
New Crystalline Salts of Nicotinamide Riboside as Food Additives

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
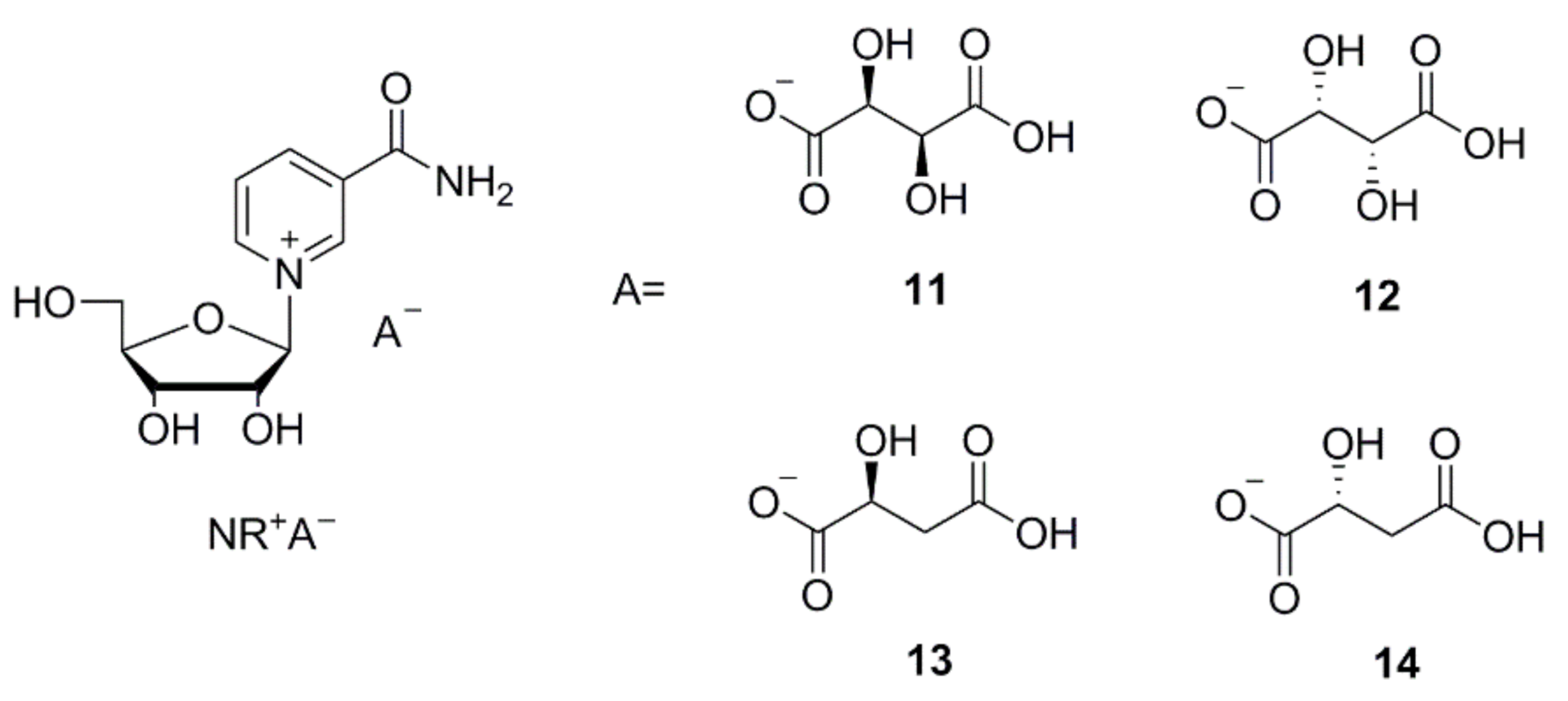
2.1. Chemical Synthesis
2.2. Single Crystal Growth Experiments
2.3. X-ray Single Crystal Diffraction
2.4. Stability Studies of NR+ Salts 1–4
3. Results
3.1. Preparation of NR+ Neutral Salts
3.2. Preparation of NR+ Acidic Salts
3.3. Single Crystal Growth of Newly Synthesized NR+ Salts
3.4. Properties of the New Crystalline NR+ Salts
3.5. Improved Synthetic Approach via the Triflate NR+
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Chini, C.C.; Tarragó, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Hirschey, M.D. Old Enzymes, New Tricks: Sirtuins Are NAD+-Dependent De-acylases. Cell Metab. 2011, 14, 718–719. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD + in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Niere, M. NAD+ surfaces again. Biochem. J. 2004, 382, E5–E6. [Google Scholar] [CrossRef] [PubMed]
- A Sauve, A.; Youn, D.Y. Sirtuins: NAD+-dependent deacetylase mechanism and regulation. Curr. Opin. Chem. Biol. 2012, 16, 535–543. [Google Scholar] [CrossRef]
- Imai, S.-I.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Sassone-Corsi, P. Minireview: NAD+, a Circadian Metabolite with an Epigenetic Twist. Endocrinol. 2012, 153, 1–5. [Google Scholar] [CrossRef]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal. 2019, 30, 251–294. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M. Vitamins and aging: Pathways to NAD+ synthesis. Cell 2007, 129, 453–454. [Google Scholar] [CrossRef]
- Hosseini, L.; Vafaee, M.S.; Mahmoudi, J.; Badalzadeh, R. Nicotinamide adenine dinucleotide emerges as a therapeutic target in aging and ischemic conditions. Biogerontology 2019, 20, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Han, J.; Xia, W.; Shi, S.; Liu, J.; Ying, W. NAD+ administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci. Lett. 2012, 512, 67–71. [Google Scholar] [CrossRef]
- Cattelan, A.; Ceolotto, G.; Bova, S.; Albiero, M.; Kuppusamy, M.; De Martin, S.; Semplicini, A.; Fadini, G.P.; de Kreutzenberg, S.V.; Avogaro, A. NAD+-dependent SIRT1 deactivation has a key role on ischemia–reperfusion-induced apoptosis. Vasc. Pharmacol. 2015, 70, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, R.; Pirnes-Karhu, S.; Pietiläinen, K.H.; Pirinen, E. Adipose tissue NAD+-homeostasis, sirtuins and poly(ADP-ribose) polymerases -important players in mitochondrial metabolism and metabolic health. Redox Biol. 2017, 12, 246–263. [Google Scholar] [CrossRef]
