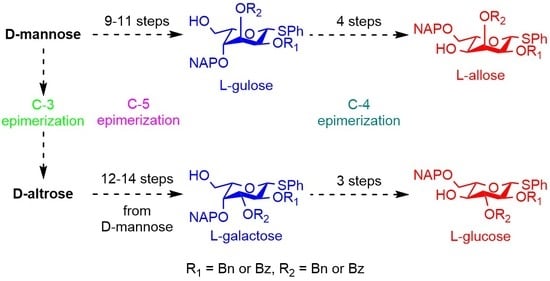
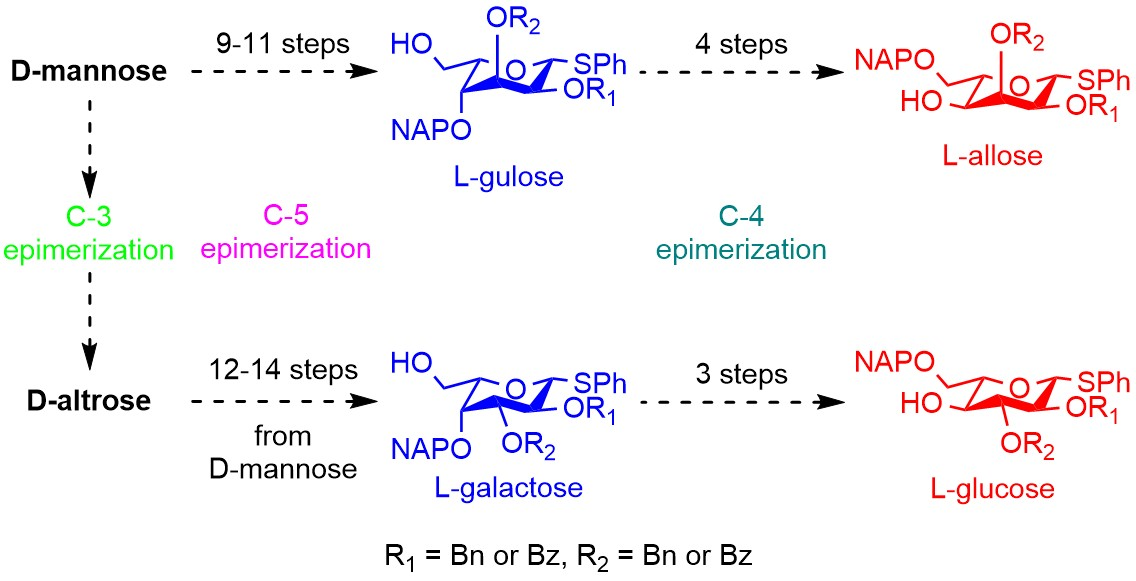
3.2.12. Synthesis of l-Gulose and l-Allose Derivatives from d-Mannose
Phenyl 2,4-di-O-benzyl-3-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (
24). The acetal derivative
23 [
79] (2.53 g, 4.272 mmol) was dissolved in anhydrous CH
2Cl
2 (38.5 mL) and anhydrous Et
2O (13 mL). LiAlH
4 (729 mg, 19.224 mmol, 4.5 equiv.) was added, and then a solution of AlCl
3 (2.57 g, 19.224 mmol, 4.5 equiv.) in anhydrous Et
2O (13 mL) was added. The reaction mixture was stirred at 0 °C for 1 h. The reaction mixture was diluted with EtOAc (86 mL) and H
2O (21.5 mL), the precipitated solid was filtered through a pad of Celite, and the filter cake was washed with ethyl acetate. The filtrate was washed with water (2 × 25 mL), dried over MgSO
4, filtered and concentrated. The crude product was purified by silica gel chromatography (7:3
n-hexane/EtOAc) to give
24 (2.01 g, 79%) as a colorless syrup. [α]
D25 +51.2 (
c 0.54, CHCl
3);
Rf 0.38 (7:3
n-hexane/EtOAc);
1H NMR (500 MHz, CDCl
3)
δ = 7.84–7.26 (22H, arom.), 5.51 (s, 1H, H-1), 4.99 (d,
J = 10.9 Hz, 1H, BnC
H2a), 4.81–4.67 (m, 5H, BnC
H2b, BnC
H2, NAPC
H2), 4.14–4.12 (m, 1H, H-5), 4.07 (t,
J = 9.3 Hz, 1H, H-4), 4.02 (s, 1H, H-2), 3.96 (d,
J = 8.9 Hz, 1H, H-3), 3.86–3.80 (m, 2H, H-6-a,b), 1.83 (t,
J = 6.2 Hz, 1H, C-6-OH) ppm;
13C NMR (100 MHz, CDCl
3)
δ = 138.4, 137.9, 135.7, 134.0, 133.4, 133.1 (6C, 6 × C
q arom.), 132.0–126.0 (22C, arom.), 86.1 (1C, C-1), 80.2 (1C, C-3), 76.5 (1C, C-2), 75.5 (1C, NAP
CH
2), 74.9 (1C, C-4), 73.4 (1C, C-5), 72.4 (2C, 2 × Bn
CH
2), 62.3 (1C, C-6) ppm; ESI-TOF-MS:
m/z calcd for C
37H
36NaO
5S [M + Na]
+ 615.2176; found: 615.2168.
Phenyl 2,4-di-O-benzyl-6-deoxy-6-iodo-3-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (25). Compound 24 (1.94 g, 3.270 mmol) was converted to 25 according to general Method A. The crude product was purified by silica gel chromatography (8:2 n-hexane/EtOAc) to give 25 (1.52 g, 66%) as a colorless syrup. [α]D25 + 50.5 (c 0.21, CHCl3); Rf 0.72 (7:3 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.84–7.22 (22H, arom.), 5.58 (s, 1H, H-1), 5.04 (d, J = 10.9 Hz, 1H, BnCH2a), 4.74–4.72 (m, 4H, NAPCH2, BnCH2), 4.64 (d, J = 12.3 Hz, 1H, BnCH2b), 4.04 (s, 1H, H-2), 3.96–3.93 (m, 3H, H-3, H-4, H-5), 3.53 (d, J = 10.3 Hz, 1H, H-6a), 3.43 (dd, J = 5.1 Hz, J = 10.4 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.4, 137.9, 135.5, 134.2, 133.4, 133.1 (6C, 6 × Cq arom.), 131.8–126.0 (22C, arom.), 85.9 (1C, C-1), 79.9, 78.7 (2C, C-3, C-4), 76.5 (1C, C-2), 75.7 (1C, NAPCH2), 72.2 (1C, C-5), 72.2, 72.1 (2C, 2 × BnCH2), 7.1 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H35INaO4S [M + Na]+ 725.1193; found: 725.1195.
Phenyl 2,4-di-O-benzyl-3-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (26) and 8-N-[phenyl 2,4-di-O-benzyl-6-deoxy-6-yl-3-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside]-1,8-diazabicyclo(5.4.0)undec-7-ene-iodide (27).
Reaction I.: Compound 25 (439 mg, 0.625 mmol) was converted to 26 according to general Method B using DBU (372 µL, 2.500 mmol, 4.0 equiv.) and refluxed (70 °C) for 5 h. The crude product was purified by silica gel chromatography (8:2 n-hexane/acetone) to give 26 (155 mg, 43%) as a colorless syrup and 27 (45 mg, 12%) as a colorless syrup.
Reaction II.: To a solution of compound 25 (323 mg, 0.460 mmol) in dry toluene (4.0 mL), DBU (274 µL, 1.839 mmol, 4.0 equiv.) was added and the mixture was stirred for 2 h at 110 °C. The reaction mixture was diluted with CH2Cl2 (100 mL), washed with 10% aqueous solution of Na2S2O3 (20 mL), 1M aqueous solution of HCl (2 × 20 mL), saturated aqueous solution of NaHCO3 (20 mL), and H2O (2 × 20 mL) until neutral pH. The organic layer was dried over MgSO4, filtered, and concentrated. The crude product was purified by silica gel chromatography (8:2 n-hexane/acetone → 9:1 CH2Cl2/MeOH) to give 26 (107 mg, 40%) as a colorless syrup and 27 (73 mg, 22%) as a light yellow syrup.
Reaction III.: Compound 25 (328 mg, 0.467 mmol) was converted to 26 according to general Method B using DBU (278 µL, 1.868 mmol, 4.0 equiv.) and refluxed for 24 h. The crude product was purified by silica gel chromatography (8:2 n-hexane/acetone → 9:1 CH2Cl2/MeOH) to give 26 (141 mg, 53%) as a colorless syrup and 27 (94 mg, 28%) as a light yellow syrup.
Data of 26: [α]D25 +53.5 (c 0.14, CHCl3); Rf 0.43 (8:2 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 7.81–7.22 (m, 22H, arom), 5.48 (d, J = 4.7 Hz, 1H, H-1), 4.86–4.60 (m, 8H, H-6a,b, 2 × BnCH2, NAPCH2), 4.29 (d, J = 7.5 Hz, 1H, H-4), 4.02 (d, J = 3.3 Hz, 1H, H-2), 3.94 (dd, J = 1.6 Hz, J = 7.2 Hz, 1H, H-3) ppm; 13C NMR (125 MHz, CDCl3) δ = 154.5 (1C, C-5), 138.2, 137.9, 135.8, 133.7, 133.3, 133.1 (6C, 6 × Cq arom.), 132.0–125.9 (22C, arom.), 99.1 (1C, C-6), 86.4 (1C, C-1), 77.3 (1C, C-3), 76.3 (1C, C-4), 76.0 (1C, C-2), 73.1, 72.8 (3C, 2 × BnCH2, NAPCH2) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO4S [M + Na]+ 597.2070; found: 597.2070.
Data of 27: [α]D25 +66.7 (c 0.24, CHCl3); Rf 0.49 (9:1 CH2Cl2/MeOH); 1H NMR (400 MHz, MeOD) δ = 7.85–7.26 (m, 22H, arom.), 5.83 (s, 1H, H-1), 5.00–4.63 (m, 6H, 2 × BnCH2, NAPCH2), 4.17–4.15 (m, 1H, H-2), 4.06 (td, J = 1.7 Hz, J = 9.4 Hz, 1H, H-5), 3.97 (dd, J = 3.0 Hz, J = 9.1 Hz, 1H, H-3), 3.75 (t, J = 9.5 Hz, 1H, H-4), 3.69–3.60 (m, 2H, H-6a,b), 3.54–3.46 (m, 2H, NCH2 DBU), 3.25–3.16 (m, 4H, 2 × NCH2 DBU), 2.70–2.59 (m, 2H, CH2 DBU), 1.67–1.27 (m, 8H, 4 × CH2 DBU) ppm; 13C NMR (100 MHz, MeOD) δ = 168.5 (1C, Cq DBU), 139.5, 139.3, 136.7, 134.7, 134.5, 134.1 (6C, 6 × Cq arom.), 132.3–127.2 (22C, arom.), 85.2 (1C, C-1), 80.9 (1C, C-3), 77.1, 76.9 (2C, C-2, C-4), 76.1, 73.8, 72.7 (3C, 2 × BnCH2, NAPCH2), 72.3 (1C, C-5), 56.0, 55.7, 50.0, 49.6 (4C, C-6, 3 × NCH2 DBU), 29.2, 29.1, 26.8, 23.8, 20.7 (5C, 5 × CH2 DBU) ppm; UHR ESI-QTOF: m/z calcd for C46H51N2O4S [M]+ 727.3564; found: 727.3568.
Phenyl 2,4-di-O-benzyl-3-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (28). Compound 26 (190 mg, 0.331 mmol) was converted to 28 according to general Method C. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 28 (104 mg, 53%) as a colorless syrup and 24 (24 mg, 12%) as a colorless syrup. [α]D25 +11.4 (c 0.21, CHCl3); Rf 0.33 (6:4 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 7.82–6.94 (m, 22H, arom.), 5.27 (d, J = 9.8 Hz, 1H, H-1), 4.84–4.14 (m, 6H, 2 × BnCH2, NAPCH2), 3.98 (d, J = 4.5 Hz, 1H, H-5), 3.84–3.80 (m, 1H, H-6a), 3.76 (s, 2H, H-2, H-3), 3.50 (d, J = 8.4 Hz, 1H, H-6b), 3.39 (s, 1H, H-4), 1.91 (s, 1H, C-6-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.0, 137.6, 135.6, 134.1, 133.2, 133.1 (6C, 6 × Cq arom.), 131.7–126.1 (22C, arom.), 84.2 (1C, C-1), 76.0 (1C, C-5), 75.3 (1C, C-4), 75.1 (1C, C-3), 73.6, 73.1, 72.2 (3C, 2 × BnCH2, NAPCH2), 72.7 (1C, C-2), 62.5 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H36NaO5S [M + Na]+ 615.2176; found: 615.2174.
Phenyl 3-O-benzyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-mannopyranoside (
30). To a solution of compound
29 [
91] (4.09 g, 10.00 mmol) in dry toluene (71 mL), dibutyltin oxide (3.644 g, 15.00 mmol, 1.5 equiv.) was added. The solution was refluxed (110 °C) under argon atmosphere for 4 h. Then, after cooling to room temperature, the solvent was evaporated to dryness and the crude product was dried on high vacuum for 2 h. The residue was dissolved in dry DMF (36 mL), and CsF (3.04 g, 2.001 mmol) and BnBr (1.78 mL, 15.00 mmol, 1.5 equiv.) were added. The reaction mixture was stirred at 90 °C for 24 h. The reaction mixture was filtered and evaporated to dryness. The residue was dissolved in EtOAc (250 mL) and washed with H
2O (2 × 25 mL). The organic layer was separated, dried over MgSO
4, filtered, and concentrated under reduced pressure. The crude product was purified by crystallization from acetone/
n-hexane to give
30 (3.59 g, 73%) as white crystals. [α]
D25 +244.2 (
c 0.19, CHCl
3); M.p. = 194–197 °C (acetone/
n-hexane);
Rf 0.32 (7:3
n-hexane/acetone);
1H NMR (400 MHz, CDCl
3)
δ = 7.98–7.20 (m, 17H, arom.), 5.75 (s, 1H, H
ac), 5.59 (s, 1H, H-1), 4.88 (d,
J = 11.8 Hz, 1H, BnC
H2a), 4.74 (d,
J = 11.8 Hz, 1H, BnC
H2b), 4.38 (td,
J = 4.8 Hz,
J = 9.7 Hz, 1H, H-5), 4.26–4.21 (m, 3H), 3.98 (dd,
J = 3.2 Hz,
J = 9.5 Hz, 1H), 3.90 (t,
J = 10.2 Hz, 1H), 3.02 (d,
J = 5.3 Hz, 1H, C-2-OH) ppm;
13C NMR (100 MHz, CDCl
3)
δ = 137.8, 134.9, 133.7, 133.4, 133.0 (5C, 5 × C
q arom.), 131.8–123.9 (17C, arom.), 101.9 (1C, C
ac), 88.0 (1C, C-1), 79.2, 75.9, 71.5, 64.8 (4C, C-2, C-3, C-4, C-5), 73.3 (1C, Bn
CH
2), 68.7 (1C, C-6) ppm; ESI-TOF-MS:
m/z calcd for C
30H
28NaO
5S [M + Na]
+ 523.1550; found: 523.1545.
Phenyl 2-O-benzoyl-3-O-benzyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-mannopyranoside (31). To a stirred solution of compound 30 (3.48 g, 5.807 mmol) in dry pyridine (15 mL), BzCl (0.85 mL, 7.259 mmol, 1.25 equiv./OH) was added at 0 °C and the reaction mixture was stirred for 24 h at room temperature. After 24 h, the mixture was diluted with CH2Cl2 (300 mL), washed with H2O (2 × 75 mL), 1M aqueous solution of H2SO4 (2 × 75 mL), H2O (2 × 75 mL), saturated aqueous solution of NaHCO3 (2 × 75 mL), and H2O (2 × 75 mL) until neutral pH. The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (7:3 n-hexane/acetone) to give 31 (3.56 g, 72%) as a colorless syrup. [α]D25 + 73.3 (c 0.34, CHCl3); Rf 0.52 (7:3 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 8.12–7.20 (m, 22H, arom.), 5.87 (s, 1H, Hac), 5.85 (s, 1H, H-2), 5.64 (s, 1H, H-1), 4.76 (q, J = 12.2 Hz, 2H, BnCH2), 4.49–4.47 (m, 1H, H-5), 4.36–4.32 (m, 2H, H-4, H-6a), 4.17 (d, J = 9.6 Hz, 1H, H-3), 3.97 (t, J = 10.1 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.8 (1C, Cq Bz), 137.8, 134.9, 133.8, 133.2, 133.1, 129.7 (6C, 6 × Cq arom.), 133.5–123.9 (22C, arom.), 102.0 (1C, Cac), 87.4 (1C, C-1), 79.1 (1C, C-4), 74.4 (1C, C-3), 72.3 (1C, BnCH2), 72.0 (1C, C-2), 68.7 (1C, C-6), 65.4 (1C, C-5) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO6S [M + Na]+ 627.1812; found: 627.1819.
Phenyl 2,3-di-O-benzyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-mannopyranoside (
32). To a solution of compound
29 [
91] (3.99 g, 9.840 mmol) in dry DMF (43 mL), NaH (60%, 984 mg, 24.60 mmol, 1.25 equiv./OH) was added in portions at 0 °C. After 30 min at that temperature, BnBr (2.8 mL, 23.61 mmol, 1.25 equiv./OH) was added and the mixture was stirred for 24 h at room temperature. Next, MeOH (5.0 mL) was added, the reaction mixture was stirred for 15 min, and the solvents were evaporated. The residue was dissolved in CH
2Cl
2 (300 mL) and washed in H
2O (3 × 50 mL) until neutral pH. The organic layer was dried over MgSO
4 and concentrated. The crude product was purified by silica gel chromatography (9:1
n-hexane/acetone) to give
32 (3.75 g, 65%) as a colorless syrup. [α]
D25 + 116.5 (
c 0.36, CHCl
3);
Rf 0.29 (9:1
n-hexane/acetone);
1H NMR (400 MHz, CDCl
3)
δ = 8.00–7.22 (m, 22H, arom.), 5.80 (s, 1H, H
ac), 5.53 (d,
J = 1.2 Hz, 1H, H-1), 4.83 (d,
J = 12.2 Hz, 1H, BnC
H2a), 4.73 (s, 2H, BnC
H2), 4.67 (d,
J = 12.2 Hz, 1H, BnC
H2b), 4.41–4.33 (m, 2H, H-5, H-6a), 4.27 (dd,
J = 4.4 Hz,
J = 10.2 Hz, 1H, H-6b), 4.07 (dd,
J = 1.3 Hz,
J = 3.1 Hz, 1H, H-2), 4.00 (dd,
J = 3.2 Hz,
J = 9.4 Hz, 1H, H-3), 3.94 (t,
J = 9.8 Hz, 1H, H-4) ppm;
13C NMR (100 MHz, CDCl
3)
δ = 138.5, 137.9, 135.1, 133.9, 133.7, 133.1 (6C, 6 × C
q arom.), 131.8–123.9 (22C, arom.), 101.8 (1C, C
ac), 87.3 (1C, C-1), 79.3, 78.2, 76.4 (3C, C-2, C-3, C-4), 73.2, 73.1 (2C, 2 × Bn
CH
2), 68.7 (1C, C-6), 65.6 (1C, C-5) ppm; ESI-TOF-MS:
m/z calcd for: C
37H
34NaO
5S [M + Na]
+ 613.2019; found: 613.2039.
