
Redox-Switchable Behavior of Transition-Metal Complexes Supported by Amino-Decorated N-Heterocyclic Carbenes

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
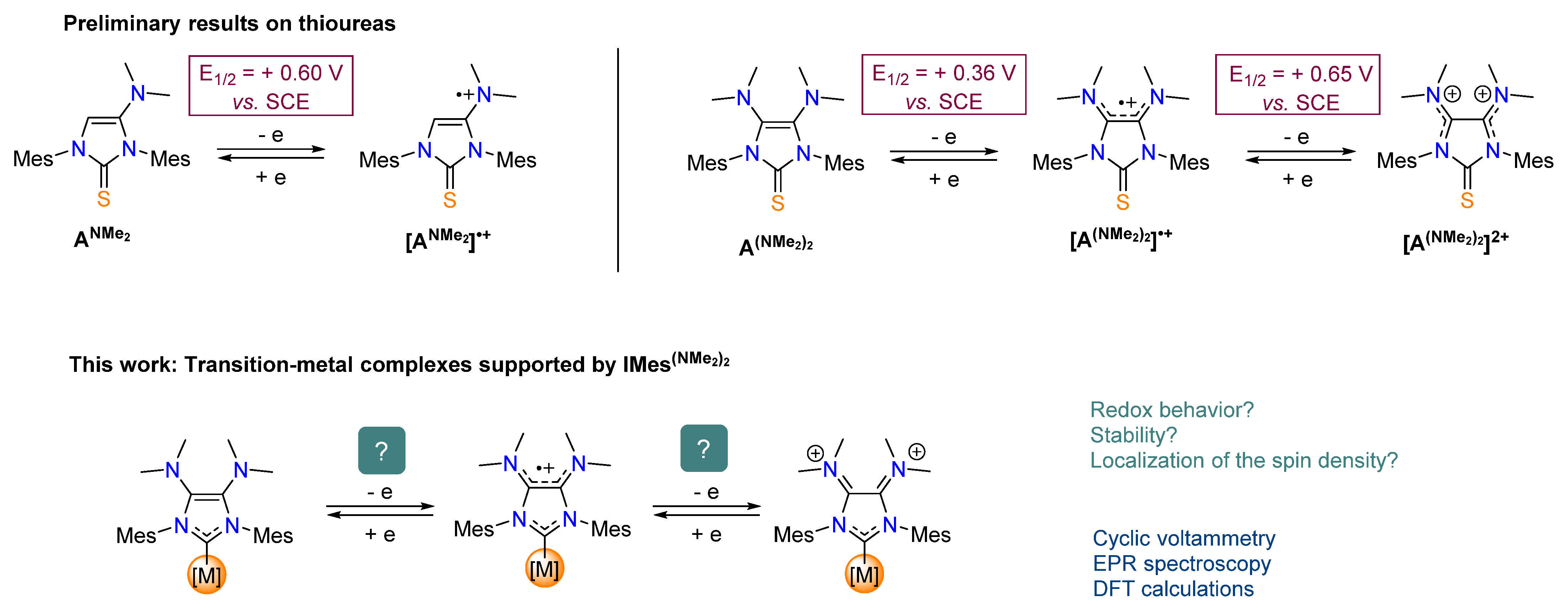
:1. Introduction
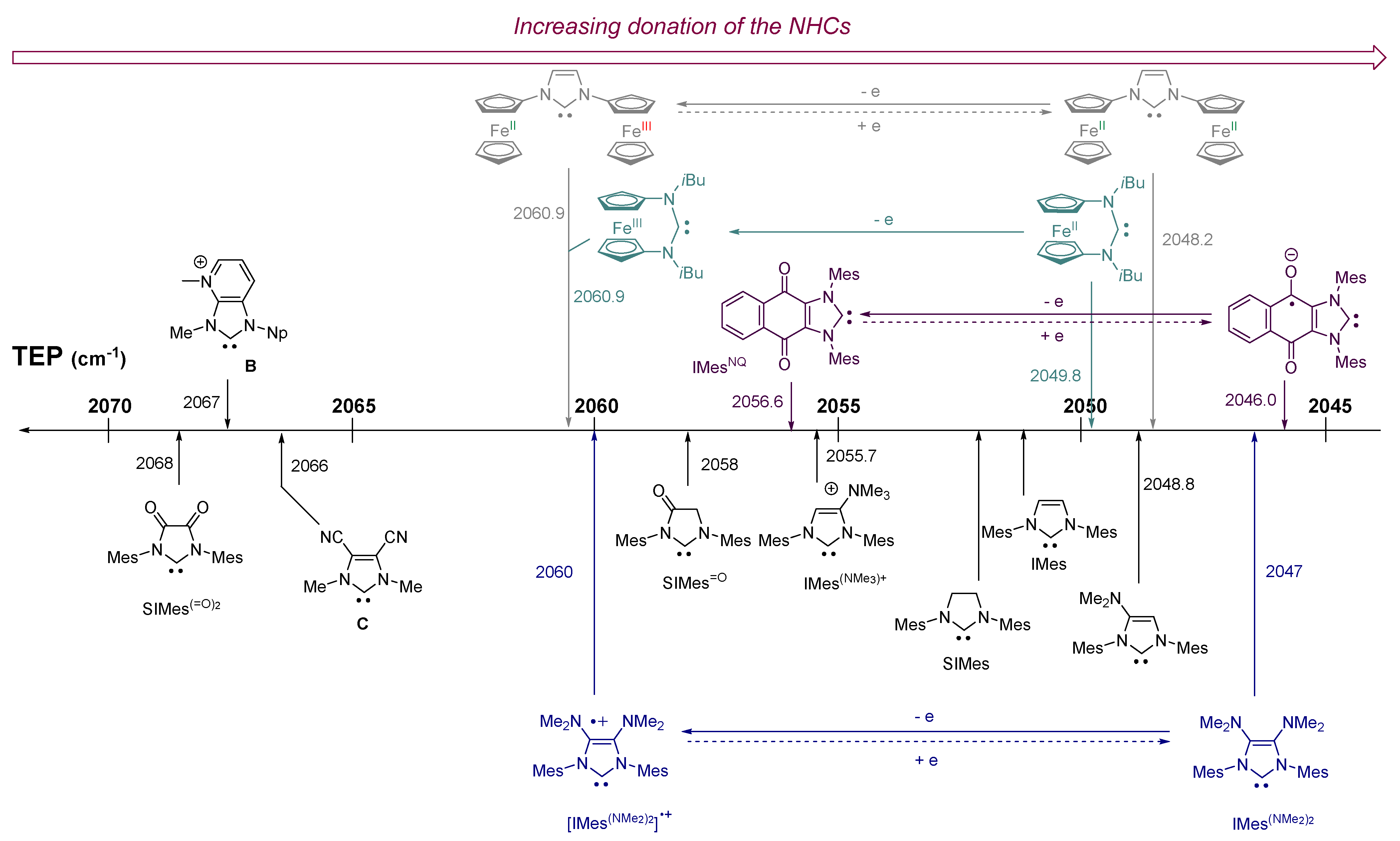
2. Results and Discussion
2.1. Redox Properties of IMes(NMe2)2 Copper(I) Complex 2
2.2. Redox Properties of IMes(NMe2)2 Dicarbonyl Rhodium(I) Complex 3
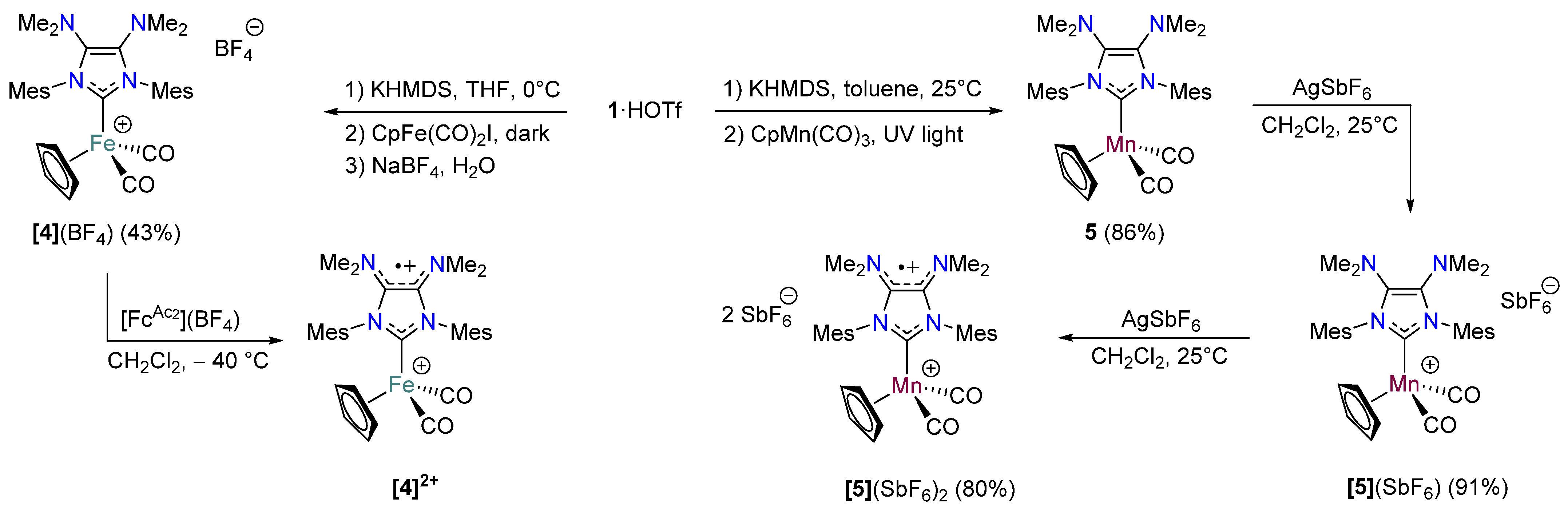
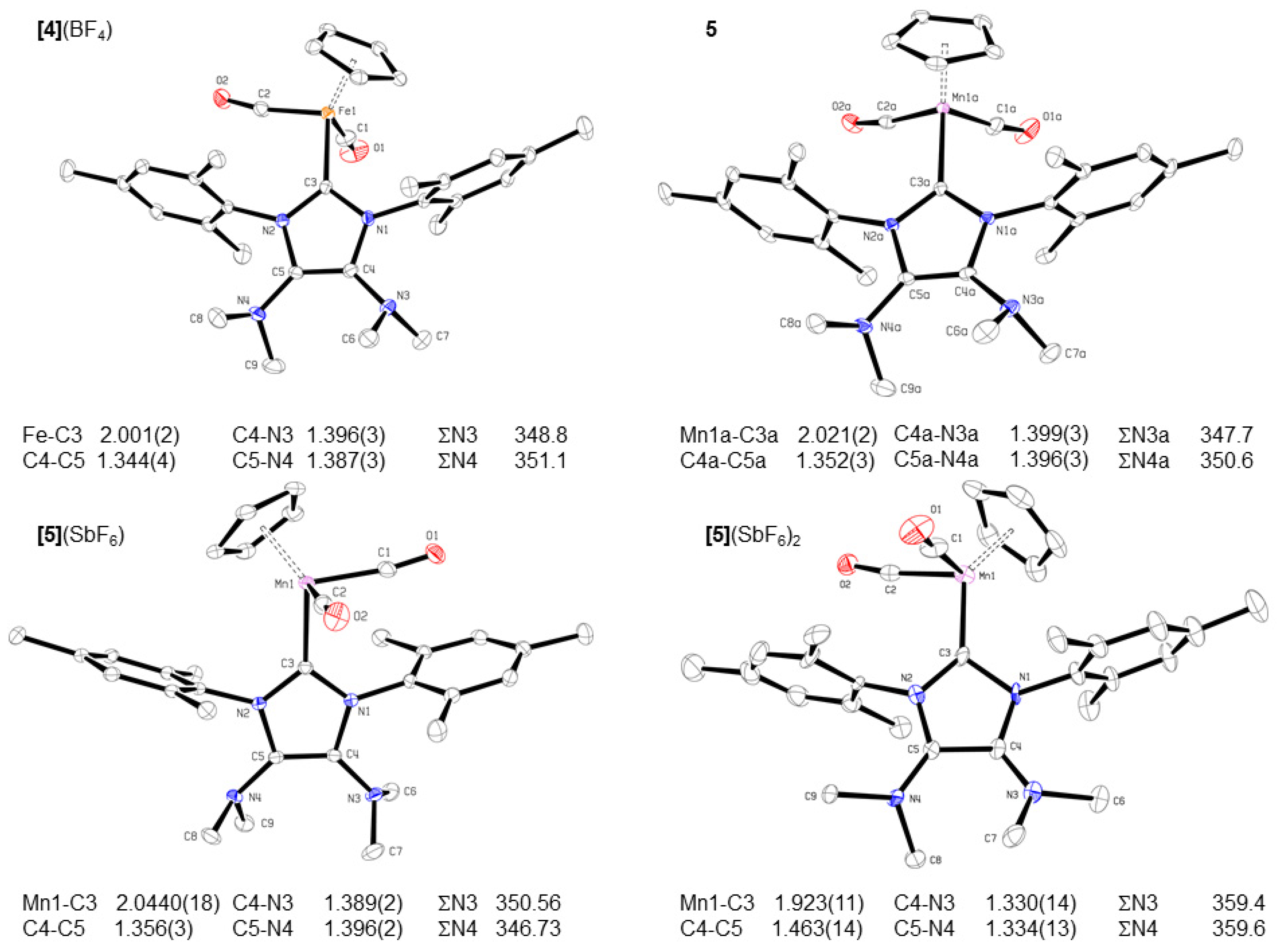
2.3. Redox Properties of Half-Sandwich IMes(NMe2)2 Fe(II) and Mn(I) Complexes [4](BF4) and 5
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Elison, M.; Fischer, J.; Köcher, C.; Artus, G.R.J. Metal Complexes of N-Heterocyclic Carbenes—A New Structural Principle for Catalysts in Homogeneous Catalysis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2371–2374. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C–H Activation Reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar] [CrossRef] [PubMed]
- Romain, C.; Bellemin-Laponnaz, S.; Dagorne, S. Recent progress on NHC-stabilized early transition metal (group 3–7) complexes: Synthesis and applications. Coord. Chem. Rev. 2020, 422, 213411. [Google Scholar] [CrossRef]
- Foster, D.; Borhanuddin, S.M.; Dorta, R. Designing successful monodentate N-heterocyclic carbene ligands for asymmetric metal catalysis. Dalton Trans. 2021, 50, 17467–17477. [Google Scholar] [CrossRef] [PubMed]
- Peris, E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. [Google Scholar] [CrossRef]
- Visbal, R.; Gimeno, M.C. N-heterocyclic carbene metal complexes: Photoluminescence and applications. Chem. Soc. Rev. 2014, 43, 3551–3574. [Google Scholar] [CrossRef]
- Mora, M.; Gimeno, M.C.; Visbal, R. Recent advances in gold–NHC complexes with biological properties. Chem. Soc. Rev. 2019, 48, 447–462. [Google Scholar] [CrossRef]
- Smith, C.A.; Narouz, M.R.; Lummis, P.A.; Singh, I.; Nazemi, A.; Li, C.-H.; Crudden, C.M. N-Heterocyclic Carbenes in Materials Chemistry. Chem. Rev. 2019, 119, 4986–5056. [Google Scholar] [CrossRef]