- Nielsen, K.N.; Peics, J.; Ma, T.; Karavaeva, I.; Dall, M.; Chubanava, S.; Basse, A.L.; Dmytriyeva, O.; Treebak, J.T.; Gerhart-Hines, Z. NAMPT-mediated NAD biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol. Metab. 2018, 11, 178–188. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Yoshino, J. Adipose tissue NAD+biology in obesity and insulin resistance: From mechanism to therapy. BioEssays 2017, 39, 1600227. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J.; Wu, J.; Jin, Z.; Zheng, H. Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes, Metab. Syndr. Obesity Targets Ther. 2016, 9, 145–153. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD+/NADH Redox State and Diabetic Cardiomyopathy. Antioxidants Redox Signal. 2019, 30, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Elhassan, Y.S.; Philp, A.A.; Lavery, G.G. Targeting NAD+ in Metabolic Disease: New Insights Into an Old Molecule. J. Endocr. Soc. 2017, 1, 816–835. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Figley, M.D.; DiAntonio, A. The SARM1 axon degeneration pathway: Control of the NAD+ metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol. 2020, 63, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S.-I. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Mehmel, M.; Jovanović, N.; Spitz, U. Nicotinamide Riboside—The Current State of Research and Therapeutic Uses. Nutr. 2020, 12, 1616. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Bieganowski, P.; Brenner, C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Douek, I.F.; Moore, W.P.T.; Gillmor, H.A.; McLean, A.E.M.; Bingley, P.J.; Gale, E.A.M. Safety of high-dose nicotinamide: A review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Conze, D.B.; Crespo-Barreto, J.; Kruger, C.L. Safety assessment of nicotinamide riboside, a form of vitamin B3. Hum. Exp. Toxicol. 2016, 35, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Ishtiaq, Y.; Nkrumah-Elie, Y.; Idoine, R.; Roberts, M.; Shao, A. Application of the Frameworks of Intrinsic Capacity and the Hallmarks of Aging to Validate Nicotinamide Riboside as a Healthy Aging Supplement—A Meta-Analysis. Curr. Dev. Nutr. 2020, 4, 36. [Google Scholar] [CrossRef]
- Fletcher, R.S.; Ratajczak, J.; Doig, C.L.; Oakey, L.A.; Callingham, R.; Xavier, G.D.S.; Garten, A.; Elhassan, Y.S.; Redpath, P.; Migaud, M.; et al. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol. Metab. 2017, 6, 819–832. [Google Scholar] [CrossRef]
- Kulikova, V.; Shabalin, K.; Nerinovski, K.; Dölle, C.; Niere, M.; Yakimov, A.; Redpath, P.; Khodorkovskiy, M.; Migaud, M.E.; Ziegler, M.; et al. Generation, Release, and Uptake of the NAD Precursor Nicotinic Acid Riboside by Human Cells. J. Biol. Chem. 2015, 290, 27124–27137. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Sci. 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Makarov, M.V.; E Migaud, M. Syntheses and chemical properties of β-nicotinamide riboside and its analogues and derivatives. Beilstein J. Org. Chem. 2019, 15, 401–430. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Schabert, G.; Soydemir, A.; Wick, L.; Spitz, U.; Spingler, B. Nicotinamide Riboside Derivatives: Single Crystal Growth and Determination of X-ray Structures. Cryst. Growth Des. 2019, 19, 4019–4028. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Gowda, G.A.N.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ Precursor Nicotinamide Riboside Enhances Oxidative Metabolism and Protects against High-Fat Diet-Induced Obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Racette, F.G.; Bogan, K.L.; McClure, J.M.; Smith, J.S.; Brenner, C. Nicotinamide Riboside Promotes Sir2 Silencing and Extends Lifespan via Nrk and Urh1/Pnp1/Meu1 Pathways to NAD+. Cell 2007, 129, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Tempel, W.; Rabeh, W.M.; Bogan, K.L.; Belenky, P.; Wójcik, M.; Seidle, H.F.; Nedyalkova, L.; Yang, T.; A Sauve, A.; Park, H.-W.; et al. Nicotinamide Riboside Kinase Structures Reveal New Pathways to NAD+. PLoS Biol. 2007, 5, e263. [Google Scholar] [CrossRef] [PubMed]
- Dollerup, O.L.; Christensen, B.; Svart, M.; Schmidt, M.S.; Sulek, K.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Møller, N.; Brenner, C.; Treebak, J.T.; et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: Safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 2018, 108, 343–353. [Google Scholar] [CrossRef]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Philip, R.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Haynes, L.J.; Todd, A.R. 66. Codehydrogenases. Part I. The synthesis of dihydronicotinamide-D-ribofuranoside [N-D-ribofuranosidyl-1: 2(or 6)-dihydronicotinamide]. J. Chem. Soc. 1950, 303–308. [Google Scholar] [CrossRef]
- Haykes, L.J.; Hughes, N.A.; Kenner, G.W.; Todd, A. 734. Codehydrogenases. Part II. A synthesis of nicotinamide nucleotide. J. Chem. Soc. 1957, 3727–3732. [Google Scholar] [CrossRef]
- Mikhailopulo, I.A.; Pricota, T.I.; Timoshchuk, V.A.; Akhrem, A.A. Synthesis of Glycosides of Nicotinamide and Nicotinamide Mononucleotide. Synthesis 1981, 1981, 388–389. [Google Scholar] [CrossRef]
- Lee, J.; Churchil, H.; Choi, W.-B.; Lynch, J.E.; Roberts, F.E.; Volante, R.P.; Reider, P.J. A chemical synthesis of nicotinamide adenine dinucleotide (NAD+). Chem. Commun. 1999, 729–730. [Google Scholar] [CrossRef]
- Jarman, M.; Ross, W.C.J. 4-Substituted nicotinic acids and nicotinamides. Part II. The preparation of 4-methylnicotinamide riboside. J. Chem. Soc. C 1969, 199–203. [Google Scholar] [CrossRef]
- Migaud, M.E.; Redpath, P.; Crossey, K.; Cunningham, R.; Erickson, A.; Nygaard, R.; Storjohann, A. Efficient and Scalable Syntheses of Nicotinoyl Ribosides and Reduced Nicotinoyl Ribosides, Modified Derivatives Thereof, Phosphorylated Analogs Thereof, Adenylyl Dinucleotide Conjugates Thereof, and Novel Crystalline Forms Thereof. WO Patent 2018/089830 A1, 17 May 2018. [Google Scholar]