Phenyl 2,3-di-O-benzoyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-mannopyranoside (
33). To a stirred solution of compound
29 [
91] (4.10 g, 10.10 mmol) in pyridine (25 mL), BzCl (2.92 mL, 25.20 mmol, 1.25 equiv./OH) was added at 0 °C and the reaction mixture was stirred for 24 h at room temperature. After 24 h, the mixture was diluted with CH
2Cl
2 (300 mL), washed with H
2O (150 mL), 1M aqueous solution of H
2SO
4 (2 × 150 mL), H
2O (150 mL), saturated aqueous solution of NaHCO
3 (2 × 150 mL), and H
2O (150 mL) until neutral pH. The organic layer was separated, dried over MgSO
4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (7:3
n-hexane/acetone) to give
33 (4.38 g, 71%) as a colorless syrup. [α]
D25 − 15.7 (
c 0.22, CHCl
3);
Rf 0.46 (7:3
n-hexane/acetone);
1H NMR (500 MHz, CDCl
3)
δ = 8.10–7.23 (m, 22H, arom.), 5.99 (dd,
J = 1.3 Hz,
J = 3.4 Hz, 1H, H-2), 5.86 (dd,
J = 3.4 Hz,
J = 10.3 Hz, 1H, H-3), 5.84 (s, 1H, H
ac), 5.70 (d,
J = 1.1 Hz, 1H, H-1), 4.69 (td,
J = 4.8 Hz,
J = 9.8 Hz, 1H, H-5), 4.48 (t,
J = 9.9 Hz, 1H, H-6a), 4.38 (dd,
J = 4.9 Hz,
J = 10.4 Hz, 1H, H-4), 4.03 (t,
J = 10.3 Hz, 1H, H-6b) ppm;
13C NMR (125 MHz, CDCl
3)
δ = 165.6, 165.4 (2C, 2 × C
q Bz), 134.5, 133.8, 133.0, 129.7, 129.6 (6C, 6 × C
q arom.), 133.8–123.9 (22C, arom.), 102.3 (1C, C
ac), 87.2 (1C, C-1), 77.2, 72.6, 69.4, 65.5 (4C, C-2, C-3, C-4, C-5), 68.8 (1C, C-6) ppm; ESI-TOF-MS:
m/z calcd for: C
37H
30NaO
7S [M + Na]
+ 641.1604; found: 641.1603.
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (34). Compound 32 (3.46 g, 5.850 mmol) was converted to 34 according to general Method D. The crude product was purified by silica gel chromatography (7:3 n-hexane/acetone) to give 34 (3.44 g, 99%) as a colorless syrup. [α]D25 + 81.4 (c 0.37, CHCl3); Rf 0.39 (7:3 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 7.81–7.27 (m, 22H, arom.), 5.52 (s, 1H, H-1), 5.10 (d, J = 11.1 Hz, 1H, BnCH2a), 4.83 (d, J = 11.0 Hz, 1H, BnCH2b), 4.69 (s, 2H, NAPCH2), 4.65 (s, 2H, BnCH2), 4.17–4.15 (m, 1H, H-5), 4.10 (t, J = 9.3 Hz, 1H, H-4), 4.02 (s, 1H, H-2), 3.92 (d, J = 8.9 Hz, 1H, H-3), 3.86–3.82 (m, 2H, H-6-a,b), 1.88 (s, 1H, C-6-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 138.2, 137.9, 135.9, 134.0, 133.4, 133.1 (6C, 6 × Cq arom.), 132.0–126.0 (22C, arom.), 86.2 (1C, C-1), 80.2 (1C, C-3), 76.5 (1C, C-2), 75.4 (1C, NAPCH2), 74.5 (1C, C-4), 73.4 (1C, C-5), 72.5, 72.3 (2C, 2 × BnCH2), 62.3 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H36NaO5S [M + Na]+ 615.2176; found: 615.2196.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (35). Compound 33 (4.83 g, 7.788 mmol) was converted to 35 according to general Method D. The crude product was purified by silica gel chromatography (65:35 n-hexane/acetone) to give 35 (4.16 g, 95%) as a colorless syrup. [α]D25 +8.1 (c 0.32, CHCl3); Rf 0.46 (6:4 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 8.01–7.24 (m, 22H, arom.), 5.86 (d, J = 1.3 Hz, 1H, H-2), 5.79 (dd, J = 3.2 Hz, J = 8.9 Hz, 1H, H-3), 5.65 (s, 1H, H-1), 4.89 (s, 2H, NAPCH2), 4.44–4.41 (m, 2H, H-4, H-5), 3.96 (s, 2H, H-6a,b), 1.92 (s, 1H, H-6-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.5, 165.4 (2C, 2 × Cq Bz), 135.1, 133.3, 133.2, 133.1, 129.6 (6C, 6 × Cq arom.), 133.6–126.1 (22C, arom.), 86.2 (1C, C-1), 75.3 (1C, NAPCH2), 73.3, 73.0 (2C, C-4, C-5), 72.7 (1C, C-3), 72.6 (1C, C-2), 61.8 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO7S [M + Na]+ 643.1761; found: 643.1742.
Phenyl 2-O-benzoyl-3-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (36). Compound 31 (3.56 g, 5.899 mmol) was converted to 36 according to general Method D. The crude product was purified by silica gel chromatography (7:3 n-hexane/acetone) to give 36 (2.82 g, 79%) as a colorless syrup. [α]D25 + 68.6 (c 0.29, CHCl3); Rf 0.42 (7:3 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 8.06–7.24 (m, 22H, arom.), 5.86 (s, 1H, H-2), 5.59 (d, J = 0.9 Hz, 1H, H-1), 5.11–4.62 (m, 4H, NAPCH2, BnCH2), 4.29–4.27 (m, 1H, H-5), 4.12–4.11 (m, 2H, H-3, H-4), 3.90–3.87 (m, 2H, H-6a,b), 1.82 (t, J = 6.6 Hz, 1H, C-6-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.7 (1C, Cq Bz), 137.8, 135.7, 133.4, 133.2, 129.8 (6C, 6 × Cq arom.), 133.5–126.1 (22C, arom.), 86.6 (1C, C-1), 78.6 (1C, C-3), 75.5 (1C, NAPCH2), 74.2 (1C, C-4), 73.2 (1C, C-5), 71.9 (1C, BnCH2), 70.9 (1C, C-2), 62.2 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO6S [M + Na]+ 629.1968; found: 629.2028.
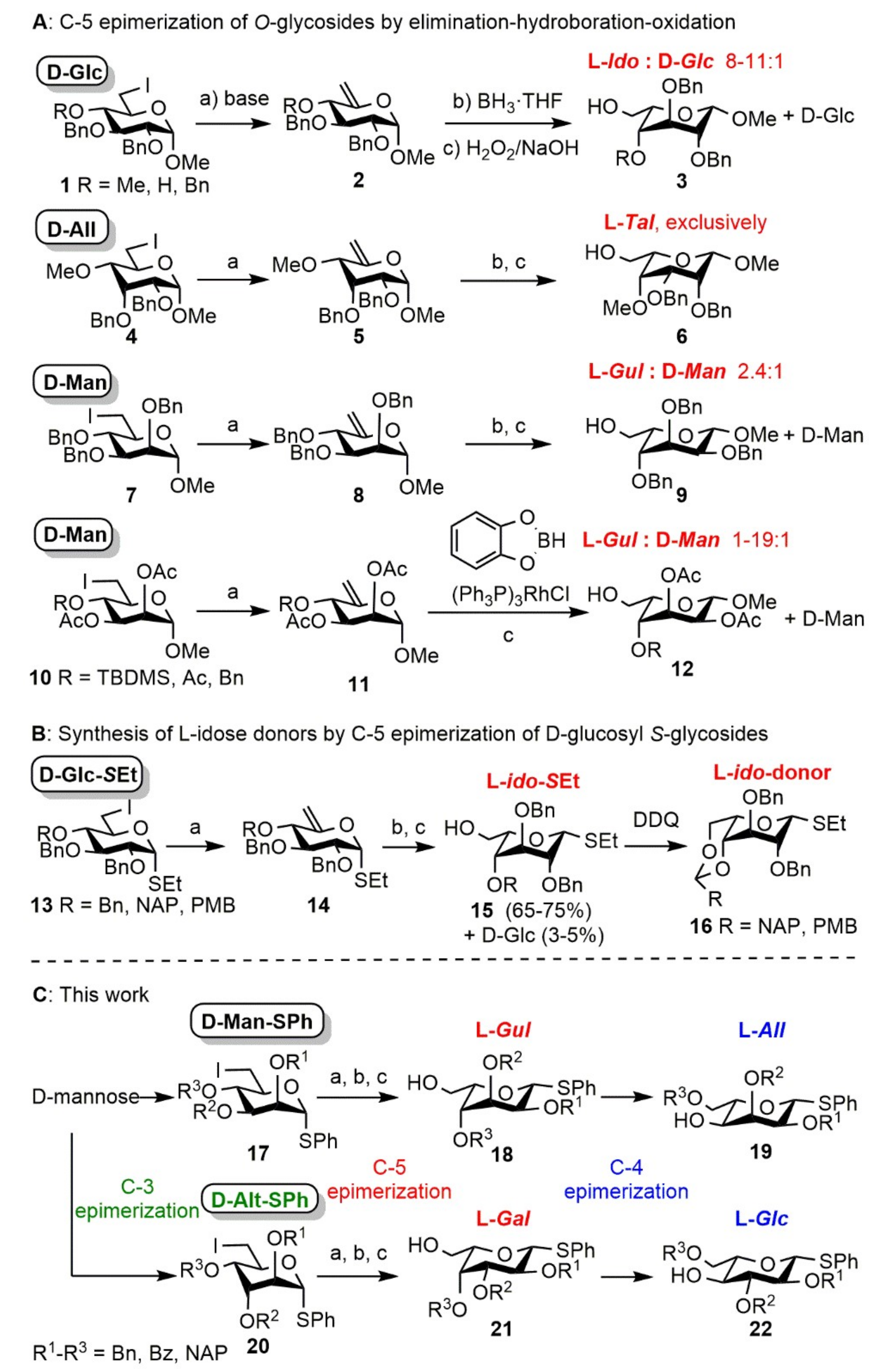
Phenyl 2,3-di-O-benzyl-6-deoxy-6-iodo-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (37). Compound 34 (3.38 g, 5.702 mmol) was converted to 37 according to general Method A. The crude product was purified by silica gel chromatography (8:2 n-hexane/acetone) to give 37 (3.12 g, 78%) as a colorless syrup. [α]D25 +57.3 (c 0.63, CHCl3); Rf 0.42 (8:2 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 7.82–7.22 (m, 22H, arom.), 5.59 (s, 1H, H-1), 5.15 (d, J = 11.1 Hz, 1H, BnCH2a), 4.88 (d, J = 11.1 Hz, 1H, BnCH2b), 4.73 (d, J = 12.3 Hz, 1H, BnCH2a), 4.65 (d, J = 12.2 Hz, 1H, BnCH2b), 4.61 (s, 2H, NAPCH2), 4.03 (s, 1H, H-2), 3.97–3.94 (m, 2H, H-4, H-5), 3.91–3.90 (m, 1H, H-3), 3.54 (d, J = 10.3 Hz, 1H, H-6a), 3.44 (dd, J = 5.4 Hz, J = 10.5 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.1, 137.9, 135.8, 134.3, 133.4, 133.2 (6C, 6 × Cq arom.), 131.7–126.1 (22C, arom.), 86.0 (1C, C-1), 79.9 (1C, C-3), 78.8 (1C, C-4), 76.4 (1C, C-2), 75.7 (1C, NAPCH2), 72.2 (1C, C-5), 72.1, 72.1 (2C, 2 × BnCH2), 7.1 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H35INaO4S [M + Na]+ 725.1193; found: 725.1183.
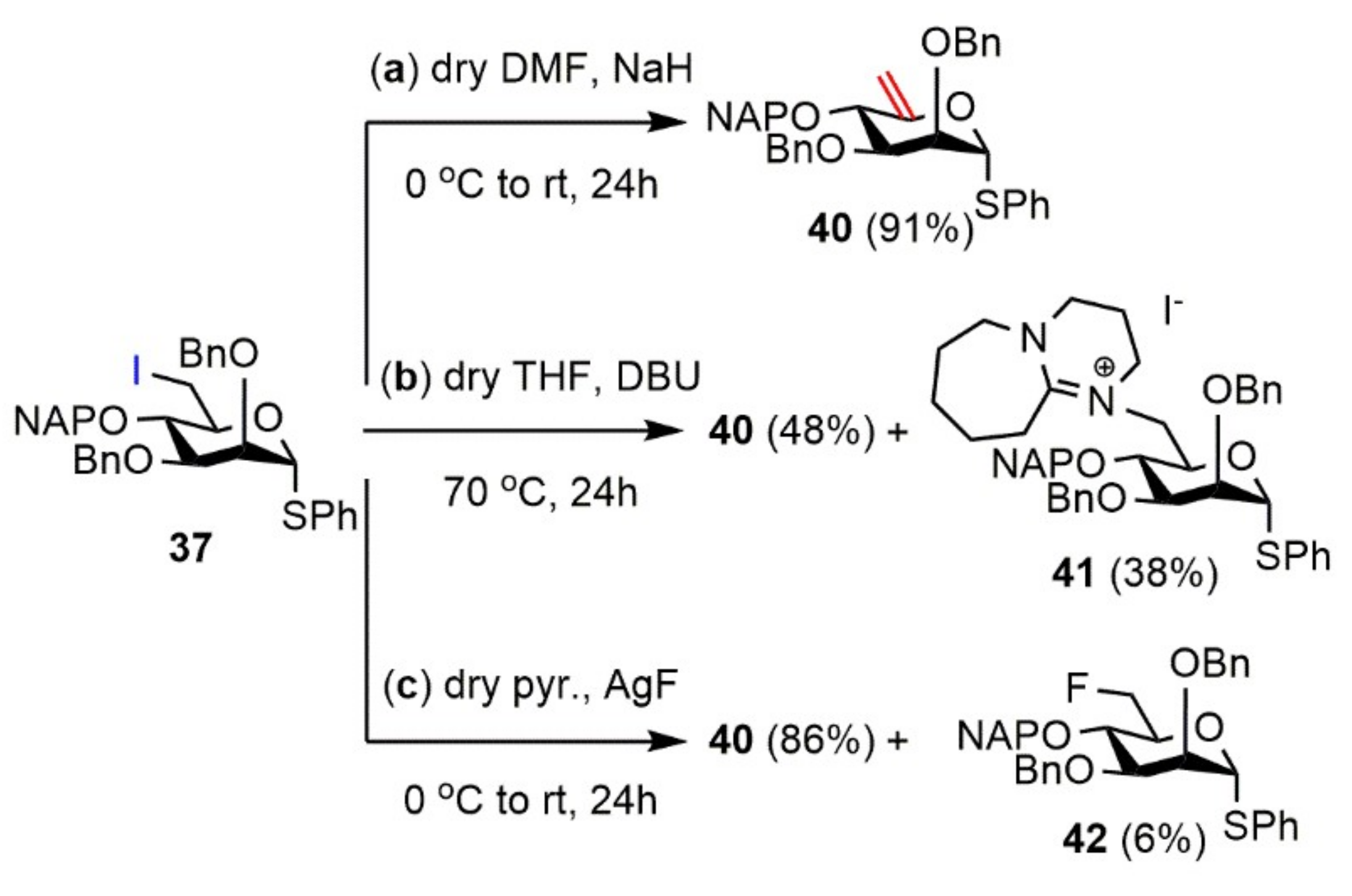
Phenyl 2,3-di-O-benzoyl-6-deoxy-6-iodo-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (38). Compound 35 (4.08 g, 5.588 mmol) was converted to 38 according to general Method A. The crude product was purified by silica gel chromatography (7:3 n-hexane/acetone) to give 38 (3.77 g, 79%) as a colorless syrup. [α]D25 + 24.1 (c 0.22, CHCl3); Rf 0.53 (7:3 n-hexane/acetone); 1H NMR (400 MHz, CDCl3) δ = 8.13–7.23 (m, 22H, arom.), 5.92 (s, 1H), 5.80 (dd, J = 2.6 Hz, J = 9.5 Hz, 1H), 5.68 (s, 1H), 4.95 (s, 2H, NAPCH2), 4.32 (d, J = 9.4 Hz, 1H), 4.14–4.09 (m, 1H), 3.69 (dd, J = 3.8 Hz, J = 10.9 Hz, 1H, H-6a), 3.59–3.57 (m, 1H, H-6b) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.4, 165.3 (2C, 2 × Cq Bz), 134.9, 133.2, 133.1, 133.0, 129.5, 129.4 (6C, 6 × Cq arom.), 133.6–125.9 (22C, arom.), 86.1 (1C, C-1), 75.7 (1C, NAPCH2), 77.7, 72.5, 72.3, 70.9 (4C, C-2, C-3, C-4, C-5), 8.6 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H31INaO6S [M + Na]+ 753.0778; found: 753.0758.