- Huynh, H.V. Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination. Chem. Rev. 2018, 118, 9457–9492. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suarez, A.; Nelson, D.J.; Nolan, S.P. Quantifying and understanding the steric properties of N-heterocyclic carbenes. Chem. Commun. 2017, 53, 2650–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumanskii, B.; Sheberla, D.; Molev, G.; Apeloig, Y. Dual Character of Arduengo Carbene–Radical Adducts: Addition versus Coordination Product. Angew. Chem. Int. Ed. 2007, 46, 7408–7411. [Google Scholar] [CrossRef] [PubMed]
- Chirila, A.; Das, B.G.; Kuijpers, P.F.; Sinha, V.; de Bruin, B. Application of Stimuli-Responsive and “Non-innocent” Ligands in Base Metal Catalysis. In Non-Noble Metal Catalysis; Wiley: Weinheim, Germany, 2019; pp. 1–31. [Google Scholar] [CrossRef]
- Broere, D.L.J.; Plessius, R.; van der Vlugt, J.I. New avenues for ligand-mediated processes—Expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. [Google Scholar] [CrossRef]
- Luca, O.R.; Crabtree, R.H. Redox-active ligands in catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef] [PubMed]
- Lyaskovskyy, V.; de Bruin, B. Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Dzik, W.I.; van der Vlugt, J.I.; Reek, J.N.H.; de Bruin, B. Ligands that Store and Release Electrons during Catalysis. Angew. Chem. Int. Ed. 2011, 50, 3356–3358. [Google Scholar] [CrossRef]
- Klenk, S.; Rupf, S.; Suntrup, L.; van der Meer, M.; Sarkar, B. The Power of Ferrocene, Mesoionic Carbenes, and Gold: Redox-Switchable Catalysis. Organometallics 2017, 36, 2026–2035. [Google Scholar] [CrossRef]
- Hettmanczyk, L.; Suntrup, L.; Klenk, S.; Hoyer, C.; Sarkar, B. Heteromultimetallic Complexes with Redox-Active Mesoionic Carbenes: Control of Donor Properties and Redox-Induced Catalysis. Chem. Eur. J. 2017, 23, 576–585. [Google Scholar] [CrossRef]
- Labande, A.; Debono, N.; Sournia-Saquet, A.; Daran, J.-C.; Poli, R. Oxidation-promoted activation of a ferrocene C-H bond by a rhodium complex. Dalton Trans. 2013, 42, 6531–6537. [Google Scholar] [CrossRef]
