fate and possible influence on dissolved organic carbon and nitrogen cycling. Limnol. Oceanogr. 2008, 53, 160–171. [Google Scholar] [CrossRef]
- De Souza, L.M.; Cipriani, T.R.; Iacomini, M.; Gorin, P.A.; Sassaki, G.L. HPLC/ESI-MS and NMR analysis of flavonoids and tannins in bioactive extract from leaves of Maytenus ilicifolia. J. Pharm. Biomed. Anal. 2008, 47, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Rubino, M.T.; Maggi, D.; Laghezza, A.; Loiodice, F.; Tortorella, P. Identification of Novel Matrix Metalloproteinase Inhibitors by Screening of Phenol Fragments Library. Arch. Pharm. 2011, 344, 557–563. [Google Scholar] [CrossRef]
- Wang, F.; Li, R.; Long, L.; Tian, X.; Xiao, Z.; Zhang, S.; Yin, H. A Three-Phase Solvent System in High-Speed Counter-Current Chromatographic for the Separation and Purification of Bioactive Constituents from Acanthus ilicifolius. Chromatographia 2015, 78, 1401–1407. [Google Scholar] [CrossRef]
- Wu, X.; Chao, Z.; Wang, C.; Yu, L. Separation of chemical constituents from three plant medicines by counter-current chro-matography using a three-phase solvent system at a novel ratio. J. Chromatogr. A 2015, 1384, 107–114. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | CCC-Fraction | MS [M – H]−(m/z) MS/MS [M – H]− (m/z) | LC-RT(min) | ESI/TOF MS Formula (Error in ppm) | Identification |
|---|---|---|---|---|---|
| Flavonoids and derivatives | |||||
| 1 | 11–15 | 255 237, 226, 209, 156 | n.d. | - | Dihydrocrysin |
| 2 | 21 | 269 151 | 13.7 | 269.04593 C15H9O5 (1.4) | Apigenin |
| 3 | 19–21 | 271 177, 151 | 1.9 | 271.04885 C15H11O5 (45.6) * | Naringenin |
| 4 | 23 | 273 167 | 29.1 | 273.08108 C15H13O5 (15.5) | Afzelechin |
| 5 | 19 | 285 257, 151 | 40.8 | 285.04413 C15H9O6 (12.6) | Kaempferol |
| 6 | 31–41 | 287 259, 151 | 12.1 | 287.05883 C15H11O6 (9.5) | Dihydrokaempferol |
| 7 | 97–115 | 289 245, 205 | 6.2 | 289.07438 C15H13O6 (9.0) | (Epi)-catechin |
| 8 | 29–33 | 301 179, 151 | 35.4 | 301.03937 C15H9O7 (13.3) | Quercetin |
| 9 | 17–19 | 305 287, 249 | 2.4 | 305.0706 C15H13O7 (12.8) | (Epi)-gallocatechin |
| 10 | 21–23 | 315 300 | 42.8 | 315.0466 C16H11O7 (14.1) | Isorhamnetin |
| 11 | 25–27 | 317 179, 151 | 28.9 | 317.03536 C15H9O8 (16.0) | Myricetin |
| 12 | 81–93 | 319 193 | 9.1 | 319.04883 C15H11O8 (9.0) | Dihydromyricetin |
| 13 | 33–41 | 329 314, 299 | 21.7 | 329.05816 C17H13O7 (25.9) * | Tricin |
| 14 | 133–149 | 393 317, 241, 169 | - | - | Myricetin derivative |
| 15 | 29–33 | 415 301 | - | - | Quercetin alkyl derivative |
| 16 | 127–131 | 419 305 | - | - | (Epi)-gallocatechin alkyl derivative |
| 17 | 55–57 | 431 317 | - | - | Myricetin alkyl derivative |
| 18 | 97–105 | 433 301, 179, 151 | 27.5 | 433.08215 C20H17O11 (10.4) | Quercetin pentoside |
| 19 | 89–91 | 433 319, 193 | - | - | Dihydromyricetin alkyl derivative |
| 20 | 91–95 | 441 289 | 19.3 | 441.08208 C22H18O10 (1.5) | (Epi)-catechin gallate |
| 21 | 105–119 | 447 301 | 29.3 | 447.09851 C21H19O11 (11.7) | Quercetin desoxyhexoside |