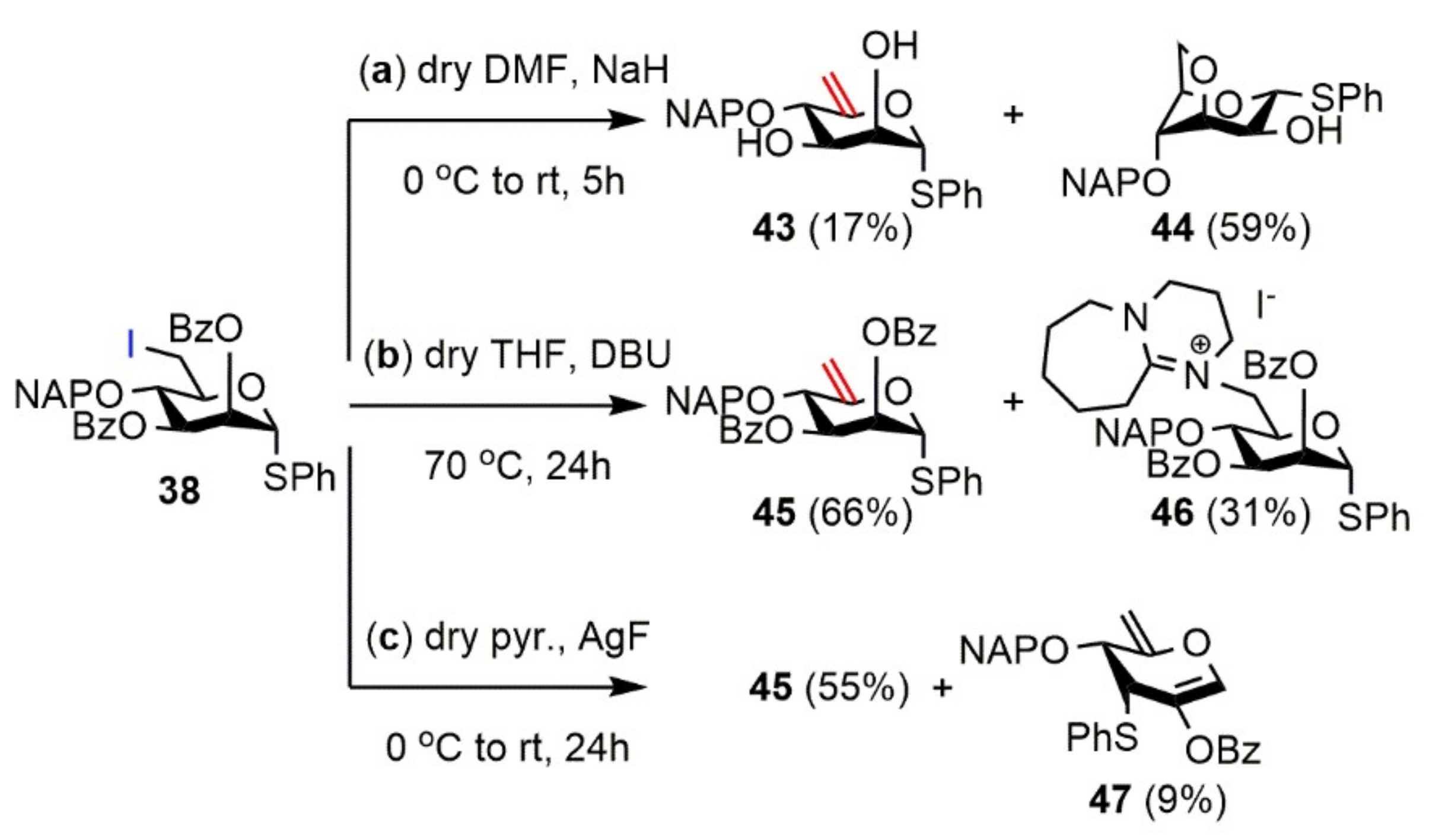
Phenyl 2-O-benzoyl-3-O-benzyl-6-deoxy-6-iodo-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (39). Compound 36 (2.77 g, 3.868 mmol) was converted to 39 according to general Method A. The crude product was purified by silica gel chromatography (8:2 n-hexane/acetone) to give 39 (3.23 g, 99%) as a colorless syrup. [α]D25 + 0.6 (c 0.14, CHCl3); Rf 0.43 (8:2 n-hexane/acetone); 1H NMR (500 MHz, CDCl3) δ = 8.16–7.24 (m, 22H, arom.), 5.90 (s, 1H, H-2), 5.62 (s, 1H, H-1), 5.15–4.60 (m, 4H, NAPCH2, BnCH2), 4.11 (d, J = 8.9 Hz, 1H, H-3), 4.02 (t, J = 9.1 Hz, 1H, H-4), 3.95 (d, J = 8.0 Hz, 1H, H-5), 3.61 (dd, J = 4.2 Hz, J = 10.8 Hz, 1H, H-6a), 3.54 (dd, J = 2.1 Hz, J = 10.7 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.7 (1C, Cq Bz), 137.6, 135.6, 133.5, 133.2, 129.8 (6C, 6 × Cq arom.), 133.5–126.1 (22C, arom.), 86.5 (1C, C-1), 78.4 (1C, C-4), 78.3 (1C, C-3), 75.8 (1C, NAPCH2), 71.8 (1C, BnCH2), 71.1 (1C, C-5), 70.6 (1C, C-2), 8.5 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H33INaO5S [M + Na]+ 739.0985; found: 739.0992.
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (40). Compound 37 (300 mg, 0.427 mmol) was converted to 40 according to general Method E. The crude product was purified by silica gel chromatography (8:2 n-hexane/acetone) to give 40 (224 mg, 91%) as a colorless syrup. [α]D25 + 62.6 (c 0.19, CHCl3); Rf 0.38 (8:2 n-hexane/acetone); 1H NMR (400 MHz, CDCl3) δ = 7.79–7.18 (m, 22H, arom.), 5.51 (d, J = 4.7 Hz, 1H, H-1), 4.86–4.57 (m, 8H, H-6a,b, 2 × BnCH2, NAPCH2), 4.32 (d, J = 7.6 Hz, 1H, H-4), 4.04 (t, J = 3.5 Hz, 1H, H-2), 3.92 (dd, J = 2.1 Hz, J = 7.6 Hz, 1H, H-3) ppm; 13C NMR (100 MHz, CDCl3) δ = 154.4 (1C, C-5), 138.2, 137.8, 135.6, 133.7, 133.3, 133.0 (6C, 6 × Cq arom.), 131.8–125.8 (22C, arom.), 99.1 (1C, C-6), 86.3 (1C, C-1), 77.3 (1C, C-3), 76.1 (1C, C-4), 75.8 (1C, C-2), 72.8, 72.7, 72.6 (3C, 2 × BnCH2, NAPCH2) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO4S [M + Na]+ 597.2070; found: 597.2058.
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (40) and 8-N-[phenyl 2,3-di-O-benzyl-6-deoxy-6-yl-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside]-1,8-diazabicyclo(5.4.0)undec-7-ene-iodide (41). Compound 37 (300 mg, 0.427 mmol) was converted to 40 according to general Method B. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane → 9:1 CH2Cl2/MeOH) to give 40 (117 mg, 48%) as a colorless syrup and 41 (175 mg, 38%) as a light yellow syrup.
Data of 41: [α]D25 + 67.7 (c 0.30, CHCl3); Rf 0.49 (9:1 CH2Cl2/MeOH); 1H NMR (500 MHz, CDCl3) δ = 7.86–7.27 (m, 22H, arom.), 5.60 (s, 1H, H-1), 5.11–4.61 (m, 6H, 2 × BnCH2, NAPCH2), 4.07 (t, J = 9.3 Hz, 1H, H-5), 3.97 (s, 1H, H-2), 3.92–3.87 (m, 2H, H-3, H-6a), 3.77 (t, J = 9.3 Hz, 1H, H-4), 3.63–3.53 (m, 3H, H-6b, NCH2 DBU), 3.46–3.40 (m, 4H, 2 × NCH2 DBU), 2.77–2.63 (m, 2H, CH2 DBU), 1.77–1.43 (m, 8H, 4 × CH2 DBU) ppm; 13C NMR (125 MHz, CDCl3) δ = 167.1 (1C, Cq DBU), 137.5, 134.9, 133.2, 133.0, 132.7 (6C, 6 × Cq arom.), 130.8–126.1 (22C, arom.), 84.5 (1C, C-1), 79.8 (1C, C-3), 75.6 (2C, C-2, C-4), 75.4, 72.7, 72.1 (3C, 2 × BnCH2, NAPCH2), 55.7 (1C, NCH2 DBU), 55.0 (1C, C-6), 49.4, 48.2 (2C, 2 × NCH2 DBU), 28.8, 28.2, 25.7, 22.5, 19.7 (5C, 5 × CH2 DBU) ppm; UHR ESI-QTOF: m/z calcd for C46H51N2O4S [M]+ 727.3564; found: 727.3562.
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (40) and Phenyl 2,3-di-O-benzyl-6-deoxy-6-fluoro-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (42). Compound 37 (300 mg, 0.427 mmol) was converted to 40 according to general Method F. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane) to give 40 (210 mg, 86%) as a colorless syrup and 42 (14 mg, 6%) as a colorless syrup.
Data of 42: [α]D25 + 60.6 (c 1.44, CHCl3); Rf 0.33 (7:3 CH2Cl2/n-hexane); 1H NMR (500 MHz, CDCl3) δ = 7.84–7.25 (m, 22H, arom.), 5.59 (d, J = 1.3 Hz, 1H, H-1), 5.14–4.54 (m, 8H, H-6a,b, 2 × BnCH2, NAPCH2), 4.26 (ddd, J = 2.7 Hz, J = 10.0 Hz, J = 27.4 Hz, 1H, H-5), 4.12 (t, J = 9.6 Hz, 1H, H-4), 4.03 (dd, J = 1.8 Hz, J = 2.9 Hz, 1H, H-2), 3.91 (dd, J = 3.0 Hz, J = 9.3 Hz, 1H, H-3) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.1, 137.8, 135.8, 134.3, 133.4, 133.1 (6C, 6 × Cq arom.), 131.4–126.1 (22C, arom.), 86.0 (1C, C-1), 82.3 (1C, JC,F = 172.9 Hz, H-6), 80.2, 76.2 (2C, C-2, C-3), 75.2 (1C, NAPCH2), 74.9 (1C, JC,F = 6.6 Hz, C-5), 72.4 (1C, C-4), 72.2 (2C, 2 × BnCH2) ppm; ESI-TOF-MS: m/z calcd for C37H35FNaO4S [M + Na]+ 617.2132; found: 617.2123.
Phenyl 4-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (43) and Phenyl 3,6-anhydro-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (44) Compound 38 (300 mg, 0.411 mmol) was converted to 43 according to general Method E. The crude product was purified by silica gel chromatography (1:1 n-hexane/EtOAc) to give 43 (21 mg, 17%) as a colorless syrup and 44 (95 mg, 59%) as a colorless syrup.
Data of 43: [α]D25 +70.4 (c 0.28, CHCl3); Rf 0.42 (1:1 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.84–7.26 (m, 12H, arom.), 5.29 (d, J = 6.2 Hz, 1H, H-1), 4.95 (s, 1H, H-6a), 4.89 (d, J = 11.9 Hz, 1H, NAPCH2a), 4.79 (s, 1H, H-6b), 4.59 (d, J = 11.9 Hz, 1H, NAPCH2b), 4.15–4.13 (m, 2H, H-2, H-3), 4.10 (d, J = 5.6 Hz, 1H, H-4), 2.53, 2.49 (2 × s, 2H, H-2-OH, H-3-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 153.8 (1C, C-5), 135.2, 133.4, 133.2, 132.5 (4C, 4 × Cq arom.), 132.3–125.8 (12C, arom.), 101.1 (1C, C-6), 87.6 (1C, C-1), 76.9 (1C, C-4), 71.5 (1C, NAPCH2), 70.3 (1C, C-3), 68.5 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for C23H22NaO4S [M + Na]+ 417.1131; found: 417.1136.
Data of 44: [α]D25 + 40.7 (c 0.28, CHCl3); Rf 0.28 (1:1 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.83–7.09 (m, 12H, arom.), 4.93 (d, J = 8.7 Hz, 1H, H-1), 4.78 (d, J = 11.9 Hz, 1H, NAPCH2a), 4.61 (d, J = 11.9 Hz, 1H, NAPCH2b), 4.47 (t, J = 2.8 Hz, 1H, H-5), 4.32 (dd, J = 0.8 Hz, J = 6.0 Hz, 1H, H-3), 4.12 (d, J = 10.9 Hz, 1H, H-6a), 3.99 (dd, J = 2.7 Hz, J = 6.0 Hz, 1H, H-4), 3.93 (dd, J = 2.7 Hz, J = 10.8 Hz, 2H, H-2, H-6b), 3.91 (t, J = 8.0 Hz, H-2), 2.46 (s, 1H, H-2-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 134.7, 133.2, 133.1, 132.7 (4C, 4 × Cq arom.), 132.5–125.7 (12C, arom.), 85.6 (1C, C-1), 77.4 (1C, C-4), 76.9 (1C, C-3), 74.2 (1C, C-5), 71.9 (1C, NAPCH2), 69.5 (1C, C-6), 67.6 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for C23H22NaO4S [M + Na]+ 417.1131; found: 417.1129.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (45) and 8-N-[phenyl 2,3-di-O-benzoyl-6-deoxy-6-yl-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside]-1,8-diazabicyclo(5.4.0)undec-7-ene-iodide (46). Compound 38 (2.3 g, 3.150 mmol) was converted to 45 according to general Method B. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane → 95:5 CH2Cl2/MeOH) to give 45 (1.25 g, 66%) as a colorless syrup and 46 (773 mg, 31%) as a light yellow syrup.
Data of 45: [α]D25 − 13.7 (c 0.27, CHCl3); Rf 0.28 (7:3 CH2Cl2/n-hexane); 1H NMR (400 MHz, CDCl3) δ = 7.89–7.25 (m, 22H, arom.), 5.84–5.82 (m, 1H, H-2), 5.78 (dd, J = 3.2 Hz, J = 8.1 Hz, 1H, H-3), 5.63 (d, J = 4.6 Hz, 1H, H-1), 5.03 (s, 1H, H-6a), 5.00 (s, 1H, H-6b), 4.96 (d, J = 12.0 Hz, 1H, NAPCH2a), 4.75 (d, J = 12.0 Hz, 1H, NAPCH2b), 4.50 (d, J = 8.1 Hz, 1H, H-4) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.2 (2C, 2 × Cq Bz), 153.4 (1C, C-5) 134.9, 133.3, 133.2, 132.5, 129.5, 129.4 (6C, 6 × Cq arom.), 133.5–126.1 (22C, arom.), 100.9 (1C, C-6), 86.1 (1C, C-1) 74.0 (1C, C-4), 72.6 (1C, NAPCH2), 70.6 (2C, C-2, C-3) ppm; ESI-TOF-MS: m/z calcd for C37H30NaO6S [M + Na]+ 625.1655; found: 625.1665.
Data of 46: [α]D25 + 27.4 (c 0.19, CHCl3); Rf 0.31 (95:5 CH2Cl2/MeOH); 1H NMR (500 MHz) δ = 8.05–7.26 (m, 22H, arom.), 5.86 (dd, J = 1.6 Hz, J = 3.1 Hz, 1H, H-2), 5.81 (d, J = 1.1 Hz, 1H, H-1), 5.78 (dd, J = 3.2 Hz, J = 9.4 Hz, 1H, H-3), 4.90 (d, J = 3.2 Hz, 2H, NAPCH2), 4.42 (t, J = 9.3 Hz, 1H, H-5), 4.23 (d, J = 15.0 Hz, 1H, H-6a), 4.12 (t, J = 9.6 Hz, 1H, H-4), 3.97 (dd, J = 9.7 Hz, J = 15.8 Hz, 1H, H-6b), 3.68–3.41 (m, 6H, 3 × NCH2 DBU), 2.80–2.79 (m, 2H, CH2 DBU), 1.94–1.56 (m, 8H, 4 × CH2 DBU) ppm; 13C NMR (125 MHz, CDCl3) δ = 167.6 (1C, Cq DBU), 165.4, 165.3 (2C, 2 × Cq Bz), 134.2, 133.1, 132.3, 129.0, 128.9 (6C, 6 × Cq arom.), 133.9–126.1 (22C, arom.), 84.6 (1C, C-1), 75.9 (1C, NAPCH2), 75.0 (1C, C-4), 72.5 (1C, C-3), 72.1 (1C, C-5), 71.6 (1C, C-2), 55.8 (1C, NCH2 DBU), 55.6 (1C, C-6), 49.5, 48.5 (2C, 2 × NCH2 DBU), 29.3, 28.5, 25.8, 22.7, 19.9 (5C, 5 × CH2 DBU) ppm; UHR ESI-QTOF: m/z calcd for C46H47N2O6S [M]+ 755.3149; found: 755.3147.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (45) and 1,5-anhydro-2-O-benzoyl-3,6-dideoxy-4-O-(2′-naphthyl)methyl-3-S-phenyl-3-thio-α-d-erythro-hex-1,5-dienitol (47). Compound 38 (300 mg, 0.411 mmol) was converted to 45 according to general Method F. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane) to give 47 (24 mg, 9%) as a colorless syrup and 45 (154 mg, 55%) as a colorless syrup.
Data of 47: [α]D25 −18.8 (c 0.25, CHCl3); Rf 0.60 (7:3 CH2Cl2/n-hexane); 1H NMR (400 MHz, CDCl3) δ = 7.94–7.27 (m, 17H, arom.), 6.54 (s, 1H, H-1), 5.19 (d, J = 6.3 Hz, 1H, H-4), 5.08 (s, 2H, NAPCH2), 5.00 (s, 1H, H-6a), 4.69 (s, 1H, H-6b), 4.09 (dd, J = 1.0 Hz, J = 6.3 Hz, 1H, H-3) ppm; 13C NMR (100 MHz, CDCl3) δ = 164.9 (1C, Cq Bz), 148.5 (1C, C-5), 147.0 (1C, C-2), 133.8, 133.4, 133.2, 133.0, 129.5 (5C, 5 × Cq arom.), 133.6–125.1 (17C, arom.), 94.1 (1C, C-6), 93.6 (1C, C-4), 92.8 (1C, C-1), 69.7 (1C, NAPCH2), 46.4 (1C, C-3) ppm; ESI-TOF-MS: m/z calcd for C30H24NaO4S [M + Na]+ 503.1288; found: 503.1281.
Phenyl 2,6-anhydro-3-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside (48). Compound 39 (310 mg, 0.433 mmol) was converted to 48 according to general Method E. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 48 (209 mg, 99%) as a colorless syrup. [α]D25 + 64.7 (c 0.15, CHCl3); Rf 0.37 (7:3 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 8.09–7.20 (m, 17H, arom.), 5.68 (s, 1H, H-1), 4.81–4.52 (m, 4H, NAPCH2, BnCH2), 4.20–4.12 (m, 4H, H-2, H-3, H-5, H-6a), 3.813 (d, J = 9.8 Hz, 1H, H-6b), 3.73 (s, 1H, H-4) ppm; 13C NMR (125 MHz, CDCl3) δ = 137.6, 135.3, 134.3, 133.3, 133.1 (5C, 5 × Cq arom.), 133.7–126.0 (17C, arom.), 86.9 (1C, C-1), 80.6 (1C, C-4), 78.9 (1C, C-3), 71.0, 70.6 (2C, NAPCH2, BnCH2), 69.9 (1C, C-2), 69.7 (1C, C-5), 66.8 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C30H28NaO4S [M + Na]+ 507.1601; found: 507.1626.