- Rosen, E.L.; Varnado, C.D.; Tennyson, A.G.; Khramov, D.M.; Kamplain, J.W.; Sung, D.H.; Cresswell, P.T.; Lynch, V.M.; Bielawski, C.W. Redox-Active N-Heterocyclic Carbenes: Design, Synthesis, and Evaluation of Their Electronic Properties. Organometallics 2009, 28, 6695–6706. [Google Scholar] [CrossRef]
- Vanicek, S.; Podewitz, M.; Stubbe, J.; Schulze, D.; Kopacka, H.; Wurst, K.; Müller, T.; Lippmann, P.; Haslinger, S.; Schottenberger, H.; et al. Highly Electrophilic, Catalytically Active and Redox-Responsive Cobaltoceniumyl and Ferrocenyl Triazolylidene Coinage Metal Complexes. Chem. Eur. J. 2018, 24, 3742–3753. [Google Scholar] [CrossRef] [PubMed]
- Tennyson, A.G.; Ono, R.J.; Hudnall, T.W.; Khramov, D.M.; Er, J.A.V.; Kamplain, J.W.; Lynch, V.M.; Sessler, J.L.; Bielawski, C.W. Quinobis(imidazolylidene): Synthesis and Study of an Electron-Configurable Bis(N-Heterocyclic Carbene) and Its Bimetallic Complexes. Chem. Eur. J. 2010, 16, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Ahumada, G.; Ryu, Y.; Bielawski, C.W. Potentiostatically Controlled Olefin Metathesis. Organometallics 2020, 39, 1744–1750. [Google Scholar] [CrossRef] [Green Version]
- Lastovickova, D.N.; Teator, A.J.; Shao, H.; Liu, P.; Bielawski, C.W. A redox-switchable ring-closing metathesis catalyst. Inorg. Chem. Front. 2017, 4, 1525–1532. [Google Scholar] [CrossRef]
- Harris, C.F.; Bayless, M.B.; van Leest, N.P.; Bruch, Q.J.; Livesay, B.N.; Bacsa, J.; Hardcastle, K.I.; Shores, M.P.; de Bruin, B.; Soper, J.D. Redox-Active Bis(phenolate) N-Heterocyclic Carbene [OCO] Pincer Ligands Support Cobalt Electron Transfer Series Spanning Four Oxidation States. Inorg. Chem. 2017, 56, 12421–12435. [Google Scholar] [CrossRef]
- Harris, C.F.; Kuehner, C.S.; Bacsa, J.; Soper, J.D. Photoinduced Cobalt(III)−Trifluoromethyl Bond Activation Enables Arene C−H Trifluoromethylation. Angew. Chem. Int. Ed. 2018, 57, 1311–1315. [Google Scholar] [CrossRef]