| 22 | 115–133 | 449 317, 316 | 22.4 | 449.07395 C20H17O12 (3.1) | Myricetin pentoside |
| 23 | 113–127 | 457 331, 305, 169 | 12.1 | 457.07859 C22H18O11 (2.1) | (Epi)-gallocatechin gallate |
| 24 | 21–23 | 461 443, 381, 301, 193 | - | - | Quercetin derivative |
| 25 | 123–131 | 463 317, 316 | 24.6 | 463.09083 C21H19O12 (5.7) | Myricetin desoxyhexoside |
| 26 | 141–145 | 463 301 | 25.9 | 463.09187 C21H19O12 (7.9) | Quercetin hexoside |
| 27 | 153–165 | 467 458, 391, 301, 169 | - | - | Quercetin derivative |
| 28 | 67–79 | 469 317 | n.d. | - | Myricetin galatte |
| 29 | 85–91 | 471 319, 301, 193 | - | - | Dihydromyricetin alkyl derivative |
| 30 | 115–131 | 477 301, 179 | 15.2 | 477.06812 C21H17O13 (1.4) | Quercetin glucuronide |
| 31 | 133–163 | 479 317, 316 | 22.1 | 479.08428 C21H19O14 (2.4) | Myricetin hexoside |
| 32 | 89–97 | 585 433, 301 | 32.7 | 585.09204 C27H21O15 (5.9) | Quercetin pentoside gallate |
| 33 | 85–95 | 599 447, 301 | 26.8 | 599.10714 C28H23O15 (4.8) | Quercetin desoxyhexoside gallate |
| 34 | 95–113 | 601 449, 317 | 28.6 | 601.08787 C27H21O16 (7.3) | Myricetin pentoside gallate |
| 35 | 29–33 | 603 301 | n.d. | - | Quercetin [2M − H]− |
| 36 | 125–131 | 611 305 | - | - | (Epi)-gallocatechin [2M − H]− |
| 37 | 91–115 | 615 463, 317, 179 | 23.4 | 615.1014 C28H23O16 (3.6) | Myricetin desoxyhexoside gallate |
| 38 | 137–153 | 615 463, 301 | 31.6 | 615.10412 C28H23O16 (8.1) | Quercetin hexoside gallate |
| 39 | 113–127 | 629 477, 317, 316, 289 | 21.5 | 629.07893 C28H21O17 (0.8) | Quercetin glucuronide gallate |
| 40 | 133–139 | 631 479, 317 | 28.9 | 631.09859 C28H23O17 (7.2) | Myricetin hexoside gallate |
| 41 | 45–57 | 635 317 | n.d. | - | Myricetin [2M − H]− |
| 42 | 87–91 | 639 319, 301 | 11.2 | 639.05562 C29H19O17 (11.2) | HHDP Dihydromyricetin |
| 43 | 25 | 657 317 | - | - | Myricetin derivative |
| 44 | 89 | 697 599 | - | - | Quercetin desoxyhexoside gallate derivative |
| 45 | 89–93 | 737 585, 301 | n.d. | - | Quercetin pentoside digalloyl |
| 46 | 93–103 | 753 601, 449, 317 | 32.5 | 753.09740 C34H25O20 (3.9) | Myricetin pentoside digalloyl |
| 47 | 85–89 | 773 471, 301 | - | - | Quercetin derivative |
| 48 | 97–103 | 867 433, 301 | n.d. | - | Quercetin pentoside [2M − H]− |
| 49 | 117–127 | 883 449, 317 | - | - | Myricetin pentoside derivative |
| 50 | 115–123 | 892 457, 433 | - | - | (Epi)-gallocatechin gallate derivative |
| 51 | 117–127 | 899 463, 449, 317 | - | - | Myricetin pentoside derivative |
| 52 | 87–93 | 901 599, 301 | - | - | Quercetin desoxyhexoside gallate derivative |
| 53 | 111–121 | 905 469, 457, 447, 425, 301 | - | - | Quercetin desoxyhexoside derivative |
| 54 | 113–125 | 907 449, 317 | - | - | Myricetin pentoside derivative |
| 55 | 113–123 | 915 457 | - | - | (Epi)-gallocatechin gallate [2M − H]− |
| 56 | 129–131 | 927 463, 317 | n.d. | - | Myricetin desoxyhexoside [2M − H]− |
| Hydrolisable tannins and deivatives | |||||
| 57 | 93–115 | 169 125 | 12.1 | 169.01664 C7H5O5 (14.2) | Gallic acid |