Phenyl 2-O-benzoyl-3-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-lyxo-hex-5-enopyranoside (49) and 8-N-[phenyl 2-O-benzoyl-3-O-benzyl-6-deoxy-6-yl-4-O-(2′-naphthyl)methyl-1-thio-α-d-mannopyranoside]-1,8-diazabicyclo(5.4.0)undec-7-ene-iodide (50).
Reaction I.: Compound 39 (336 mg, 0.469 mmol) was converted to 49 according to general Method B. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane → 95:5 CH2Cl2/MeOH) to give 49 (149 mg, 54%) as a colorless syrup and 50 (118 mg, 29%) as a light yellow syrup.
Reaction II.: Compound 39 (1.388 g, 1.938 mmol) was converted to 49 according to general Method F. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 49 (991 mg, 87%) as a colorless syrup.
Data of 49: [α]D25 + 40.9 (c 0.35, CHCl3); Rf 0.60 (7:3 CH2Cl2/n-hexane); 1H NMR (400 MHz, CDCl3) δ = 8.03–7.19 (m, 22H, arom.), 5.80 (d, J = 3.5 Hz, 1H, H-2), 5.62 (d, J = 4.1 Hz, H-1), 5.00 (s, 1H, H-6a), 4.94–4.62 (m, 5H, H-6b, NAPCH2, BnCH2), 4.35 (d, J = 8.1 Hz, 1H, H-4), 4.12 (dd, J = 2.9 Hz, J = 8.1 Hz, 1H, H-3) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.5 (1C, Cq Bz), 154.2 (1C, C-5), 137.7, 135.4, 133.4, 133.1, 133.0, 129.7 (6C, 6 × Cq arom.), 133.4–126.0 (22C, arom.), 99.8 (1C, C-6), 86.3 (1C, C-1), 76.8 (1C, C-3), 75.5 (1C, C-4), 73.2, 72.5 (2C, NAPCH2, BnCH2), 70.6 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO5S [M + Na]+ 611.1863; found: 611.1897.
Data of 50: [α]D25 + 67.3 (c 0.26, CHCl3); Rf 0.63 (9:1 CH2Cl2/MeOH); 1H NMR (400 MHz) δ = 8.08–7.25 (m, 22H, arom.), 5.82 (dd, J = 1.5 Hz, J = 2.7 Hz, 1H, H-2), 5.73 (s, 1H, H-1), 5.10–4.59 (m, 4H, NAPCH2, BnCH2), 4.23 (t, J = 9.4 Hz, 1H, H-5), 4.10 (dd, J = 3.1 Hz, J = 9.0 Hz, 1H, H-3), 4.04 (d, J = 15.0 Hz, 1H, H-6a), 3.79 (t, J = 9.4 Hz, 1H, H-4), 3.72 (dd, J = 9.7 Hz, J = 15.0 Hz, 1H, H-6b), 3.65–3.38 (m, 6H, 3 × NCH2 DBU), 2.79–2.65 (m, 2H, CH2 DBU), 1.84–1.47 (m, 8H, 4 × CH2 DBU) ppm; 13C NMR (100 MHz, CDCl3) δ = 167.1 (1C, Cq DBU), 165.4 (1C, Cq Bz), 137.0, 134.6, 133.1, 132.9, 132.1, 129.1 (6C, 6 × Cq arom.), 133.6–126.1 (22C, arom.), 84.7 (1C, C-1), 78.2 (1C, C-3), 75.4 (1C, NAPCH2), 75.1 (1C, C-4), 71.7 (1C, C-5), 71.6 (1C, BnCH2), 69.6 (1C, C-2), 55.6 (1C, NCH2 DBU), 55.3 (1C, C-6), 49.3, 48.2 (2C, 2 × NCH2 DBU), 29.0, 28.2, 25.6, 22.4, 19.7 (5C, 5 × CH2 DBU) ppm; UHR ESI-QTOF: m/z calcd for C46H49N2O5S [M]+ 741.3357; found: 741.3357.
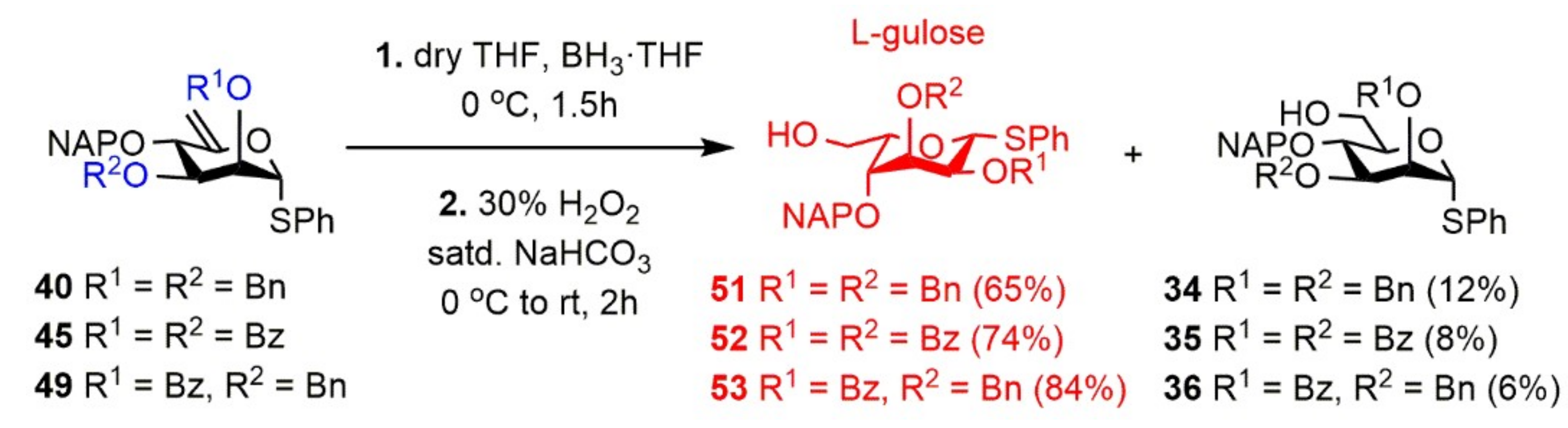
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (51). Compound 40 (574 mg, 1.000 mmol) was converted to 51 according to general Method C. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 34 (70 mg, 12%) as a colorless syrup and 51 (385 mg, 65%) as a colorless syrup.
Data of 51: [α]D25 + 3.1 (c 0.35, CHCl3); Rf 0.40 (65:35 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.85–7.17 (m, 22H, arom.), 5.24 (d, J = 9.6 Hz, 1H, H-1), 4.68–4.33 (m, 6H, 2 × BnCH2, NAPCH2), 3.98–3.95 (m, 1H, H-5), 3.89–3.86 (m, 1H, H-6a), 3.80–3.76 (m, 2H, H-2, H-3), 3.53 (td, J = 4.2 Hz, J = 11.5 Hz, 1H, H-6b), 3.45 (dd, J = 1.2 Hz, J = 3.5 Hz, 1H, H-4), 1.78 (d, J = 8.5 Hz, 1H, H-6-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 138.0, 137.8, 135.1, 134.1, 133.0 (6C, 6 × Cq arom.), 131.5–125.8 (22C, arom.), 84.1 (1C, C-1), 76.0 (1C, C-5), 75.2 (1C, C-4), 74.8 (1C, C-2), 73.2, 72.8, 72,4 (3C, 2 × BnCH2, NAPCH2), 72.6 (1C, C-3), 62.3 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H36NaO5S [M + Na]+ 615.2176; found: 615.2183.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (52). Compound 45 (1.15 g, 1.909 mmol) was converted to 52 according to general Method C. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 35 (100 mg, 8%) as a colorless syrup and 52 (870 mg, 74%) as a colorless syrup.
Data of 52: [α]D25 + 108.0 (c 0.10, CHCl3); Rf 0.32 (65:35 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 8.01–7.26 (m, 22H, arom.), 6.02 (s, 1H, H-3), 5.57 (dd, J = 2.8 Hz, J = 10.3 Hz, 1H, H-2), 5.39 (d, J = 10.3 Hz, 1H, H-1), 5.07 (d, J = 11.9 Hz, 1H, NAPCH2a), 4.74 (d, J = 11.9 Hz, 1H, NAPCH2b), 4.10–4.08 (m, 1H, H-5), 3.99–3.96 (m, 1H, H-6a), 3.79 (d, J = 1.5 Hz, 1H, H-4), 3.60–3.55 (m, 1H, H-6b), 1.79 (d, J = 6.1 Hz, 1H, C-6-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.3, 165.1 (2C, 2 × Cq Bz), 134.5, 133.2, 132.3, 129.6, 129.3 (6C, 6 × Cq arom.), 133.7–126.0 (22C, arom.), 83.2 (1C, C-1), 77.1 (1C, C-5), 73.5 (1C, C-4), 72.4 (1C, NAPCH2), 67.8 (1C, C-2), 67.7 (1C, C-3), 62.3 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO7S, [M + Na]+ 643.1761; found: 643.1743.
Phenyl 2-O-benzoyl-3-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (53). Compound 49 (325 mg, 0.553 mmol) was converted to 53 according to general Method C. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 36 (20 mg, 6%) as a colorless syrup and 53 (285 mg, 84%) as a colorless syrup.
Data of 53: [α]D25 + 35.8 (c 0.19, CHCl3); Rf 0.41 (65:35 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 8.09–7.10 (m, 22H, arom.), 5.42–5.36 (m, 2H, H-1, H-2), 4.80–4.43 (m, 4H, NAPCH2, BnCH2), 4.27–4.24 (m, 1H, H-3), 4.12–4.09 (m, 1H, H-5), 3.94 (dd, J = 7.6 Hz, J = 11.2 Hz, 1H, H-6a), 3.59 (d, J = 9.1 Hz, 1H, H-6b), 3.55 (s, 1H, H-4), 1.96 (s, 1H, C-6-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.5 (1C, Cq Bz), 137.5, 134.8, 133.2, 133.1, 129.9 (6C, 6 × Cq arom.), 133.4–126.0 (22C, arom.), 83.0 (1C, C-1), 76.5 (1C, C-5), 74.4 (1C, C-4), 73.7 (1C, NAPCH2), 72.9 (1C, C-3), 72.3 (1C, BnCH2), 70.1 (1C, C-2), 62.6 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO6S, [M + Na]+ 629.1968; found: 629.1961.
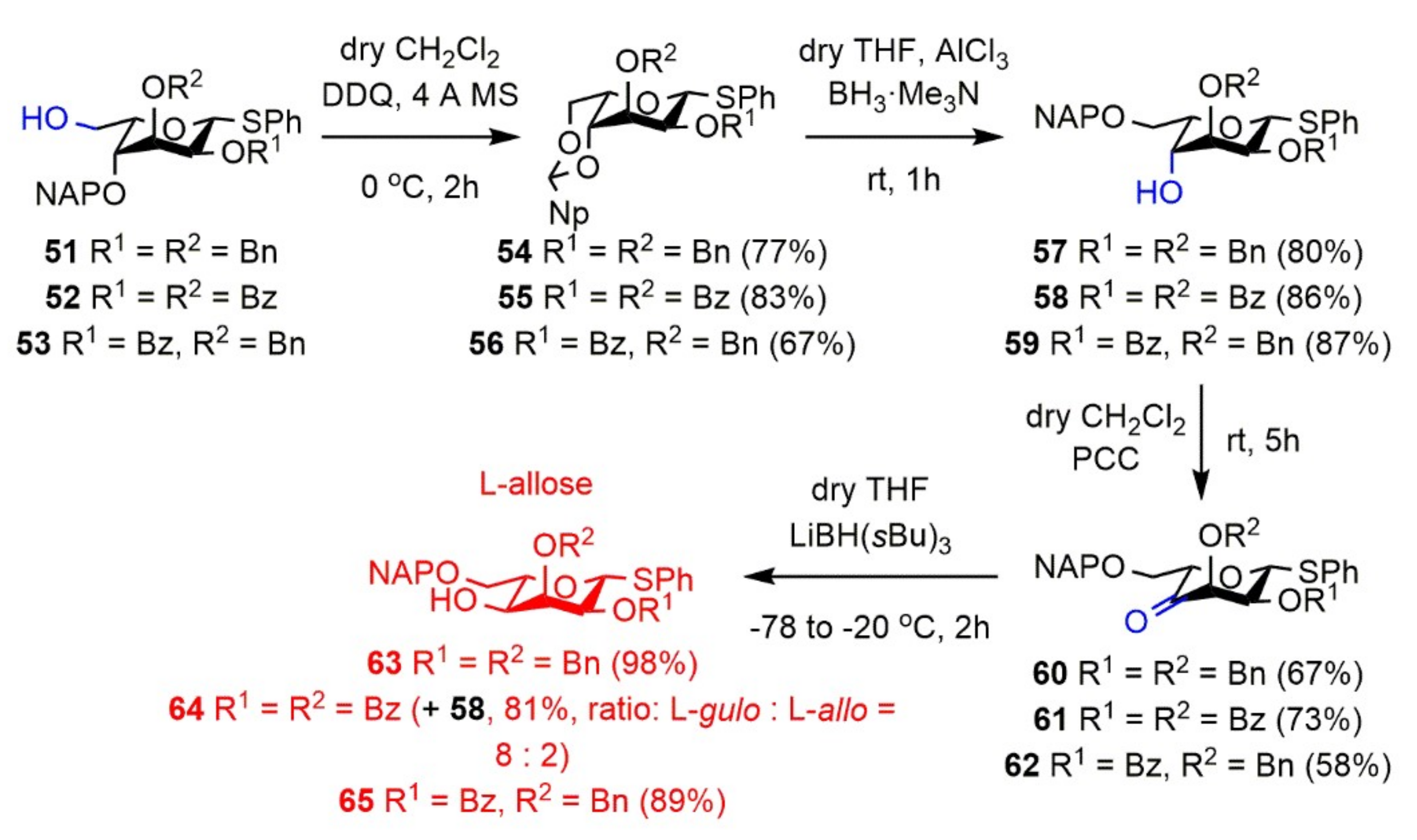
Phenyl 2,3-di-O-benzyl-4,6-O-(2′-naphthyl)methylidene-1-thio-β-l-gulopyranoside (54). Compound 51 (350 mg, 0.591 mmol) was converted to 54 according to general Method G. The crude product was purified by silica gel chromatography (7:3 n-hexane/acetone) to give 54 (267 mg, 77%) as a colorless syrup. [α]D25 + 7.5 (c 0.16, CHCl3); Rf 0.36 (8:2 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.89–7.21 (m, 22H, arom.), 5.63 (s, 1H, Hac), 5.30 (d, J = 9.8 Hz, 1H, H-1), 4.83–4.60 (m, 5H, H-6a, 2 × BnCH2), 4.11 (d, J = 3.7 Hz, 1H, H-5), 4.04 (dd, J = 1.4 Hz, J = 12.4 Hz, 1H, H-6b), 3.95–3.93 (m, 1H, H-4), 3.88 (s, 1H, H-3), 3.85 (dd, J = 2.6 Hz, J = 10.0 Hz, 1H, H-2) ppm; 13C NMR (100 MHz, CDCl3) δ = 138.3, 138.1, 135.4, 133.9, 133.2, 133.0 (6C, 6 × Cq arom.), 132.9–124.2 (22C, arom.), 101.3 (1C, Cac), 83.4 (1C, C-1), 77.3 (1C, C-5), 74.6 (1C, C-4), 74.2 (1C, C-2), 73.8, 72.7 (2C, 2 × BnCH2), 69.8 (1C, C-6), 67.7 (1C, C-3) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO5S [M + Na]+ 613.2019; found: 613.2019.
Phenyl 2,3-di-O-benzoyl-4,6-O-(2′-naphthyl)methylidene-1-thio-β-l-gulopyranoside (55). Compound 52 (870 mg, 1.402 mmol) was converted to 55 according to general Method G. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 55 (719 mg, 83%) as a colorless syrup. [α]D25 + 35.0 (c 0.14, CHCl3); Rf 0.48 (65:35 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 8.04–7.23 (m, 22H, arom.), 5.82 (s, 1H, H-3), 5.72 (s, 1H, Hac), 5.48 (dd, J = 2.7 Hz, J = 10.2 Hz, 1H, H-2), 5.40 (d, J = 10.2 Hz, 1H, H-1), 4.50 (d, J = 12.4 Hz, 1H, H-6a), 4.32 (d, J = 2.9 Hz, 1H, H-4), 4.15 (d, J = 12.3 Hz, 1H, H-6b), 4.01 (s, 1H, H-5) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.0, 164.7 (2C, 2 × Cq Bz), 134.9, 134.0, 133.0, 130.9, 129.7, 129.5 (6C, 6 × Cq arom.), 134.2–124.2 (22C, arom.), 101.6 (1C, Cac), 82.2 (1C, C-1), 74.2 (1C, C-4), 69.6 (1C, C-3), 69.5 (1C, C-6), 68.5 (1C, C-5), 66.7 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for C37H30NaO7S [M + Na]+ 641.1604; found: 641.1606.
Phenyl 2-O-benzoyl-3-O-benzyl-4,6-O-(2′-naphthyl)methylidene-1-thio-β-l-gulopyranoside (56). Compound 53 (270 mg, 0.445 mmol) was converted to 56 according to general Method G. The crude product was purified by silica gel chromatography (85:15 CH2Cl2/n-hexane) to give 56 (179 mg, 67%) as a colorless syrup. [α]D25 + 40.9 (c 0.21, CHCl3); Rf 0.63 (85:15 CH2Cl2/n-hexane); 1H NMR (400 MHz, CDCl3) δ = 8.07–7.15 (m, 22H, arom.), 5.61 (s, 1H, Hac), 5.44 (dd, J = 2.4 Hz, J = 10.3 Hz, 1H, H-1), 5.33 (dt, J = 3.2 Hz, J = 10.3 Hz, 1H, H-2), 4.58 (s, 2H, BnCH2), 4.37 (d, J = 12.3 Hz, 1H, H-6a), 4.18 (s, 1H, H-3), 4.11 (d, J = 3.5 Hz, 1H, H-4), 4.01–3.98 (m, 1H, H-6b), 3.89 (s, 1H, H-5) ppm; 13C NMR (100 MHz, CDCl3) δ = 164.9 (1C, Cq Bz), 137.6, 135.2, 133.8, 132.8, 131.6 (6C, 6 × Cq arom.), 133.5–124.1 (22C, arom.) 101.2 (1C, Cac), 81.9 (1C, C-1), 75.0 (1C, C-3), 74.6 (1C, C-4), 73.8 (1C, BnCH2), 69.5 (1C, C-6), 68.9 (1C, C-2), 67.9 (1C, C-5) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO6S, [M + Na]+ 627.1812; found: 627.1822.