- Gravogl, L.; Heinemann, F.W.; Munz, D.; Meyer, K. An Iron Pincer Complex in Four Oxidation States. Inorg. Chem. 2020, 59, 5632–5645. [Google Scholar] [CrossRef]
- Kunert, R.; Philouze, C.; Jarjayes, O.; Thomas, F. Stable M(II)-Radicals and Nickel(III) Complexes of a Bis(phenol) N-Heterocyclic Carbene Chelated to Group 10 Metal Ions. Inorg. Chem. 2019, 58, 8030–8044. [Google Scholar] [CrossRef]
- Romain, C.; Choua, S.; Collin, J.-P.; Heinrich, M.; Bailly, C.; Karmazin-Brelot, L.; Bellemin-Laponnaz, S.; Dagorne, S. Redox and Luminescent Properties of Robust and Air-Stable N-Heterocyclic Carbene Group 4 Metal Complexes. Inorg. Chem. 2014, 53, 7371–7376. [Google Scholar] [CrossRef]
- Ruiz-Zambrana, C.; Gutiérrez-Blanco, A.; Gonell, S.; Poyatos, M.; Peris, E. Redox-Switchable Cycloisomerization of Alkynoic Acids with Napthalenediimide-Derived N-Heterocyclic Carbene Complexes. Angew. Chem. Int. Ed. 2021, 60, 20003–20011. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Zambrana, C.; Poyatos, M.; Peris, E. A Redox-Switchable Gold(I) Complex for the Hydroamination of Acetylenes: A Convenient Way for Studying Ligand-Derived Electronic Effects. ACS Catal. 2022, 12, 4465–4472. [Google Scholar] [CrossRef]
- Crabtree, R.H. Multifunctional ligands in transition metal catalysis. New J. Chem. 2011, 35, 18–23. [Google Scholar] [CrossRef]
- Ruamps, M.; Bastin, S.; Rechignat, L.; Sournia-Saquet, A.; Valyaev, D.A.; Mouesca, J.-M.; Lugan, N.; Maurel, V.; Cesar, V. Unveiling the redox-active character of imidazolin-2-thiones derived from amino-substituted N-heterocyclic carbenes. Chem. Commun. 2018, 54, 7653–7656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; César, V.; Storch, G.; Lugan, N.; Lavigne, G. Skeleton Decoration of NHCs by Amino Groups and its Sequential Booster Effect on the Palladium-Catalyzed Buchwald–Hartwig Amination. Angew. Chem. Int. Ed. 2014, 53, 6482–6486. [Google Scholar] [CrossRef]
- Zhang, Y.; César, V.; Lavigne, G. Efficient and Versatile Buchwald–Hartwig Amination of (Hetero)aryl Chlorides Using the Pd–PEPPSI-IPr(NMe2)2 Precatalyst in the Presence of Carbonate Base. Eur. J. Org. Chem. 2015, 2015, 2042–2050. [Google Scholar] [CrossRef]