| 58 | 61–81 | 183 124 | 5.9 | 183.01418 C8H7O5 (12.2) | Methyl gallate |
| 59 | 43–57 | 197 169, 125 | 14.3 | 197.04741 C9H9O5 (9.5) | Ethyl gallate |
| 60 | 81–93 | 301 283, 257, 229, 163 | 18.5 | 300.99939 C14H5O8 (1.3) | Ellagic acid |
| 61 | 19–21 | 315 300 | 6.1 | 315.01809 C15H7O8 (10.9) | Ellagic acid methyl ether |
| 62 | 97–103 | 321 169 | 3.9 | 321.03300 C14H9O9 (24.3) * | Galloyl gallate |
| 63 | 133–151 | 325 169 | 3.3 | 325.06016 C14H13O9 (11.3) | Galloyl shikimate |
| 64 | 33–39 | 329 314 | 44.2 | 329.02154 C16H9O8 (8.9) | Ellagic acid dimethyl ether |
| 65 | 163 | 331 271, 169, 125 | 11.5 | 331.06888 C13H15O10 (5.5) | Galloyl hexoside |
| 66 | 81–87 | 335 183 | 9.2 | 335.02817 C15H11O9 (37.9) * | Galloyl methyl gallate |
| 67 | 21–23 | 343 328 | 44.1 | 343.04787 C17H11O8 (5.6) | Ellagic acid trimethyl ether |
| 68 | 59–79 | 349 197 | 13.9 | 349.0416 C16H13O9 (42.7) * | Galloyl ethyl gallate |
| 69 | 105–119 | 425 301 | 15.8 | 425.01469 C20H9O11 (0.8) | Ellagic acid pyrogallol ether |
| 70 | 103–119 | 469 425 | 15.7 | 469.0039 C21H9O13 (2.1) | Valoneic acid dilactone |
| 71 | 161–165 | 481 439, 331, 301, 169 | 1.2 | 481.06556 C20H17O14 (6.6) | HHDP hexoside |
| 72 | 155–165 | 483 439, 331, 313, 169 | 11.5 | 483.07806 C20H19O14 (0.1) | Digalloyl hexoside |
| 73 | 85–89 | 497 301 | 23.4 | 497.03631 C23H13O13 (0.3) | Valoneic acid dilactone ethyl ether |
| 74 | 83–91 | 625 471, 301 | 28.6 | 625.07458 C26H25O18 (28.1) * | Ellagic acid dihexoside |
| 75 | 147–155 | 631 479, 301 | 19.4 | 631.09323 C27H19O18 (26.3) * | NHDP hexoside |
| 76 | 155–165 | 633 479, 301 | 7.8 | 633.07511 C27H21O18 (2.8) | HHDP galloyl hexoside |
| 77 | 133–151 | 635 483, 465, 313 | 15.6 | 635.08832 C27H23O18 (1.0) | Trigalloyl hexoside |
| 78 | 135–139 | 733 635 | n.d. | - | Trigalloyl hexoside derivative |
| 79 | 149 | 781 631, 301 | 3.7 | 781.06132 C34H21O22 (10.7) | Punicalin |
| 80 | 155–167 | 783 481, 301 | 2.7 | 783.07063 C34H23O22 (2.5) | DiHHDP hexoside |
| 81 | 133–165 | 785 633, 481, 301, 275 | 9.6 | 785.08378 C34H25O22 (0.7) | HHDP digalloyl hexoside |
| 82 | 133–155 | 787 635, 617, 483, 465, 301 | 21.1 | 787.09741 C34H27O22 (3.2) | Tetragalloyl hexoside |
| 83 | 145–167 | 935 917, 633, 571, 365, 329, 299, 275 | 6.0 | 935.07728 C41H27O26 (2.5) | Galloyl diHHDP hexoside |
| 84 | 131–169 | 937 785, 769, 633, 617, 301 | 6.0 | 937.28345 C41H29O26 (12.6) * | HHDP trigalloyl hexoside |
| 85 | 155–167 | 939 787, 769, 617, 465 | 26.2 | 939.11228 C41H31O26 (1.5) | Pentagalloyl hexoside |
| 86 | 155–169 | 951 907, 783, 605 | - | - | DiHHDP hexoside derivative |
| Condensed tannins | |||||
| 87 | 121–133 | 577 463, 425, 313, 289 | 3.9 | 577.13515 C30H25O12 (2.9) | (Epi)-catechin dimer |
| 88 | 123–125 | 593 575, 467, 441, 425, 305 | 2.6 | 593.15119 C30H25O13 (4.3) | (Epi)-catechin-(epi)-gallocatechin dimer |
| 89 | 107–119 | 609 457, 439, 321, 169 | 4.1 | 609.12858 C30H25O14 (5.9) | (Epi)-gallocatechin dimer |