Phenyl 2,3-di-O-benzyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (57). Compound 54 (250 mg, 0.423 mmol) was converted to 57 according to general Method H. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 57 (200 mg, 80%) as a colorless syrup. [α]D25 + 24.4 (c 0.18, CHCl3); Rf 0.46 (65:35 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.79–7.16 (m, 22H, arom.), 5.26 (d, J = 10.0 Hz, 1H, H-1), 4.74–4.48 (m, 6H, 2 × BnCH2, NAPCH2), 4.06 (t, J = 3.8 Hz, 1H, H-5), 3.96 (s, 1H, H-4), 3.90 (t, J = 3.6 Hz, 1H, H-3), 3.83 (dd, J = 2.9 Hz, J = 9.9 Hz, 1H, H-2), 3.81 (dd, J = 3.4 Hz, J = 10.3 Hz, 1H, H-6a), 3.75 (dd, J = 4.4 Hz, J = 10.6 Hz, 1H, H-6b), 3.43 (s, 1H, C-4-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 138.2, 137.9, 134.9, 134.0, 133.1, 132.9 (6C, 6 × Cq arom.), 131.5–125.5 (22C, arom.), 84.7 (1C, C-1), 75.4 (1C, C-2), 74.7 (1C, C-3), 73.9 (1C, C-5), 73.7, 73.3, 72.5 (3C, 2 × BnCH2, NAPCH2), 70.9 (1C, C-6), 70.0 (1C, C-4) ppm; ESI-TOF-MS: m/z calcd for C37H36NaO5S [M + Na]+ 615.2176; found: 615.2181.
Phenyl 2,3-di-O-benzoyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (58). Compound 55 (90 mg, 0.145 mmol) was converted to 58 according to general Method H. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 58 (78 mg, 86%) as a colorless syrup. [α]D25 + 43.5 (c 0.17, CHCl3); Rf 0.34 (7:3 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.99–7.21 (m, 22H, arom.), 5.77 (t, J = 3.5 Hz, 1H, H-3), 5.60 (dd, J = 3.3 Hz, J = 10.4 Hz, 1H, H-2), 5.36 (d, J = 10.4 Hz, 1H, H-1), 4.76 (d, J = 3.3 Hz, 2H, NAPCH2), 4.23 (t, J = 4.4 Hz, 1H, H-5), 4.14 (d, J = 3.5 Hz, 1H, H-4), 3.91 (d, J = 4.7 Hz, 2H, H-6a,b), 3.47 (s, 1H, H-4-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.1, 164.9 (2C, 2 × Cq Bz), 135.0, 133.3, 133.1, 132.5, 129.5, 129.4 (6C, 6 × Cq arom.), 133.6–125.8 (22C, arom.), 83.8 (1C, C-1), 75.5 (1C, C-5), 74.1 (1C, NAPCH2), 70.7 (1C, C-3), 70.3 (1C, C-6), 69.1 (1C, C-4), 67.1 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO7S [M + Na]+ 643.1761; found: 643.1762.
Phenyl 2-O-benzoyl-3-O-benzyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (59). Compound 56 (428 mg, 0.708 mmol) was converted to 59 according to general Method H. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 59 (373 mg, 87%) as a colorless syrup. [α]D25 + 20.4 (c 0.23, CHCl3); Rf 0.44 (65:35 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 8.08–7.19 (m, 22H, arom.), 5.39 (dd, J = 2.7 Hz, J = 10.4 Hz, 1H, H-2), 5.36 (d, J = 10.4 Hz, 1H, H-1), 4.82–4.56 (m, 4H, NAPCH2, BnCH2), 4.20 (t, J = 4.2 Hz, 1H, H-5), 4.12–4.11 (m, 1H, H-3), 4.03 (t, J = 4.1 Hz, 1H, H-4), 3.90 (dd, J = 4.1 Hz, J = 10.6 Hz, 1H, H-6a), 3.85 (dd, J = 4.4 Hz, J = 10.6 Hz, 1H, H-6b), 3.23 (d, J = 4.3 Hz, 1H, C-4-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.1 (1C, Cq Bz), 137.8, 135.1, 133.1, 132.9, 129.7 (6C, 6 × Cq arom.), 133.2–125.6 (22C, arom.) 83.6 (1C, C-1), 76.5 (1C, C-3), 74.6 (1C, C-5), 73.8, 73.6 (2C, NAPCH2, BnCH2), 70.6 (1C, C-6), 69.5 (1C, C-4), 69.1 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO6S, [M + Na]+ 629.1968; found: 629.1954.
Phenyl 2,3-di-O-benzyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-ribo-hexopyranos-4-uloside (60). Compound 57 (160 mg, 0.270 mmol) was converted to 60 according to general Method I. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 60 (107 mg, 67%) as a colorless syrup. [α]D25 + 1.2 (c 0.17, CHCl3); Rf 0.55 (7:3 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.81–7.17 (m, 22H, arom.), 5.16 (d, J = 2.6 Hz, 1H, H-1), 4.73–4.54 (m, 6H, 2 × BnCH2, NAPCH2), 4.46 (d, J = 3.5 Hz, 1H, H-3), 4.32–4.29 (m, 1H, H-5), 4.19 (t, J = 3.0 Hz, 1H, H-2), 3.88 (qd, J = 3.4 Hz, J = 10.8 Hz, 2H, H-6a,b) ppm; 13C NMR (100 MHz, CDCl3) δ = 206.0 (1C, C-4), 137.4, 137.3, 135.5, 133.3, 133.1, 132.2 (6C, 6 × Cq arom.), 133.0–125.7 (22C, arom.), 86.4 (1C, C-1), 82.5 (1C, C-5), 82.1 (1C, C-3), 80.1 (1C, C-2), 73.7, 73.3, 72.8 (3C, 2 × BnCH2, NAPCH2), 69.6 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO5S [M + Na]+ 613.2019; found: 613.2011.
Phenyl 2,3-di-O-benzoyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-ribo-hexopyranos-4-uloside (61). Compound 58 (69 mg, 0.112 mmol) was converted to 61 according to general Method I. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 61 (50 mg, 73%) as a colorless syrup. [α]D25 − 17.5 (c 0.12, CHCl3); Rf 0.34 (7:3 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.98–7.24 (m, 22H, arom.), 6.12 (dd, J = 2.8 Hz, J = 4.2 Hz, 1H, H-2), 6.07 (d, J = 4.2 Hz, 1H, H-3), 5.31 (d, J = 2.7 Hz, 1H, H-1), 4.77 (s, 2H, NAPCH2), 4.51 (t, J = 3.8 Hz, 1H, H-5), 4.00 (d, J = 3.9 Hz, 2H, H-6a,b) ppm; 13C NMR (100 MHz, CDCl3) δ = 201.0 (1C, C-4), 165.0, 164.7 (2C, 2 × Cq Bz), 135.3, 133.4, 133.2, 131.3, 128.9 (6C, 6 × Cq arom.), 133.9–125.7 (22C, arom.), 85.9 (1C, C-1), 83.4 (1C, C-5), 74.7, 74.4 (2C, C-2, C-3), 73.9 (1C, NAPCH2), 69.5 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H30NaO7S [M + Na]+ 641.1604; found: 641.1607.
Phenyl 2-O-benzoyl-3-O-benzyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-ribo-hexopyranos-4-uloside (62). Compound 59 (342 mg, 0.563 mmol) was converted to 62 according to general Method I. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 62 (196 mg, 58%) as a colorless syrup. [α]D25 − 16.2 (c 0.08, CHCl3); Rf 0.42 (7:3 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.97–7.16 (m, 22H, arom.), 5.85 (t, J = 4.0 Hz, 1H, H-2), 5.22 (d, J = 3.9 Hz, 1H, H-1), 4.67 (d, J = 2.3 Hz, 2H, NAPCH2), 4.55 (d, J = 12.2 Hz, 1H, BnCH2a), 4.46–4.40 (m, 3H, H-3, H-5, BnCH2b), 3.92 (d, J = 3.6 Hz, 2H, H-6) ppm; 13C NMR (100 MHz, CDCl3) δ = 203.9 (1C, C-4), 165.0 (1C, Cq Bz), 136.7, 135.2, 133.2, 133.0, 131.3, 129.1 (6C, 6 × Cq arom.), 133.6–125.6 (22C, arom.), 85.1 (1C, C-1), 82.4 (1C, C-5), 79.3 (1C, C-3), 73.8 (1C, C-2), 73.7, 72.2 (2C, NAPCH2, BnCH2), 69.4 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO6S, [M + Na]+ 627.1812; found: 627.1809.
Phenyl 2,3-di-O-benzyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-allopyranoside (63). Compound 60 (100 mg, 0.169 mmol) was converted to 63 according to general Method J. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 63 (98 mg, 98%) as a colorless syrup. [α]D25 + 10.7 (c 0.29, CHCl3); Rf 0.33 (65:35 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.78–7.19 (m, 22H, arom.), 5.20 (d, J = 7.1 Hz, 1H, H-1), 5.05 (d, J = 9.7 Hz, 1H, BnCH2a), 4.72–4.60 (m, 4H, NAPCH2, BnCH2), 4.56 (d, J = 10.8 Hz, 1H, BnCH2b), 4.05 (s, 1H, H-3), 3.83–3.81 (m, 2H, H-4, H-6a), 3.69–3.67 (m, 1H, H-6b), 3.54–3.51 (m, 1H, H-5), 3.44 (d, J = 8.3 Hz, 1H, H-2), 2.40 (s, 1H, C-4-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.4, 137.6, 135.9, 134.2, 133.3, 133.0 (6C, 6 × Cq arom.), 131.6–125.8 (22C, arom.), 84.0 (1C, C-1), 78.4 (1C, C-2), 77.5 (1C, C-3), 76.7 (1C, C-5), 75.3, 73.6, 73.1 (3C, 2 × BnCH2, NAPCH2), 70.2 (1C, C-6), 68.1 (1C, C-4) ppm; ESI-TOF-MS: m/z calcd for C37H36NaO5S [M + Na]+ 615.2176; found: 615.2132.
Phenyl 2,3-di-O-benzoyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-gulopyranoside (58) and Phenyl 2,3-di-O-benzoyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-allopyranoside (64). Compound 61 (27 mg, 0.044 mmol) was converted to 64 according to general Method J. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give mixture of 58 and 64 (22 mg, 81%, inseparable mixture of the l-gulo and the l-allo configured compounds, ratio of l-gulo (58): l-allo (64) = 5:1 based on the 1H NMR spectra (H-3 l-gulo: 5.75 ppm and H-3 l-allo: 6.10 ppm) as a colorless syrup.
Data of 64: Rf 0.36 (7:3 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 8.01–7.21 (m, 22H, arom.), 6.10 (t, J = 2.9 Hz, 1H, H-3), 5.27 (dd, J = 2.8 Hz, J = 10.2 Hz, 1H, H-2), 5.10 (d, J = 9.9 Hz, 1H, H-1), 4.78–4.67 (m, 2H, NAPCH2), 4.36 (ddd, J = 2.2 Hz, J = 5.4 Hz, J = 10.3 Hz, 1H, H-5), 3.82 (dd, J = 3.3 Hz, J = 6.5 Hz, 1H, H-4), 3.79 (dd, J = 2.2 Hz, J = 11.0 Hz, 1H, H-6a), 3.71 (dd, J = 5.5 Hz, J = 11.0 Hz, 1H, H-6b), 2.43 (d, J = 3.6 Hz, 1H, C-4-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 133.4–125.9 (22C, arom.), 85.5 (1C, C-1), 75.0 (1C, C-5), 74.1 (1C, NAPCH2), 70.3 (1C, C-3), 68.9 (1C, C-6), 68.1 (1C, C-4), 67.7 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for C37H32NaO7S [M + Na]+ 643.1761; found: 643.1828.
Phenyl 2-O-benzoyl-3-O-benzyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-allopyranoside (65). Compound 62 (174 mg, 0.287 mmol) was converted to 65 according to general Method J. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 65 (155 mg, 89%) as a colorless syrup. [α]D25 + 25.9 (c 0.17, CHCl3); Rf 0.38 (7:3 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 8.09–7.18 (m, 22H, arom.), 5.33 (d, J = 10.2 Hz, 1H, H-1), 4.99 (d, J = 10.0 Hz, 1H, H-2), 4.83 (d, J = 11.4 Hz, 1H, BnCH2a), 4.75 (s, 2H, NAPCH2), 4.56 (d, J = 11.4 Hz, 1H, BnCH2b), 4.30 (s, 1H, H-3), 3.94–3.93 (m, 1H, H-5), 3.88 (d, J = 10.5 Hz, 1H, H-6a), 3.79–3.72 (m, 2H, H-4, H-6b), 2.44 (d, J = 9.3 Hz, 1H, C-4-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.3 (1C, Cq Bz), 137.9, 135.7, 133.4, 133.1, 132.7, 129.6 (6C, 6 × Cq arom.), 133.6–125.8 (22C, arom.), 82.5 (1C, C-1), 77.9 (1C, C-3), 76.9 (1C, C-5), 75.8, 73.8 (2C, NAPCH2, BnCH2), 71.7 (1C, C-2), 70.4 (1C, C-6), 68.5 (1C, C-4) ppm; ESI-TOF-MS: m/z calcd for C37H34NaO6S, [M + Na]+ 629.1968; found: 629.1964.
3.2.13. Synthesis of l-Galactose and l-Glucose Derivatives from d-Altrose
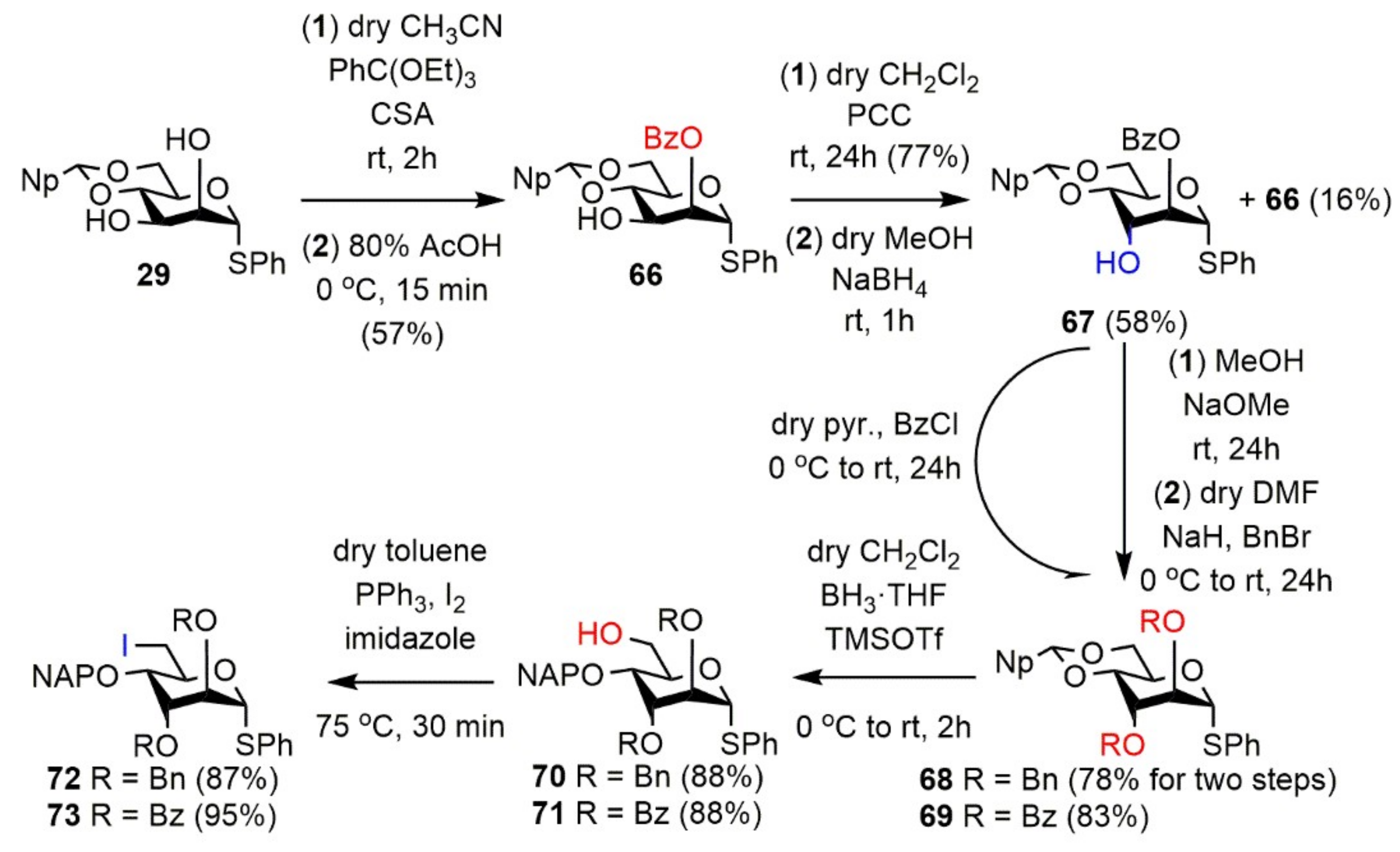
Phenyl 2-O-benzoyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-mannopyranoside (
66). To a solution of compound
29 [
91] (2.5 g, 6.096 mmol) in dry CH
3CN (85 mL), we added triethyl-orthobenzoate (2.22 mL, 9.753 mmol, 1.6 equiv.) and stirred for 15 min. After 15 min at that temperature, CSA (425 mg, 1.828 mmol, 0.30 equiv.) was added and the mixture was stirred for 2 h at room temperature. After that, the solvents were evaporated. The residue was dissolved in AcOH (80%, 40 mL) at 0 °C and stirred for 15 min. The reaction mixture was neutralized with NaHCO
3, extracted with CH
2Cl
2 (3 × 100 mL), and the organic phases was washed with H
2O (3 × 50 mL) until neutral pH. The organic layer was dried over MgSO
4 and concentrated. The crude product was purified by silica gel chromatography (7:3
n-hexane/acetone) to give
66 (1.81 g, 57%) as a colorless syrup. [α]
D25 −6.2 (
c 0.13, CHCl
3);
Rf 0.42 (7:3
n-hexane/acetone);
1H NMR (500 MHz, CDCl
3)
δ = 8.11–7.24 (m, 17H, arom.), 5.83 (s, 1H, H
ac), 5.75 (dd,
J = 1.3 Hz,
J = 3.5 Hz, 1H, H-2), 5.64 (d,
J = 1.0 Hz, 1H, H-1), 4.49 (td,
J = 4.9 Hz,
J = 9.8 Hz, 1H, H-5), 4.39 (dt,
J = 3.7 Hz,
J = 9.8 Hz, 1H, H-3), 4.33 (dd,
J = 4.9 Hz,
J = 10.3 Hz, 1H, H-6a), 4.18 (t,
J = 9.7 Hz, 1H, H-4), 3.95 (t,
J = 10.3 Hz, 1H, H-6b), 2.56 (dd,
J = 4.1 Hz,
J = 9.4 Hz, 1H, C-3-O
H) ppm;
13C NMR (125 MHz, CDCl
3)
δ = 166.1 (1C, C
q Bz), 134.5, 133.9, 133.2, 133.1, 129.6 (5C, 5 × C
q arom.), 133.7–123.8 (17C, arom.), 102.6 (1C, C
ac), 87.2 (1C, C-1), 79.8 (1C, C-4), 74.3 (1C, C-2), 68.7 (1C, C-6), 68.3 (1C, C-3), 64.9 (1C, C-5) ppm; ESI-TOF-MS:
m/z calcd for: C
30H
26NaO
6S [M + Na]
+ 537.1342; found: 537.1348.