- Zhang, Y.; Lavigne, G.; César, V. Buchwald–Hartwig Amination of (Hetero)Aryl Tosylates Using a Well-Defined N-Heterocyclic Carbene/Palladium(II) Precatalyst. J. Org. Chem. 2015, 80, 7666–7673. [Google Scholar] [CrossRef]
- César, V.; Zhang, Y.; Kośnik, W.; Zieliński, A.; Rajkiewicz, A.A.; Ruamps, M.; Bastin, S.; Lugan, N.; Lavigne, G.; Grela, K. Ruthenium Catalysts Supported by Amino-Substituted N-Heterocyclic Carbene Ligands for Olefin Metathesis of Challenging Substrates. Chem. Eur. J. 2017, 23, 1950–1955. [Google Scholar] [CrossRef]
- Zhang, Y.; Lavigne, G.; Lugan, N.; César, V. Buttressing Effect as a Key Design Principle towards Highly Efficient Palladium/N-Heterocyclic Carbene Buchwald–Hartwig Amination Catalysts. Chem. Eur. J. 2017, 23, 13792–13801. [Google Scholar] [CrossRef]
- Broggi, J.; Terme, T.; Vanelle, P. Organic Electron Donors as Powerful Single-Electron Reducing Agents in Organic Synthesis. Angew. Chem. Int. Ed. 2014, 53, 384–413. [Google Scholar] [CrossRef]
- Doni, E.; Murphy, J.A. Evolution of neutral organic super-electron-donors and their applications. Chem. Commun. 2014, 50, 6073–6087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danopoulos, A.A.; Simler, T.; Braunstein, P. N-Heterocyclic Carbene Complexes of Copper, Nickel, and Cobalt. Chem. Rev. 2019, 119, 3730–3961. [Google Scholar] [CrossRef] [PubMed]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Kelly, R.A., III; Clavier, H.; Giudice, S.; Scott, N.M.; Stevens, E.D.; Bordner, J.; Samardjiev, I.; Hoff, C.D.; Cavallo, L.; Nolan, S.P. Determination of N-Heterocyclic Carbene (NHC) Steric and Electronic Parameters using the [(NHC)Ir(CO)2Cl] System. Organometallics 2007, 27, 202–210. [Google Scholar] [CrossRef]
- Wolf, S.; Plenio, H. Synthesis of (NHC)Rh(cod)Cl and (NHC)RhCl(CO)2 complexes—Translation of the Rh- into the Ir-scale for the electronic properties of NHC ligands. J. Organomet. Chem. 2009, 694, 1487–1492. [Google Scholar] [CrossRef]