| 90 | 125–129 | 897 745, 575, 463, 449, 423 | 13.0 | 897.14880 C44H33O21 (3.6) | (Epi)-catechin gallate -(epi)-gallocatechin gallate dimer |
| 91 | 123–131 | 913 463, 449, 317 | 8.0 | 913.14548 C44H33O22 (1.5) | (Epi)-gallocatechin gallate dimer |
| 92 | 137–155 | 913 761, 573, 449, 423 | 24.6 | 913.16762 C45H37O21 (17.1) | (Epi)-gallocatechin trimer |
| Others | |||||
| 93 | 73–85 | 109 - | 3.0 | 109.02893 C6H5O2 (5.3) | Catechol |
| 94 | 67–81 | 124 - | n.d. | - | Amino catechol |
| 95 | 93–115 | 125 - | 12.1 | 125.02756 C6H5O3 (17.2) | Pyrrogallol |
| 96 | 77–83 | 153 109 | 9.1 | 153.02038 C7H5O4 (6.9) | Protocatechuic acid |
| 97 | 23–25 | 167 125 | 9.1 | 167.03454 C8H7O4 (2.6) * | Vanillic acid |
| 98 | 67–79 | 168 124 | n.d. | - | Amino protocatechuic acid |
| 99 | 85–95 | 193 111 | 9.1 | 193.01665 C9H5O5 (12.5) | Trihydroxychromone |
| 100 | 15 | 209 187, 165, 125 | n.d. | - | Jasmonic acid |
| 101 | 59–69 | 217 155 | - | - | Unknown |
| 102 | 13–15 | 279 277, 243, 237 | 73.4 | 279.23401 C18H31O2 (3.8) | Linoleic acid |
| 103 | 281 277, 255 | 75.8 | 281.24987 C18H33O2 (4.5) | Oleic acid | |
| 104 | 11–15 | 295 277, 275, 265, 251, 249, 185 | 70.2 | 295.2304 C18H31O3 (8.6) | Hydroxy linoleic acid |
| 105 | 125–131 | 305 221, 219, 179, 165, 125 | 12.1 | 305.06942 C12H17O7S (2.1) | 5′-hydroxysulphonyloxy jasmonic acid |
| 106 | 11–17 | 383 337 | - | - | Unknown |
| 107 | 157–159 | 707 687, 671, 533, 359 | n.d. | - | Integracin D |
| 108 | 97–99 | 875 441, 433, 289 | - | - | Unknown |
| 109 | 89–93 | 887 585, 301 | - | - | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, F.d.N.; Jerz, G.; Hewitson, P.; Figueiredo, F.d.S.; Ignatova, S. Laguncularia racemosa Phenolics Profiling by Three-Phase Solvent System Step-Gradient Using High-Performance Countercurrent Chromatography with Off-Line Electrospray Mass-Spectrometry Detection. Molecules 2021, 26, 2284. https://doi.org/10.3390/molecules26082284
Costa FdN, Jerz G, Hewitson P, Figueiredo FdS, Ignatova S. Laguncularia racemosa Phenolics Profiling by Three-Phase Solvent System Step-Gradient Using High-Performance Countercurrent Chromatography with Off-Line Electrospray Mass-Spectrometry Detection. Molecules. 2021; 26(8):2284. https://doi.org/10.3390/molecules26082284
Chicago/Turabian StyleCosta, Fernanda das Neves, Gerold Jerz, Peter Hewitson, Fabiana de Souza Figueiredo, and Svetlana Ignatova. 2021. "Laguncularia racemosa Phenolics Profiling by Three-Phase Solvent System Step-Gradient Using High-Performance Countercurrent Chromatography with Off-Line Electrospray Mass-Spectrometry Detection" Molecules 26, no. 8: 2284. https://doi.org/10.3390/molecules26082284
APA StyleCosta, F. d. N., Jerz, G., Hewitson, P., Figueiredo, F. d. S., & Ignatova, S. (2021). Laguncularia racemosa Phenolics Profiling by Three-Phase Solvent System Step-Gradient Using High-Performance Countercurrent Chromatography with Off-Line Electrospray Mass-Spectrometry Detection. Molecules, 26(8), 2284. https://doi.org/10.3390/molecules26082284