Phenyl 2-O-benzoyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-altropyranoside (67). Compound 66 (5.68 g, 11.05 mmol) was converted to 3-ulose according to general Method I. The crude product was purified by silica gel chromatography (6:4 n-hexane/EtOAc) to give the 3-ulose derivative (4.37 g, 77%) as a colorless syrup. [α]D25 + 112.0 (c 0.15, CHCl3); Rf 0.39 (7:3 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 8.15–7.20 (m, 17H, arom.), 5.92 (s, 1H, H-1), 5.81 (s, 1H, Hac), 5.54 (s, 1H, H-2), 4.98 (d, J = 9.7 Hz, 1H, H-4), 4.77–4.75 (m, 1H, H-5), 4.41–4.30 (m, 1H, H-6a), 4.05 (t, J = 10.0 Hz, 1H, H-6b) ppm; 13C NMR (100 MHz, CDCl3) δ = 193.4 (1C, C-3), 164.8 (1C, Cq Bz), 133.8, 133.7, 132.9, 131.4 (5C, 5 × Cq arom.), 134.1–123.8 (17C, arom.), 102.2 (1C, Cac), 88.6 (1C, C-1), 81.9 (1C, C-4), 78.6 (1C, C-2), 69.1 (1C, C-6), 68.1 (1C, C-5) ppm; ESI-TOF-MS: m/z calcd for: C30H24NaO6S [M + Na]+ 535.1186; found: 535.1210.
To the solution of the 3-ulose compound (4.44 g, 8.669 mmol) in dry MeOH (53.6 mL), NaBH4 (492 mg, 13.00 mmol, 1.5 equiv.) was added and the reaction mixture was stirred for 1 h at room temperature. After 1 h the mixture was neutralized with 60% AcOH (6 mL) and concentrated, then the residue was coevaporated with MeOH (3 × 100 mL). The crude product was purified by silica gel chromatography (CH2Cl2) to give 67 (2.61 g, 58%) as a white foam and 66 (710 mg, 16%) as a colorless syrup.
Data of 67: [α]D25 +167.0 (c 0.10, CHCl3); Rf 0.40 (CH2Cl2); 1H NMR (500 MHz, CDCl3) δ = 8.08–7.25 (m, 17H, arom.), 5.90 (s, 1H, Hac), 5.62 (d, J = 3.2 Hz, 1H, H-2), 5.55 (s, 1H, H-1), 4.93 (td, J = 5.1 Hz, J = 10.1 Hz, 1H, H-5), 4.44 (dd, J = 5.1 Hz, J = 10.3 Hz, 1H, H-6a), 4.38 (s, 1H, H-3), 4.17 (dd, J = 2.9 Hz, J = 9.8 Hz, 1H, H-4), 3.97 (t, J = 10.4 Hz, 1H, H-6b), 2.58 (d, J = 1.7 Hz, 1H, C-3-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 165.0 (1C, Cq Bz), 136.1, 134.5, 133.9, 133.0, 129.3 (5C, 5 × Cq arom.), 133.7–123.8 (17C, arom.), 102.5 (1C, Cac), 86.3 (1C, C-1), 77.4 (1C, C-4), 74.1 (1C, C-2), 69.1 (1C, C-6), 66.9 (1C, C-3), 59.6 (1C, C-5) ppm; ESI-TOF-MS: m/z calcd for: C30H26NaO6S [M + Na]+ 537.1342; found: 537.1389.
Phenyl 2,3-di-O-benzyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-altropyranoside (68). To the solution of compound 67 (888 mg, 1.728 mmol) in MeOH (21 mL), NaOMe (55 mg) was added, and the reaction mixture was stirred for 24 h at room temperature. After 24 h the reaction mixture was neutralized by Amberlite IR-120 (H+) ion-exchange resin, then it was filtered, washed with MeOH, and concentrated. The crude product was reacted in the further reaction without purification (Rf 0.35 (6:4 n-hexane/EtOAc)). To a solution of the crude product (709 mg, 1.728 mmol), dry DMF (7.3 mL) at 0 °C NaH (60%, 173 mg, 4.320 mmol, 1.25 equiv./-OH) was added in portions. After 30 min stirring at this temperature, BnBr (513 μL, 4.320 mmol, 1.25 equiv./-OH) was added, and the reaction mixture was allowed to warm up to room temperature and stirred for 24 h. After completion of the reaction, MeOH (20 mL) was added. The reaction mixture was stirred for 15 min, then the solvents were evaporated. The residue was diluted with CH2Cl2 (500 mL), washed with H2O (3 × 100 mL), dried over MgSO4, filtered, and concentrated. The crude product was purified by silica gel chromatography (8:2 n-hexane/EtOAc) to give 68 (796 mg, 78% for two steps) as a yellow syrup. [α]D25 −145.0 (c 0.12, CHCl3); Rf 0.49 (8:2 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.96–7.25 (m, 22H, arom.), 5.75 (s, 1H, Hac), 5.49 (s, 1H, H-1), 4.88–4.85 (m, 2H, H-5, BnCH2a), 4.74 (d, J = 12.5 Hz, 1H, BnCH2b), 4.61 (d, J = 11.9 Hz, 1H, BnCH2a), 4.52 (d, J = 11.9 Hz, 1H, BnCH2b), 4.37 (dd, J = 5.0 Hz, J = 10.0 Hz, 1H, H-6a), 4.18 (d, J = 9.5 Hz, 1H, H-4), 4.03–4.01 (m, 2H, H-2, H-3), 3.89 (t, J = 10.3 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.5, 137.4, 137.3, 135.3, 133.8, 133.1 (6C, 6 × Cq arom.), 130.8–124.0 (22C, arom.), 102.5 (1C, Cac), 86.4 (1C, C-1), 78.9 (1C, C-2), 77.7 (1C, C-4), 73.5 (1C, C-3), 73.2, 72.4 (2C, 2 × BnCH2), 69.4 (1C, C-6), 60.3 (1C, C-5) ppm; ESI-TOF-MS: m/z calcd for: C37H34NaO5S [M + Na]+ 613.2019; found: 613.2014.
Phenyl 2,3-di-O-benzoyl-4,6-O-(2′-naphthyl)methylidene-1-thio-α-d-altropyranoside (69). To a stirred solution of compound 67 (892 mg, 1.735 mmol) in dry pyridine (4.7 mL), BzCl (252 µL, 2.169 mmol, 1.25 equiv.) was added at 0 °C and the reaction mixture was stirred for 24 h at room temperature. After 24 h the mixture was diluted with CH2Cl2 (100 mL), washed with H2O (25 mL), 1M aqueous solution of H2SO4 (2 × 25 mL), H2O (25 mL), saturated aqueous solution of NaHCO3 (2 × 25 mL), and H2O (25 mL) until neutral pH. The organic layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (7:3 → 8:2 CH2Cl2/n-hexane) to give 69 (887 mg, 83%) as a colorless syrup. [α]D25 + 174.2 (c 0.12, CHCl3); Rf 0.49 (7:3 CH2Cl2/n-hexane); 1H NMR (500 MHz, CDCl3) δ = 8.34–7.23 (m, 22H, arom.), 5.88 (s, 1H, Hac), 5.83 (s, 1H, H-3), 5.72 (s, 1H, H-2), 5.61 (s, 1H, H-1), 5.05 (td, J = 5.2 Hz, J = 9.9 Hz, 1H, H-5), 4.47 (dd, J = 5.0 Hz, J = 10.3 Hz, 1H, H-6a), 4.38 (d, J = 9.6 Hz, 1H, H-4), 3.99 (t, J = 10.4 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.4, 164.9 (2C, 2 × Cq Bz), 135.2, 134.5, 133.8, 133.0, 129.5, 129.2 (6C, 6 × Cq arom.), 133.9–123.7 (22C, arom.), 102.3 (1C, Cac), 86.5 (1C, C-1), 75.6 (1C, C-4), 72.6 (1C, C-2), 69.3 (1C, C-6), 67.3 (1C, C-3), 60.7 (1C, C-5) ppm; ESI-TOF-MS: m/z calcd for: C37H30NaO7S [M + Na]+ 641.1604; found: 641.1608.
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-altropyranoside (70). Compound 68 (750 mg, 1.270 mmol) was converted to 70 according to general Method D. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 70 (660 mg, 88%) as a colorless syrup. [α]D25 + 140.0 (c 0.17, CHCl3); Rf 0.48 (65:35 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.83–7.12 (m, 22H, arom.), 5.44 (s, 1H, H-1), 4.73–4.33 (m, 7H, H-5, NAPCH2, 2 × BnCH2), 3.96–3.94 (m, 2H, H-2, H-4), 3.93–3.88 (m, 2H, H-6a,b), 3.83 (s, 1H, H-4), 1.89 (s, 1H, C-6-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.0, 137.5, 137.4, 135.6, 133.4, 133.1 (6C, 6 × Cq arom.), 131.1–126.0 (22C, arom.), 86.2 (1C, C-1), 77.4 (1C, C-2), 72.8 (1C, C-4), 72.6 (1C, C-3), 72.2, 72.2, 71.7 (3C, NAPCH2, 2 × BnCH2), 68.6 (1C, C-5), 62.7 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C37H36NaO5S [M + Na]+ 615.2176; found: 615.2173.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-altropyranoside (71). Compound 69 (823 mg, 1.331 mmol) was converted to 71 according to general Method D. The crude product was purified by silica gel chromatography (65:35 n-hexane/EtOAc) to give 71 (728 mg, 88%) as a colorless syrup. [α]D25 +131.8 (c 0.11, CHCl3); Rf 0.49 (6:4 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 8.35–7.25 (m, 22H, arom.), 5.94 (s, 1H, H-3), 5.59 (d, J = 3.1 Hz, 1H, H-2), 5.56 (s, 1H, H-1), 4.93 (d, J = 11.5 Hz, 1H, NAPCH2a), 4.77–4.72 (m, 2H, H-5, NAPCH2b), 4.14 (dd, J = 2.7 Hz, J = 9.9 Hz, 1H, H-4), 3.97 (d, J = 11.6 Hz, 1H, H-6a), 3.91–3.87 (m, 1H, H-6b), 1.86 (t, J = 6.2 Hz, 1H, C-6-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.6, 164.9 (2C, 2 × Cq Bz), 135.4, 134.7, 133.3, 133.2, 129.5, 129.1 (6C, 6 × Cq arom.), 133.7–126.2 (22C, arom.), 86.1 (1C, C-1), 72.5 (1C, C-2), 71.5 (1C, NAPCH2), 70.5 (1C, C-4), 68.6 (1C, C-5), 66.1 (1C, C-3), 62.5 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C37H32NaO7S [M + Na]+ 643.1761; found: 643.1821.
Phenyl 2,3-di-O-benzyl-6-deoxy-6-iodo-4-O-(2′-naphthyl)methyl-1-thio-α-d-altropyranoside (72). Compound 70 (650 mg, 1.095 mmol) was converted to 72 according to general Method A. The crude product was purified by silica gel chromatography (4:6 → 8:2 CH2Cl2/n-hexane) to give 72 (665 mg, 87%) as a white foam. [α]D25 + 93.3 (c 0.12, CHCl3); Rf 0.57 (7:3 CH2Cl2/n-hexane); 1H NMR (500 MHz, CDCl3) δ = 7.85–7.19 (m, 22H, arom.), 5.51 (s, 1H, H-1), 4.72–4.48 (m, 5H, NAPCH2, 2 × BnCH2a, BnCH2b), 4.40 (t, J = 6.8 Hz, 1H, H-5), 4.36 (d, J = 12.2 Hz, 1H, BnCH2b), 4.01 (d, J = 1.9 Hz, 1H, H-2), 3.81–3.79 (m, 2H, H-3, H-4), 3.64–3.62 (m, 1H, H-6a), 3.47 (dd, J = 6.5 Hz, J = 10.6 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 137.8, 137.7, 137.6, 135.3, 133.3, 133.1 (6C, 6 × Cq arom.), 131.0–126.1 (22C, arom.), 86.2 (1C, C-1), 77.2 (1C, C-2), 76.7 (1C, C-3), 72.5, 72.0, 71.6 (3C, NAPCH2, 2 × BnCH2), 72.1 (1C, C-4), 67.5 (1C, C-5), 8.7 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C37H35INaO4S [M + Na]+ 725.1193; found: 725.1195.
Phenyl 2,3-di-O-benzoyl-6-deoxy-6-iodo-4-O-(2′-naphthyl)methyl-1-thio-α-d-altropyranoside (73). Compound 71 (676 mg, 1.089 mmol) was converted to 73 according to general Method A. The crude product was purified by silica gel chromatography (1:1 → 8:2 CH2Cl2/n-hexane) to give 73 (759 mg, 95%) as a colorless syrup. [α]D25 + 99.2 (c 0.13, CHCl3); Rf 0.46 (8:2 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 8.34–7.26 (m, 22H, arom.), 5.95 (s, 1H, H-3), 5.64 (s, 1H, H-2), 5.59 (s, 1H, H-1), 4.96 (d, J = 11.1 Hz, 1H, NAPCH2a), 4.72 (d, J = 11.1 Hz, 1H, NAPCH2b), 4.46–4.43 (m, 1H, H-5), 4.02 (d, J = 9.2 Hz, 1H, H-4), 3.64 (d, J = 10.6 Hz, 1H, H-6a), 3.52 (dd, J = 5.7 Hz, J = 10.5 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.5, 164.8 (2C, 2 × Cq Bz), 135.6, 134.4, 133.3, 129.3, 129.1 (6C, 6 × Cq arom.), 133.7–126.3 (22C, arom.), 86.3 (1C, C-1), 74.5 (1C, C-4), 72.4 (1C, C-2), 71.7 (1C, NAPCH2), 67.3 (1C, C-5), 65.6 (1C, C-3), 8.3 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C37H31INaO6S [M + Na]+ 753.0778; found: 753.0792.
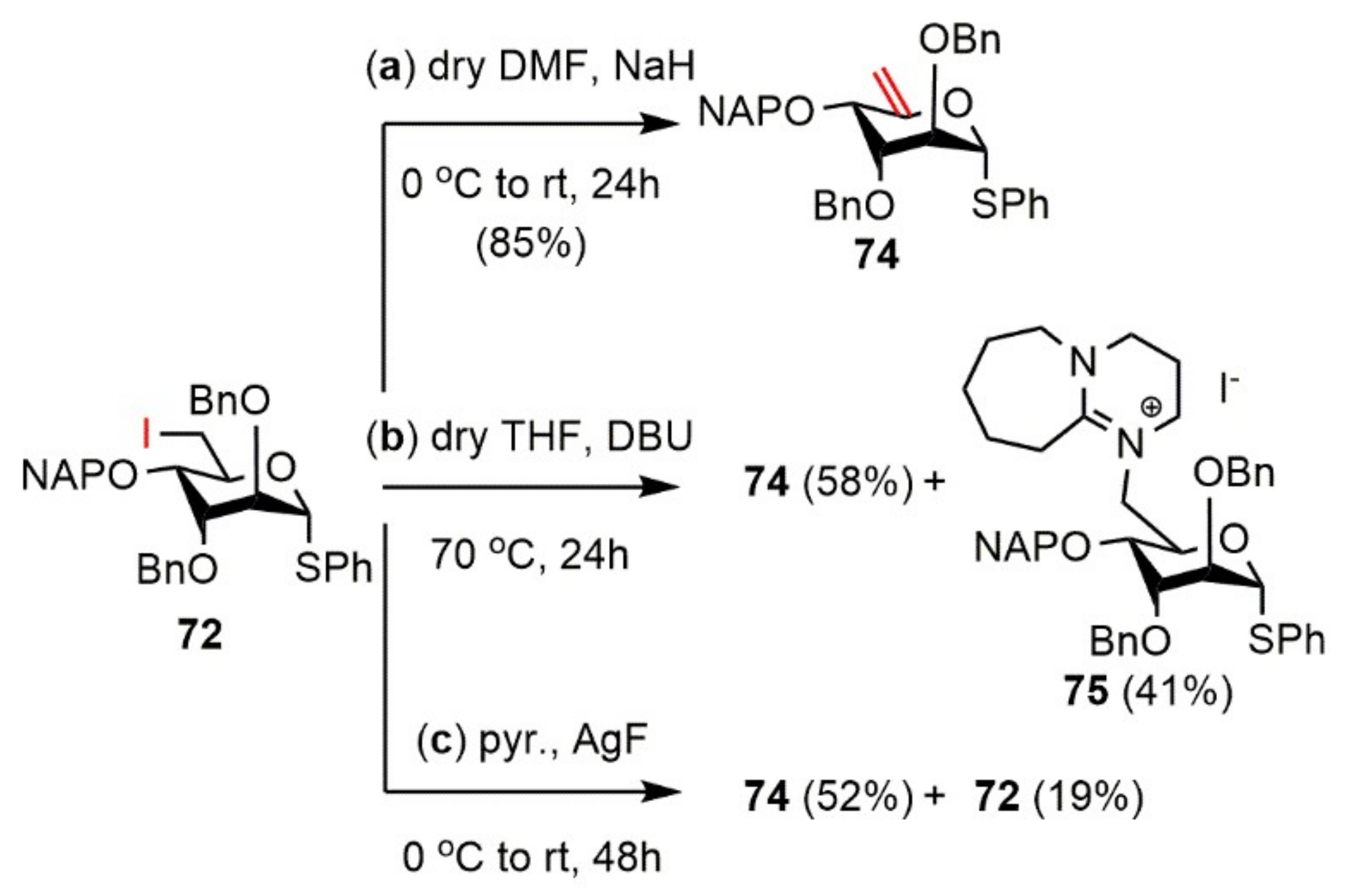
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-arabino-hex-5-enopyranoside (74).