- Sanderson, M.D.; Kamplain, J.W.; Bielawski, C.W. Quinone-Annulated N-Heterocyclic Carbene−Transition-Metal Complexes: Observation of π-Backbonding Using FT-IR Spectroscopy and Cyclic Voltammetry. J. Am. Chem. Soc. 2006, 128, 16514–16515. [Google Scholar] [CrossRef]
- Benhamou, L.; Vujkovic, N.; César, V.; Gornitzka, H.; Lugan, N.l.; Lavigne, G. Facile Derivatization of a “Chemo-active” NHC Incorporating an Enolate Backbone and Relevant Tuning of Its Electronic Properties. Organometallics 2010, 29, 2616–2630. [Google Scholar] [CrossRef]
- Ruamps, M.; Lugan, N.; César, V. A Cationic N-Heterocyclic Carbene Containing an Ammonium Moiety. Organometallics 2017, 36, 1049–1055. [Google Scholar] [CrossRef]
- Braun, M.; Frank, W.; Ganter, C. Reactivity of an Oxalamide-Based N-Heterocyclic Carbene. Organometallics 2012, 31, 1927–1934. [Google Scholar] [CrossRef]
- Buhl, H.; Ganter, C. Tuning the electronic properties of an N-heterocyclic carbene by charge and mesomeric effects. Chem. Commun. 2013, 49, 5417–5419. [Google Scholar] [CrossRef]
- Khramov, D.M.; Lynch, V.M.; Bielawski, C.W. N-Heterocyclic Carbene−Transition Metal Complexes: Spectroscopic and Crystallographic Analyses of π-Back-bonding Interactions. Organometallics 2007, 26, 6042–6049. [Google Scholar] [CrossRef]
- Ryu, Y.; Ahumada, G.; Bielawski, C.W. Redox- and light-switchable N-heterocyclic carbenes: A “soup-to-nuts” course on contemporary structure–activity relationships. Chem. Commun. 2019, 55, 4451–4466. [Google Scholar] [CrossRef] [PubMed]
- Buchgraber, P.; Toupet, L.; Guerchais, V. Syntheses, Properties, and X-ray Crystal Structures of Piano-Stool Iron Complexes Bearing an N-Heterocyclic Carbene Ligand. Organometallics 2003, 22, 5144–5147. [Google Scholar] [CrossRef]
- Zheng, J.; Elangovan, S.; Valyaev, D.A.; Brousses, R.; César, V.; Sortais, J.-B.; Darcel, C.; Lugan, N.; Lavigne, G. Hydrosilylation of Aldehydes and Ketones Catalyzed by Half-Sandwich Manganese(I) N-Heterocyclic Carbene Complexes. Adv. Synth. Catal. 2014, 356, 1093–1097. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- Brousses, R.; Maurel, V.; Mouesca, J.-M.; César, V.; Lugan, N.; Valyaev, D.A. Half-sandwich manganese complexes Cp(CO)2Mn(NHC) as redox-active organometallic fragments. Dalton Trans. 2021, 50, 14264–14272. [Google Scholar] [CrossRef]