- Migaud, M.E.; Redpath, P.; Crossey, K.; Cunningham, R.; Dellinger, R.; Rhonemus, T.; Venkataraman, S.; Nettles, B. B-Vitamin and Amino Acid Conjugates of Nicotinoyl Ribosides and Reduced Nicotinoyl Ribosides, Derivatives Thereof, and Methods of Preparation Thereof. WO Patent 2017/161165 A1, 21 September 2017. [Google Scholar]
- Tanimori, S.; Ohta, T.; Kirihata, M. An efficient chemical synthesis of nicotinamide riboside (NAR) and analogues. Bioorganic Med. Chem. Lett. 2002, 12, 1135–1137. [Google Scholar] [CrossRef]
- Franchetti, P.; Pasqualini, M.; Petrelli, R.; Ricciutelli, M.; Vita, P.; Cappellacci, L. Stereoselective synthesis of nicotinamide β-riboside and nucleoside analogs. Bioorganic Med. Chem. Lett. 2004, 14, 4655–4658. [Google Scholar] [CrossRef]
- Dellinger, R.; Migaud, M.E.; Redpath, P.; Rhonemus, T.; Cunningham, R. Nicotinic Acid Riboside or Nicotinamide Riboside Compositions, Reduced Derivatives Thereof, and the Use Thereof. WO Patent 2016/149395 A1, 22 September 2016. [Google Scholar]
- Szczepankiewicz, B.; Koppetsch, K.; Perni, R.B. Preparation and Use of Crystalline Beta-D-Nicotinamide Riboside. WO Patent 2015/186068 A1, 10 December 2015. [Google Scholar]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagné, J.-P.; De Moura, M.B.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 Negatively Regulates Glycolysis by Inhibiting Hexokinase 1 Independent of NAD+ Depletion. Cell Rep. 2014, 8, 1819–1831. [Google Scholar] [CrossRef]
- Yang, T.; Chan, N.Y.-K.; Sauve, A.A. Syntheses of Nicotinamide Riboside and Derivatives: Effective Agents for Increasing Nicotinamide Adenine Dinucleotide Concentrations in Mammalian Cells. J. Med. Chem. 2007, 50, 6458–6461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Sauve, A.A. Synthesis of β-Nicotinamide Riboside Using an Efficient Two-Step Methodology. Curr. Protoc. Nucleic Acid Chem. 2017, 71, 14.14.1–14.14.9. [Google Scholar] [CrossRef]
- Sauve, A.A.; Yang, T. Nicotinoyl Riboside Compositions and Methods of Use. U.S. Patent 8,106,184 B2, 31 January 2012. [Google Scholar]
- Felczak, K.Z. Synthetic Methods for the Preparation of Nicotinamide Riboside and Related Compounds. WO Patent 2017/218580 A1, 21 December 2017. [Google Scholar]
- Spingler, B.; Schnidrig, S.; Todorova, T.; Wild, F. Some thoughts about the single crystal growth of small molecules. CrystEngComm 2012, 14, 751–757. [Google Scholar] [CrossRef]