Reaction I.: Compound 72 (218 mg, 0.310 mmol) was converted to 74 according to general Method E. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane) to give 74 (151 mg, 85%) as a colorless syrup.
Reaction II.: Compound 72 (201 mg, 0.286 mmol) was converted to 74 according to general Method F. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane) to give 72 (31 mg, 19%) as a colorless syrup and 74 (85 mg, 52%) as a colorless syrup.
Data of 74: [α]D25 + 50.8 (c 0.12, CHCl3); Rf 0.47 (8:2 CH2Cl2/n-hexane); 1H NMR (500 MHz, CDCl3) δ = 7.85–7.20 (m, 22H, arom.), 4.96 (d, J = 7.5 Hz, 1H, H-1), 4.91 (s, 1H, H-6a), 4.91–4.59 (m, 6H, NAPCH2, 2 × BnCH2), 4.53 (s, 1H, H-6b), 4.13–4.10 (m, 2H, H-2, H-4), 3.67 (dd, J = 3.1 Hz, J = 8.0 Hz, 1H, H-3) ppm; 13C NMR (125 MHz, CDCl3) δ = 153.9 (1C, C-5), 138.2, 138.1, 135.4, 134.3, 133.4, 133.1 (6C, 6 × Cq arom.), 131.9–126.0 (22C, arom.), 100.2 (1C, C-6), 88.6 (1C, C-1), 80.0 (1C, C-3), 77.1 (1C, C-2), 74.9, 72.1, 69.9 (3C, NAPCH2, 2 × BnCH2), 73.3 (1C, C-4) ppm; ESI-TOF-MS: m/z calcd for: C37H34NaO4S [M + Na]+ 597.2070; found: 597.2051.
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-arabino-hex-5-enopyranoside (74) and 8-N-[phenyl 2,3-di-O-benzyl-6-deoxy-6-yl-4-O-(2′-naphthyl)methyl-1-thio-α-d-altropyranoside]-1,8-diazabicyclo(5.4.0)undec-7-ene-iodide (75). Compound 72 (215 mg, 0.306 mmol) was converted to 74 according to general Method B. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane → 95:5 CH2Cl2/MeOH) to give 74 (102 mg, 58%) as a colorless syrup and 75 (107 mg, 41%) as a light yellow syrup.
Data of 75: [α]D25 + 120.8 (c 0.13, CHCl3); Rf 0.44 (7:3 CH2Cl2/MeOH); 1H NMR (500 MHz, CDCl3) δ = 7.86–7.17 (m, 22H, arom.), 5.51 (s, 1H, H-1), 4.69–4.41 (m, 7H, H-5, NAPCH2, 2 × BnCH2), 4.00 (dd, J = 1.6 Hz, J = 15.3 Hz, 1H, H-6a), 3.94 (d, J = 3.4 Hz, 1H, H-2), 3.86 (t, J = 3.0 Hz, 1H, H-3), 3.68 (dd, J = 2.8 Hz, J = 9.9 Hz, 1H, H-4), 3.64–3.39 (m, 7H, H-6b, 3 × NCH2 DBU), 2.84–2.68 (m, 2H, CH2 DBU), 1.84–1.80 (m, 2H, CH2 DBU), 1.61–1.47 (m, 6H, 3 × CH2 DBU) ppm; 13C NMR (125 MHz, CDCl3) δ = 167.2 (1C, Cq DBU), 137.2, 137.0, 136.1, 134.4, 133.0, 132.9 (6C, 6 × Cq arom.), 129.4–125.9 (22C, arom.), 84.5 (1C, C-1), 76.4 (1C, C-2), 73.4 (1C, C-4), 72.5, 72.3, 70.9 (3C, NAPCH2, 2 × BnCH2), 71.0 (1C, C-3), 66.5 (1C, C-5), 55.5 (1C, C-6), 55.0, 49.4, 48.0 (3C, 3 × NCH2 DBU), 28.9, 28.1, 25.6, 22.5, 19.7 (5C, 5 × CH2 DBU) ppm; UHR ESI-QTOF: m/z calcd for C46H51N2O4S [M]+ 727.3564; found: 727.3572.
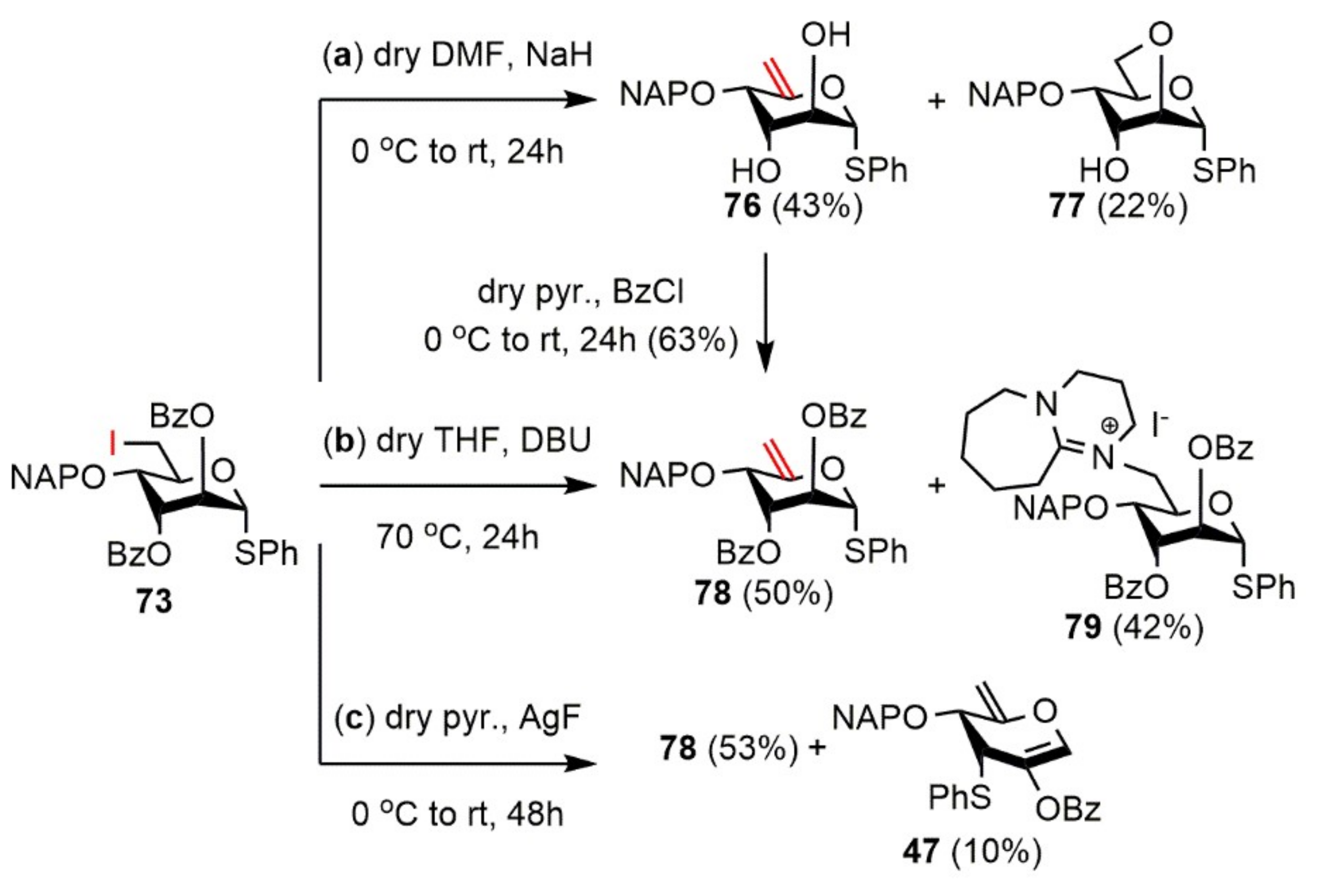
Phenyl 4-O-(2′-naphthyl)methyl-1-thio-α-d-arabino-hex-5-enopyranoside (76) and Phenyl 2,6-anhydro-4-O-(2′-naphthyl)methyl-1-thio-α-d-altropyranoside (77). Compound 73 (254 mg, 0.347 mmol) was converted to 76 according to general Method E. The crude product was purified by silica gel chromatography (97:3 → 9:1 CH2Cl2/acetone) to give 76 (60 mg, 43%) as a white foam and 77 (30 mg, 22%) as a white foam.
Data of 76: [α]D25 +34.3 (c 0.14, CHCl3); Rf 0.17 (97:3 CH2Cl2/acetone); 1H NMR (500 MHz, CDCl3) δ = 7.81–7.21 (m, 12H, arom.), 4.94 (s, 1H, H-6a), 4.77 (d, J = 11.7 Hz, 1H, NAPCH2a), 4.64 (d, J = 9.3 Hz, 1H, H-1), 4.55 (s, 1H, H-6b), 4.43 (d, J = 11.7 Hz, 1H, NAPCH2b), 4.03 (d, J = 3.5 Hz, 1H, H-4), 3.87 (t, J = 9.1 Hz, 1H, H-2), 3.65 (d, J = 6.3 Hz, 1H, H-3), 3.06 (s, 2H, C-2-OH, C-3-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 154.0 (1C, C-5), 134.9, 133.3, 133.1, 132.0 (4C, 4 × Cq arom.), 132.7–125.9 (12C, arom.), 102.0 (1C, C-6), 88.9 (1C, C-1), 76.7 (1C, C-4), 73.6 (1C, C-3), 70.1 (1C, NAPCH2), 69.5 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for: C23H22NaO4S [M + Na]+ 417.1131; found: 417.1131.
Data of 77: [α]D25 +227.3 (c 0.12, CHCl3); Rf 0.54 (97:3 CH2Cl2/acetone); 1H NMR (500 MHz, CDCl3) δ = 7.86–7.23 (m, 12H, arom.), 5.72 (t, J = 1.5 Hz, 1H, H-1), 4.93 (s, 2H, NAPCH2), 4.28–4.24 (m, 1H, H-5), 4.18 (dd, J = 2.7 Hz, J = 10.4 Hz, 1H, H-6a), 4.07 (s, 1H, H-3), 4.05 (dd, J = 1.9 Hz, J = 3.6 Hz, 1H, H-2), 3.88 (dd, J = 2.0 Hz, J = 8.8 Hz, 1H, H-4), 3.73 (s, 1H, C-3-OH), 3.71 (d, J = 4.2 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 136.5, 134.6, 133.3, 133.2 (4C, 4 × Cq arom.), 131.2–125.8 (12C, arom.), 85.8 (1C, C-1), 72.5 (1C, NAPCH2), 72.2 (1C, C-4), 70.2 (1C, C-2), 68.0 (1C, C-3), 66.3 (1C, C-6), 66.1 (1C, C-5) ppm; ESI-TOF-MS: m/z calcd for: C23H22NaO4S [M + Na]+ 417.1131; found: 417.1144.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-arabino-hex-5-enopyranoside (78). To a stirred solution of compound 76 (60 mg, 0.150 mmol) in dry pyridine (1.0 mL), BzCl (52 µL, 0.449 mmol, 1.5 equiv.) was added at 0 °C and the reaction mixture was stirred for 24 h at room temperature. After 24 h the mixture was diluted with CH2Cl2 (50 mL), washed with H2O (15 mL), 1M aqueous solution of H2SO4 (2 × 15 mL), H2O (15 mL), saturated aqueous solution of NaHCO3 (2 × 15 mL), and H2O (15 mL) until neutral pH. The organic layer was separated, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (8:2 CH2Cl2/n-hexane) to give 78 (57 mg, 63%) as a colorless syrup. [α]D25 + 10.8 (c 0.12, CHCl3); Rf 0.45 (7:3 CH2Cl2/n-hexane); 1H NMR (500 MHz, CDCl3) δ = 8.08–7.23 (m, 22H, arom.), 5.96 (dd, J = 7.2 Hz, J = 7.8 Hz, 1H, H-2), 5.48 (dd, J = 3.2 Hz, J = 8.0 Hz, 1H, H-3), 5.25 (d, J = 7.0 Hz, 1H, H-1), 5.05 (s, 1H, H-6a), 4.87 (d, J = 12.6 Hz, 1H, NAPCH2a), 4.81 (s, 1H, H-6b), 4.63 (d, J = 12.6 Hz, 1H, NAPCH2b), 4.43 (d, J = 3.2 Hz, 1H, H-4) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.8, 165.1 (2C, 2 × Cq Bz), 152.8 (1C, C-5), 135.0, 133.3, 133.1, 129.4 (6C, 6 × Cq arom.), 133.5–125.7 (22C, arom.), 101.3 (1C, C-6), 87.6 (1C, C-1), 73.1 (1C, C-4), 72.1 (1C, C-3), 70.3 (1C, NAPCH2), 69.3 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for: C37H30NaO6S [M + Na]+ 625.1655; found: 625.1654.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-arabino-hex-5-enopyranoside (78) and 8-N-[phenyl 2,3-di-O-benzoyl-6-deoxy-6-yl-4-O-(2′-naphthyl)methyl-1-thio-α-d-altropyranoside]-1,8-diazabicyclo(5.4.0)undec-7-ene-iodide (79). Compound 73 (257 mg, 0.351 mmol) was converted to 78 according to general Method B. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane → 9:1 CH2Cl2/MeOH) to give 78 (105 mg, 50%) as a white foam and 79 (130 mg, 42%) as a light yellow syrup.
Data of 79: [α]D25 + 75.0 (c 0.16, CHCl3); Rf 0.54 (9:1 CH2Cl2/MeOH); 1H NMR (500 MHz, CDCl3) δ = 8.31–7.27 (m, 22H, arom.), 5.96 (s, 1H, H-3), 5.72 (s, 1H, H-1), 5.58 (d, J = 3.1 Hz, 1H, H-2), 4.92 (d, J = 11.2 Hz, 1H, NAPCH2a), 4.77 (d, J = 11.2 Hz, 1H, NAPCH2b), 4.71 (t, J = 9.6 Hz, 1H, H-5), 4.16 (d, J = 15.5 Hz, 1H, H-6a), 3.99 (dd, J = 2.5 Hz, J = 9.8 Hz, 1H, H-4), 3.76 (dd, J = 9.7 Hz, J = 15.7 Hz, 1H, H-6b), 3.67–3.49 (m, 6H, 3 × NCH2 DBU), 2.87–2.74 (m, 2H, CH2 DBU), 1.92–1.83 (m, 2H, CH2 DBU), 1.66–1.56 (m, 6H, 3 × CH2 DBU) ppm; 13C NMR (125 MHz, CDCl3), δ = 167.5 (1C, Cq DBU), 165.0, 164.7 (2C, 2 × Cq Bz), 134.3, 133.6, 133.0, 132.9, 128.8, 128.3 (6C, 6 × Cq arom.), 133.8–126.2 (22C, arom.), 84.6 (1C, C-1), 71.6 (1C, C-4), 71.2 (1C, C-2), 71.1 (1C, NAPCH2), 67.3 (1C, C-5), 64.9 (1C, C-3), 55.2 (1C, C-6), 55.7, 49.4, 48.4 (3C, 3 × NCH2 DBU), 29.1, 28.2, 25.7, 22.7, 19.8 (5C, 5 × CH2 DBU) ppm; UHR ESI-QTOF: m/z calcd for C46H47N2O6S [M]+ 755.3149; found: 755.3154.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-α-d-arabino-hex-5-enopyranoside (78) and 1,5-anhydro-2-O-benzoyl-3,6-dideoxy-4-O-(2′-naphthyl)methyl-3-S-phenyl-3-thio-α-d-erythro-hex-1,5-dienytol (47). Compound 73 (234 mg, 0.320 mmol) was converted to 78 according to general Method F. The crude product was purified by silica gel chromatography (7:3 CH2Cl2/n-hexane) to give 78 (103 mg, 53%) as a white foam and 47 (20 mg, 10%) as a colorless syrup.
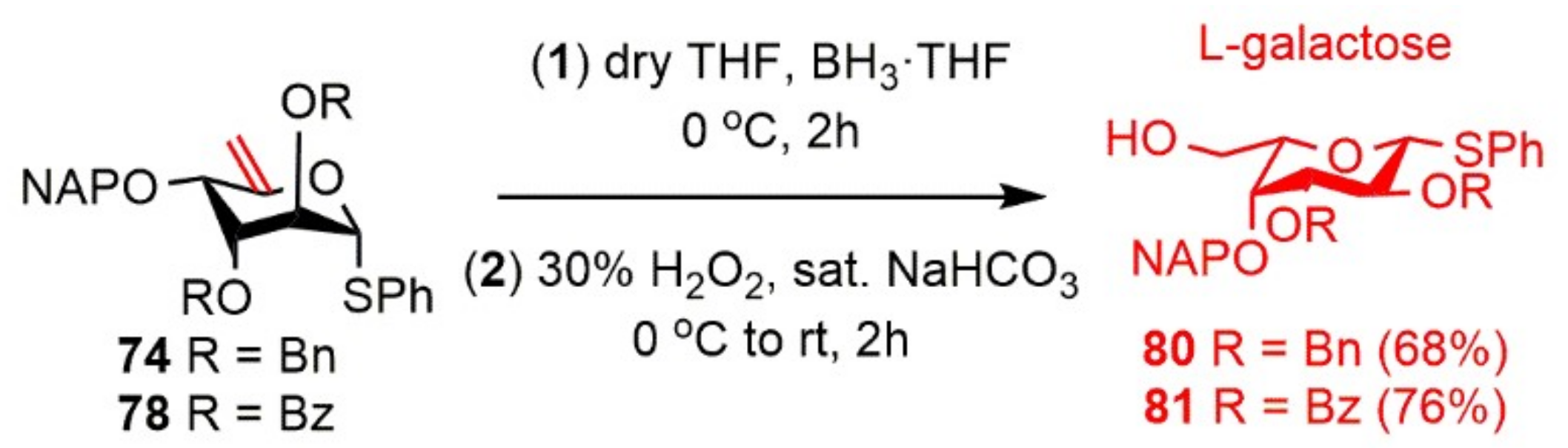
Phenyl 2,3-di-O-benzyl-4-O-(2′-naphthyl)methyl-1-thio-β-l-galactopyranoside (80). Compound 74 (143 mg, 0.249 mmol) was converted to 80 according to general Method C. The crude product was purified by silica gel chromatography (55:45 n-hexane/EtOAc) to give 80 (100 mg, 68%) as a colorless syrup. [α]D25 + 7.0 (c 0.10, CHCl3); Rf 0.41 (55:45 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.84–7.17 (m, 22H, arom.), 5.12–4.65 (m, 6H, NAPCH2, 2 × BnCH2), 4.66 (d, J = 9.6 Hz, 1H, H-1), 3.99 (t, J = 9.4 Hz, 1H, H-2), 3.88–3.84 (m, 2H, H-4, H-6a), 3.61 (dd, J = 2.7 Hz, J = 9.2 Hz, 1H, H-3), 3.53–3.49 (m, 1H, H-6b), 3.45–3.42 (m, 1H, H-5), 1.84 (d, J = 4.7 Hz, 1H, C-6-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 138.3, 138.2, 135.8, 134.1, 133.2, 133.1 (6C, 6 × Cq arom.), 131.6–126.2 (22C, arom.), 87.8 (1C, C-1), 84.4 (1C, C-3), 79.0 (1C, C-5), 77.6 (1C, C-2), 75.8, 74.4, 73.2 (3C, NAPCH2, 2 × BnCH2), 73.4 (1C, C-4), 62.4 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C37H36NaO5S [M + Na]+ 615.2176; found: 615.2189.