- Grineva, A.A.; Valyaev, D.A.; César, V.; Filippov, O.A.; Khrustalev, V.N.; Nefedov, S.E.; Lugan, N. Oxidative Coupling of Anionic Abnormal N-Heterocyclic Carbenes: Efficient Access to Janus-Type 4,4′-Bis(2H-imidazol-2-ylidene)s. Angew. Chem. Int. Ed. 2018, 57, 7986–7991. [Google Scholar] [CrossRef]
- Wu, K.; Conger, M.A.; Waterman, R.; Liptak, M.; Geiger, W.E. Electrochemical and structural characterization of a radical cation formed by one-electron oxidation of a cymantrene complex containing an N-heterocyclic carbene ligand. Polyhedron 2019, 157, 442–448. [Google Scholar] [CrossRef]
- Buhaibeh, R.; Duhayon, C.; Valyaev, D.A.; Sortais, J.-B.; Canac, Y. Cationic PCP and PCN NHC Core Pincer-Type Mn(I) Complexes: From Synthesis to Catalysis. Organometallics 2021, 40, 231–241. [Google Scholar] [CrossRef]
- Ding, M.; Cutsail, G.E., III; Aravena, D.; Amoza, M.; Rouzières, M.; Dechambenoit, P.; Losovyj, Y.; Pink, M.; Ruiz, E.; Clérac, R.; et al. A low spin manganese(iv) nitride single molecule magnet. Chem. Sci. 2016, 7, 6132–6140. [Google Scholar] [CrossRef] [Green Version]
- Telser, J. EPR Interactions—Zero-Field Splittings. eMagRes 2017, 6, 207–234. [Google Scholar] [CrossRef]
- Bertrand, P. Spectrum Intensity, Saturation, Spin-Lattice Relaxation. In Electron Paramagnetic Resonance Spectroscopy: Fundamentals; Bertrand, P., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 155–196. [Google Scholar] [CrossRef]
- Yasuda, S.; Yorimitsu, H.; Oshima, K. Synthesis of Aryliron Complexes by Palladium-Catalyzed Transmetalation between [CpFe(CO)2I] and Aryl Grignard Reagents and Their Chemistry Directed toward Organic Synthesis. Organometallics 2008, 27, 4025–4027. [Google Scholar] [CrossRef]