- Babor, M.; Nievergelt, P.P.; Čejka, J.; Zvoníček, V.; Spingler, B. Microbatch under-oil salt screening of organic cations: Single-crystal growth of active pharmaceutical ingredients. IUCrJ 2019, 6, 145–151. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro Software System, Rigaku Oxford Diffraction; Rigaku Corporation: Tokyo, Japan, 2019.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT– Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Egli, M.; Saenger, W. Principles of Nucleic Acid Structure; Springer: Berlin/Heidelberg, Germany, 1983. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A in Et3N·A | NR+Br− (g) | H2O (mL) | MeOH (mL) | S/D* | Et3N·A (mL) | Solvent Added (mL) | NR·A (g) | NR+Br− (g) | Yield % |
|---|---|---|---|---|---|---|---|---|---|
| l-Ascorbate | 5.0 | 3 | 10 | D | 16 | 375 EtOH | 3.45 | - | 54 |
| Benzenesulfonate | 0.56 | 0.4 | 1 | D | 1 | - | - | - | 0 |
| Citrate | 9.0 | - | 18 (DMSO) | D | 73 | 1125 iPrOH | 6.32 | - | 75 |
| Formiate | 10.0 | - | 25 | S | 12.5 | - | - | 6.21 | 0 |
| Fumarate | 0.50 | 0.3 | 1 | S | 0.5 | - | - | - | 0 |
| d-Glucuronate | 5.0 | 3 | 10 | D | 11.3 | 455 n-BuOH | 6.64 | - | 99 |
| Lactate | 5.80 | - | 20 | S | 10 | 400 iPrOH | - | 4.27 | 0 |
| l-Malate | 5.0 | - | 15 | S | 10 | 280 iPrOH | 4.51 | - | 100 |
| Maleate | 0.94 | - | 1 | S | 1 | - | - | 0.24 | 0 |
| Malonate | 7.10 | 4.3 | 14 | D | 7.3 | 350 iPrOH | - | 5.75 | 0 |
| Methane sulfonate | 0.62 | 0.4 | 1 | D | 1 | 24 n-BuOH | - | 0.23 | 0 |
| Salicylate | 2.75 | - | 9 | S | 5 | - | - | 1.73 | 0 |
| Sorbate | 2.90 | 1.5 | 5 | D | 5 | 100 Dioxane | - | 1.93 | 0 |
| Succinate | 0.48 | 0.3 | 1 | S | 0.5 | - | - | - | 0 |
| l-Tartrate | 9.65 | 5.8 | 20 | D | 10 | 500 EtOH | 8.06 | - | 85 |
| HA in Et3N·HA | NR+Br− (g) | H2O (mL) | MeOH (mL) | S/D* | Et3N·HA (mL) | NR·HA (g) | NR+Br− (g) | Yield % |
|---|---|---|---|---|---|---|---|---|
| Hydrogen citrate | 0.92 | - | 2 | S | 1 | - | ** | 0 |
| Dihydrogen citrate | 0.41 | - | 1 | S | 1 | 0.23 | 42 | |
| Hydrogen fumarate | 0.58 | - | 1.5 | S | 1 | - | ** | 0 |
| Hydrogen maleate | 0.58 | - | 1.5 | S | 1 | - | 0.13 | 0 |
| d-Hydrogen malate | 5.80 | - | 10 | S | 10 | 4.01 | 60 | |
| l-Hydrogen malate | 5.80 | - | 10 | S | 10 | 4.15 | 62 | |
| dl-Hydrogen malate | 5.80 | - | 10 | S | 10 | 4.52 | 67 | |
| Hydrogen malonate | 0.60 | - | 1 | S | 1 | - | 0.07 | 0 |
| H-mercaptosuccinate | 0.58 | - | 1.5 | S | 1 | - | ** | 0 |
| Hydrogen ketoglutarate | 0.58 | - | 1.5 | S | 1 | - | ** | 0 |
| Hydrogen oxalacetate | 0.60 | - | 1 | S | 1 | - | ** | 0 |
| Hydrogen oxalate | 0.62 | - | 1 | S | 1 | - | 0.15 | 0 |
| Hydrogen succinate | 0.58 | - | 1 | S | 1 | - | ** | 0 |
| d-Hydrogen tartrate | 5.80 | - | 10 | S | 10 | 4.90 | 70 | |
| l-Hydrogen tartrate | 5.80 | 3.5 | 10 | D | 10 | 6.62 | 95 | |
| dl-Hydrogen tartrate | 5.80 | 3.5 | 10 | D | 10 | 6.31 | 90 | |
| meso-Hydrogen tartrate | 0.57 | - | 1 | S | 1 | 0.44 | 62 | |
| Hydrogen tartronate | 0.60 | - | 1 | S | 1 | - | ** | 0 |
| NR+ Salt | Purity (NMR) | Residual Br (IC) |
|---|---|---|