Phenyl 2,3-di-O-benzoyl-4-O-(2′-naphthyl)methyl-1-thio-β-l-galactopyranoside (81). Compound 78 (193 mg, 0.319 mmol) was converted to 81 according to general Method C. The crude product was purified by silica gel chromatography (55:45 n-hexane/EtOAc) to give 81 (152 mg, 76%) as a colorless syrup. [α]D25 − 68.3 (c 0.12, CHCl3); Rf 0.40 (55:45 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.98–7.26 (m, 22H, arom.), 5.93 (t, J = 9.9 Hz, 1H, H-2), 5.38 (d, J = 9.9 Hz, 1H, H-3), 4.97 (d, J = 9.9 Hz, 1H, H-1), 4.90 (d, J = 11.8 Hz, 1H, NAPCH2a), 4.68 (d, J = 11.8 Hz, 1H, NAPCH2b), 4.24 (s, 1H, H-4), 3.98–3.95 (m, 1H, H-6a), 3.81 (t, J = 5.6 Hz, 1H, H-5), 3.65–3.63 (m, 1H, H-6b), 1.73 (d, J = 6.0 Hz, 1H, C-6-OH) ppm; 13C NMR (125 MHz, CDCl3) δ = 166.0, 165.4 (2C, 2 × Cq Bz), 134.9, 133.2, 133.1, 132.8, 129.7, 129.0 (6C, 6 × Cq arom.), 133.6–126.1 (22C, arom.), 86.7 (1C, C-1), 79.3 (1C, C-5), 76.1 (1C, C-3), 75.0 (1C, NAPCH2), 74.0 (1C, C-4), 68.6 (1C, C-2), 62.1 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C37H32NaO7S [M + Na]+ 643.1761; found: 643.1768.
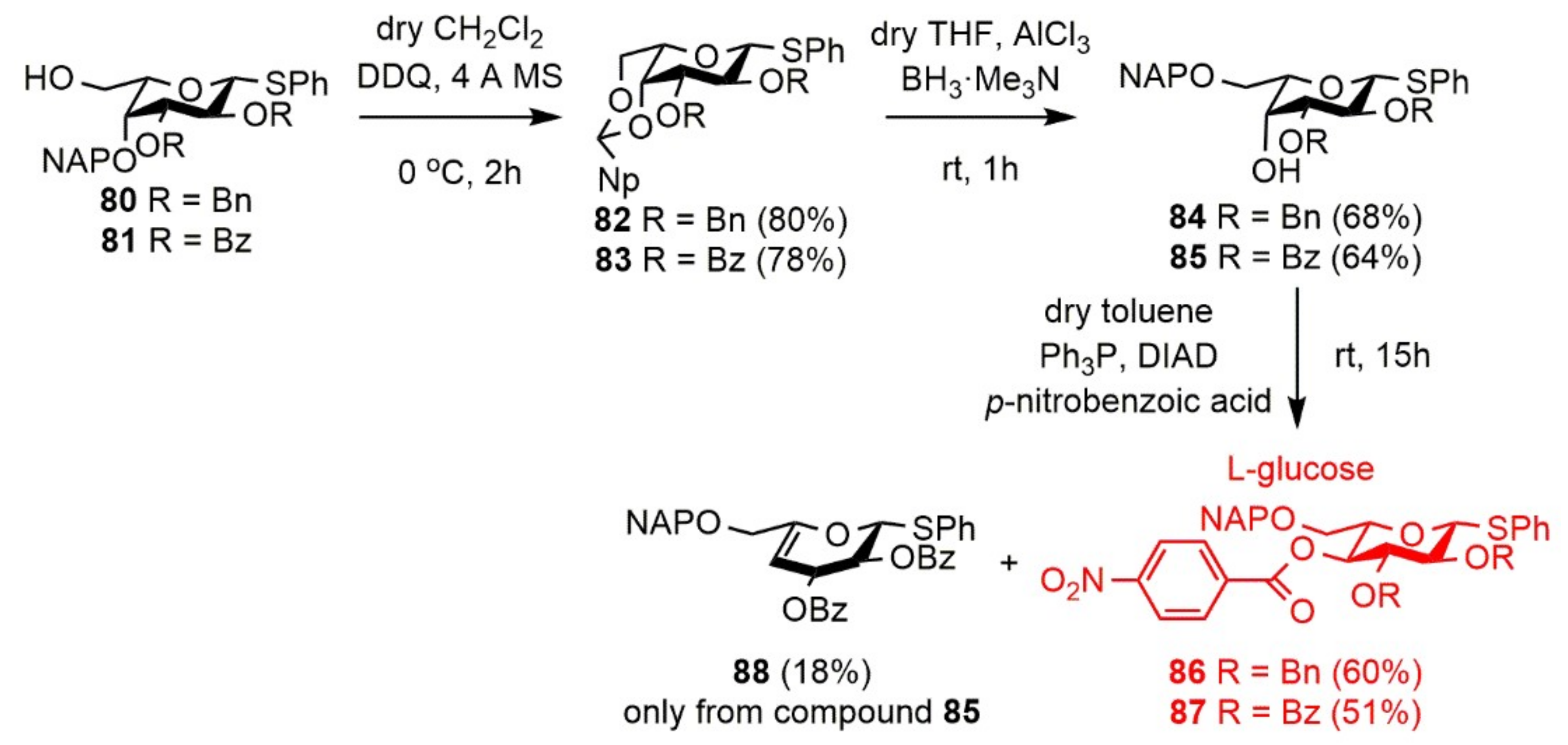
Phenyl 2,3-di-O-benzyl-4,6-O-(2′-naphthyl)methylidene-1-thio-β-l-galactopyranoside (82). Compound 80 (85 mg, 0.144 mmol) was converted to 82 according to general Method G. The crude product was purified by silica gel chromatography (CH2Cl2) to give 82 (68 mg, 80%) as a colorless syrup. [α]D25 − 30.0 (c 0.12, CHCl3); Rf 0.47 (6:4 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.98–7.14 (m, 22H, arom.), 5.61 (s, 1H, Hac), 4.73–4.69 (m, 4H, 2 × BnCH2), 4.61 (d, J = 9.6 Hz, 1H, H-1), 4.37 (dd, J = 1.2 Hz, J = 12.3 Hz, 1H, H-6a), 4.16 (d, J = 3.3 Hz, 1H, H-4), 3.97 (dd, J = 1.3 Hz, J = 12.3 Hz, 1H, H-6b), 3.93 (t, J = 9.4 Hz, 1H, H-2), 3.62 (dd, J = 3.4 Hz, J = 9.2 Hz, 1H, H-3), 3.35 (s, 1H, H-5) ppm; 13C NMR (125 MHz, CDCl3) δ = 138.6, 138.3, 135.4, 133.9, 133.0, 132.9 (6C, 6 × Cq arom.), 132.8–124.4 (22C, arom.), 101.5 (1C, Cac), 86.7 (1C, C-1), 81.5 (1C, C-3), 75.3 (2C, C-2, BnCH2), 73.9 (1C, C-4), 71.9 (1C, BnCH2), 70.0 (1C, C-5), 69.6 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C37H34NaO5S [M + Na]+ 613.2019; found: 613.2054.
Phenyl 2,3-di-O-benzoyl-4,6-O-(2′-naphthyl)methylidene-1-thio-β-l-galactopyranoside (83). Compound 81 (149 mg, 0.239 mmol) was converted to 83 according to general Method G. The crude product was purified by silica gel chromatography (6:4 n-hexane/EtOAc) to give 83 (115 mg, 78%) as a white crystal. [α]D25 − 111.0 (c 0.10, CHCl3); M.p.: 226–228 °C (EtOAc/n-hexane); Rf 0.44 (6:4 n-hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ = 7.99–7.19 (m, 22H, arom.), 5.85 (t, J = 9.9 Hz, 1H, H-2), 5.61 (s, 1H, Hac), 5.40 (dd, J = 3.4 Hz, J = 9.9 Hz, 1H, H-3), 4.94 (d, J = 9.8 Hz, 1H, H-1), 4.61 (d, J = 3.2 Hz, 1H, H-4), 4.43 (dd, J = 0.9 Hz, J = 12.2 Hz, 1H, H-6a), 4.06 (dd, J = 1.1 Hz, J = 12.3 Hz, 1H, H-6b), 3.66 (s, 1H, H-5) ppm; 13C NMR (100 MHz, CDCl3) δ = 166.3, 165.0 (2C, 2 × Cq Bz), 135.1, 133.7, 132.8, 131.2, 129.7, 129.1 (6C, 6 × Cq arom.), 133.8–124.2 (22C, arom.), 101.2 (1C, Cac), 85.3 (1C, C-1), 74.1 (1C, C-3), 73.8 (1C, C-4), 69.9 (1C, C-5), 69.2 (1C, C-6), 67.2 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for: C37H30NaO7S [M + Na]+ 641.1604; found: 641.1616.
Phenyl 2,3-di-O-benzyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-galactopyranoside (84). Compound 82 (59 mg, 0.099 mmol) was converted to 84 according to general Method H. The crude product was purified by silica gel chromatography (98:2 CH2Cl2/acetone) to give 84 (40 mg, 68%) as a colorless syrup. [α]D25 + 8.0 (c 0.15, CHCl3); Rf 0.35 (98:2 CH2Cl2/acetone); 1H NMR (400 MHz, CDCl3) δ = 7.84–7.20 (m, 22H, arom.), 4.84–4.66 (m, 6H, NAPCH2, 2 × BnCH2), 4.65 (d, J = 9.8 Hz, 1H, H-1), 4.11 (d, J = 1.7 Hz, 1H, H-4), 3.87–3.79 (m, 2H, H-6a,b), 3.76 (t, J = 9.3 Hz, 1H, H-2), 3.63 (t, J = 5.7 Hz, 1H, H-5), 3.58 (dd, J = 3.2 Hz, J = 8.9 Hz, 1H, H-3), 2.56 (s, 1H, C-4-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 138.3, 137.8, 135.5, 134.0, 133.4, 133.1 (6C, 6 × Cq arom.), 131.9–125.9 (22C, arom.), 87.8 (1C, C-1), 82.7 (1C, C-3), 77.2 (1C, C-5), 77.1 (1C, C-2), 75.9, 74.0, 72.3 (3C, NAPCH2, 2 × BnCH2), 69.9 (1C, C-6), 67.1 (1C, C-4) ppm; ESI-TOF-MS: m/z calcd for: C37H36NaO5S [M + Na]+ 615.2176; found: 615.2161.
Phenyl 2,3-di-O-benzoyl-6-O-(2′-naphthyl)methyl-1-thio-β-l-galactopyranoside (85). Compound 83 (107 mg, 0.173 mmol) was converted to 85 according to general Method H. The crude product was purified by silica gel chromatography (98:2 CH2Cl2/acetone) to give 85 (68 mg, 64%) as a white foam. [α]D25 −7 7.0 (c 0.10, CHCl3); Rf 0.32 (98:2 CH2Cl2/acetone); 1H NMR (400 MHz, CDCl3) δ = 7.98–7.19 (m, 22H, arom.), 5.82 (t, J = 9.9 Hz, 1H, H-2), 5.34 (dd, J = 2.9 Hz, J = 9.8 Hz, 1H, H-3), 4.96 (d, J = 10.0 Hz, 1H, H-1), 4.74 (s, 2H, NAPCH2), 4.43 (s, 1H, H-4), 3.94–3.92 (m, 1H, H-5), 3.89–3.87 (m, 2H, H-6a,b), 2.86 (s, 1H, C-4-OH) ppm; 13C NMR (100 MHz, CDCl3) δ = 166.0, 165.4 (2C, 2 × Cq Bz), 135.2, 133.4, 133.2, 132.7, 129.6, 129.2 (6C, 6 × Cq arom.), 133.5–125.8 (22C, arom.), 86.7 (1C, C-1), 77.4 (1C, C-5), 75.7 (1C, C-3), 74.0 (1C, NAPCH2), 69.7 (1C, C-6), 68.4 (1C, C-4), 68.1 (1C, C-2) ppm; ESI-TOF-MS: m/z calcd for: C37H32NaO7S [M + Na]+ 643.1761; found: 643.1779.
Phenyl 2,3-di-O-benzyl-6-O-(2′-naphthyl)methyl-4-O-(4′-nitrobenzoyl)-1-thio-β-l-glucopyranoside (86). Compound 84 (24 mg, 0.040 mmol) was converted to 86 according to general Method K. The crude product was purified by silica gel chromatography (7:3 n-hexane/EtOAc) to give 86 (18 mg, 60%) as a colorless syrup. [α]D25 + 12.0 (c 0.10, CHCl3); Rf 0.63 (7:3 n-hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ = 7.95–7.03 (m, 26H, arom.), 5.31 (t, J = 9.4 Hz, 1H, H-4), 4.95–4.52 (m, 7H, H-1, NAPCH2, 2 × BnCH2), 3.80 (t, J = 9.1 Hz, 1H, H-3), 3.76–3.73 (m, 1H, H-5), 3.71–3.66 (m, 2H, H-6a,b), 3.64 (t, J = 9.3 Hz, 1H, H-2) ppm; 13C NMR (125 MHz, CDCl3) δ = 163.7, 150.2 (2C, 2 × Cq p-NO2-Bz), 137.9, 137.8, 135.0, 134.9, 133.5, 133.0 (6C, 6 × Cq arom.), 132.2–123.2 (26C, arom.), 88.0 (1C, C-1), 83.5 (1C, C-3), 81.0 (1C, C-2), 77.0 (1C, C-5), 75.7, 75.5, 73.8 (3C, NAPCH2, 2 × BnCH2), 72.6 (1C, C-4), 70.1 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C44H39NNaO8S [M + Na]+ 764.2289; found: 764.2139.
Phenyl 2,3-di-O-benzoyl-6-O-(2′-naphthyl)methyl-4-O-(4′-nitrobenzoyl)-1-thio-β-l-glucopyranoside (87) and Phenyl 2,3-di-O-benzoyl-6-O-(2′-naphthyl)methyl-4-deoxy-1-thio-α-d-threo-hex-4-enopyranoside (88). Compound 85 (40 mg, 0.063 mmol) was converted to 87 according to general Method K. The crude product was purified by silica gel chromatography (CH2Cl2) to give 87 (25 mg, 51%) as a colorless syrup and 88 (7 mg, 18%) as a colorless syrup.
Data of 87: [α]D25 + 20.9 (c 0.11, CHCl3); Rf 0.55 (CH2Cl2); 1H NMR (500 MHz, CDCl3) δ = 7.96–7.26 (m, 26H, arom.), 5.85 (t, J = 9.4 Hz, 1H, H-3), 5.56 (t, J = 9.7 Hz, 1H, H-4), 5.51 (t, J = 9.7 Hz, 1H, H-2), 5.05 (d, J = 10.0 Hz, 1H, H-1), 4.64 (s, 2H, NAPCH2), 4.07–4.05 (m, 1H, H-5), 3.84–3.78 (m, 2H, H-6a,b) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.9, 165.2, 163.7, 150.4 (4C, 2 × Cq Bz, 2 × Cq p-NO2-Bz), 134.9, 134.4, 133.0, 132.1, 129.3, 128.8 (6C, 6 × Cq arom.), 133.5–123.3 (26C, arom.), 86.6 (1C, C-1), 77.4 (1C, C-5), 74.4 (1C, C-3), 73.9 (1C, NAPCH2), 71.3 (1C, C-4), 70.6 (1C, C-2), 69.5 (1C, C-6) ppm; ESI-TOF-MS: m/z calcd for: C44H35NNaO10S [M + Na]+ 792.1874; found: 792.1800.
Data of 88: [α]D25 + 49.5 (c 0.62, CHCl3); Rf 0.41 (CH2Cl2); 1H NMR (500 MHz, CDCl3) δ = 8.20–7.26 (m, 22H, arom.), 5.86 (s, 1H, H-1), 5.75 (s, 1H, H-2), 5.54 (s, 2H, H-3, H-4), 4.78 (dd, J = 11.8 Hz, J = 30.2 Hz, 2H, NAPCH2), 4.18 (d, J = 13.3 Hz, 1H, H-6a), 4.10 (d, J = 13.1 Hz, 1H, H-6b) ppm; 13C NMR (125 MHz, CDCl3) δ = 165.6, 165.5 (2C, 2 × Cq Bz), 152.0 (1C, C-5), 135.9, 135.3, 134.5, 129.9, 129.2 (6C, 6 × Cq arom.), 133.7–126.0 (22C, arom.), 97.3 (1C, C-4), 84.0 (1C, C-1), 72.5 (1C, NAPCH2), 70.0 (1C, C-2), 69.6 (1C, C-6), 64.9 (1C, C-3) ppm; ESI-TOF-MS: m/z calcd for: C37H30NaO6S [M + Na]+ 625.1655; found: 625.1632.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}