- Guillon, C.; Vierling, P. Unexpected formation of novel benzofuranyl-substituted ferrocenes by action of p-benzoquinone on 1,1′-bis- acylferrocene. J. Organomet. Chem. 1994, 464, C42–C44. [Google Scholar] [CrossRef]
- Valyaev, D.A.; Uvarova, M.A.; Grineva, A.A.; César, V.; Nefedov, S.N.; Lugan, N. Post-coordination backbone functionalization of an imidazol-2-ylidene and its application to synthesize heteropolymetallic complexes incorporating the ambidentate IMesCO2- ligand. Dalton Trans. 2016, 45, 11953–11957. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. Sect. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- te Velde, G.; Baerends, E.J. Numerical integration for polyatomic systems. J. Comput. Phys. 1992, 99, 84–98. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruamps, M.; Bastin, S.; Rechignat, L.; Sournia-Saquet, A.; Vendier, L.; Lugan, N.; Mouesca, J.-M.; Valyaev, D.A.; Maurel, V.; César, V. Redox-Switchable Behavior of Transition-Metal Complexes Supported by Amino-Decorated N-Heterocyclic Carbenes. Molecules 2022, 27, 3776. https://doi.org/10.3390/molecules27123776
Ruamps M, Bastin S, Rechignat L, Sournia-Saquet A, Vendier L, Lugan N, Mouesca J-M, Valyaev DA, Maurel V, César V. Redox-Switchable Behavior of Transition-Metal Complexes Supported by Amino-Decorated N-Heterocyclic Carbenes. Molecules. 2022; 27(12):3776. https://doi.org/10.3390/molecules27123776
Chicago/Turabian StyleRuamps, Mirko, Stéphanie Bastin, Lionel Rechignat, Alix Sournia-Saquet, Laure Vendier, Noël Lugan, Jean-Marie Mouesca, Dmitry A. Valyaev, Vincent Maurel, and Vincent César. 2022. "Redox-Switchable Behavior of Transition-Metal Complexes Supported by Amino-Decorated N-Heterocyclic Carbenes" Molecules 27, no. 12: 3776. https://doi.org/10.3390/molecules27123776
APA StyleRuamps, M., Bastin, S., Rechignat, L., Sournia-Saquet, A., Vendier, L., Lugan, N., Mouesca, J.-M., Valyaev, D. A., Maurel, V., & César, V. (2022). Redox-Switchable Behavior of Transition-Metal Complexes Supported by Amino-Decorated N-Heterocyclic Carbenes. Molecules, 27(12), 3776. https://doi.org/10.3390/molecules27123776