| d-Hydrogen tartrate 11 | >97% | 0.3% |
| l-Hydrogen tartrate 12 | >97% | 0.2% |
| dl-Hydrogen tartrate 11 + 12 | >97% | 0.1% |
| l-Hydrogen malate 13 | >97% | 0.1% |
| d-Hydrogen malate 14 | >97% | 0.9% |
| dl-Hydrogen malate 13 + 14 | >96% | 2.3% |
| Compound | Space Group | Conformation | χ(O4-C1-N1-C6) [°] | Ref. |
|---|---|---|---|---|
| Nicotinamide-β-d-riboside d-Hydrogen tartrate anhydrate (11a) | P21 | 2’-endo, Twisted (inbetween C1’-exo and C2’-endo) | −157.0(2) | This work |
| Nicotinamide-β-d-riboside d-Hydrogen tartrate hydrate (11b) | P212121 | 3’-endo, Envelope | 10.9(2) | This work |
| Nicotinamide-β d-riboside l-Hydrogen tartrate (12) | P21 | 2’-endo, Envelope | −158.10(9) | This work |
| Nicotinamide-β-d-riboside l-Hydrogen malate (13) | P21 | 3’-endo, Envelope | −154.35(7) | This work |
| Nicotinamide-β-d-riboside d-Hydrogen malate (14) | P212121 | 3’-endo, Envelope | −164.1(2) | This work |
| Nicotinamide-β-d-riboside bromide | P212121 | 3’-endo, Envelope | −137.1(2) | [34] |
| Nicotinamide-β-d-riboside chloride | P212121 | 3’-endo, Envelope | −136.1(1) | [34] |
| Example (SI) | Deacetylation | Neutralization | Salt for SM | Product | Yield % |
|---|---|---|---|---|---|
| 15a | H2SO4 | Et3N | Et3N l-H-Tartrate | NR+ l-H-Tartrate | 54 |
| 15b | HBr | - | - | NR+ Bromide | 25 |
| 15b | HBr | Et3N | Et3N l-H-Tartrate | NR+ l-H-Tartrate | 53 |
| 15c | HCl | - | - | NR+ Chloride | 2 |
| 15c | HCl | Et3N | Et3N l-H-Tartrate | NR+ l-H-Tartrate | 60 |
| 15d | Et3N | - | Et3N l-H-Tartrate | NR+ l-H-Tartrate | 63 |
| 16a | H2SO4 | Et3N | Et3N l-H-Malate | NR+ l-H-Malate | 37 |
| 16b | HBr | Bu3N | Et3N l-H-Malate | NR+ l-H-Malate | 33 |
| 16c | HCl | Et3N | Et3N l-H-Malate | NR+ l-H-Malate | 43 |
| 16d | Et3N | - | Et3N l-H-Malate | NR+ l-H-Malate | 43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schabert, G.; Haase, R.; Parris, J.; Pala, L.; Hery-Barranco, A.; Spingler, B.; Spitz, U. New Crystalline Salts of Nicotinamide Riboside as Food Additives. Molecules 2021, 26, 2729. https://doi.org/10.3390/molecules26092729
Schabert G, Haase R, Parris J, Pala L, Hery-Barranco A, Spingler B, Spitz U. New Crystalline Salts of Nicotinamide Riboside as Food Additives. Molecules. 2021; 26(9):2729. https://doi.org/10.3390/molecules26092729
Chicago/Turabian StyleSchabert, Günter, Robert Haase, Jaclyn Parris, Laura Pala, Adrian Hery-Barranco, Bernhard Spingler, and Urs Spitz. 2021. "New Crystalline Salts of Nicotinamide Riboside as Food Additives" Molecules 26, no. 9: 2729. https://doi.org/10.3390/molecules26092729
APA StyleSchabert, G., Haase, R., Parris, J., Pala, L., Hery-Barranco, A., Spingler, B., & Spitz, U. (2021). New Crystalline Salts of Nicotinamide Riboside as Food Additives. Molecules, 26(9), 2729. https://doi.org/10.3390/molecules26092729