
Structural Investigation of Magnesium Complexes Supported by a Thiopyridyl Scorpionate Ligand

 , , , and
, , , and
Abstract
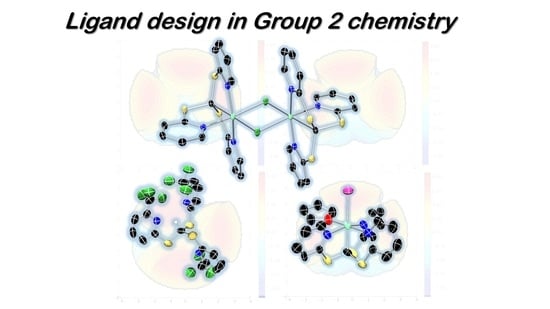
:
1. Introduction
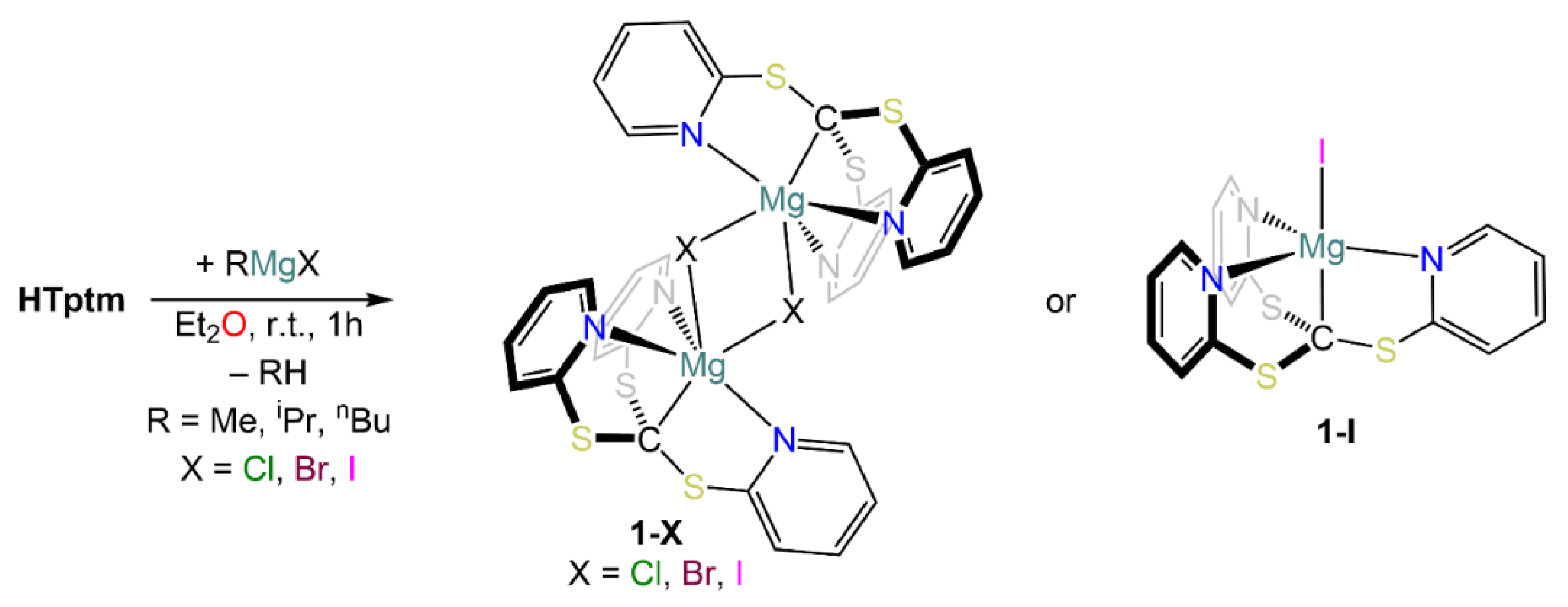
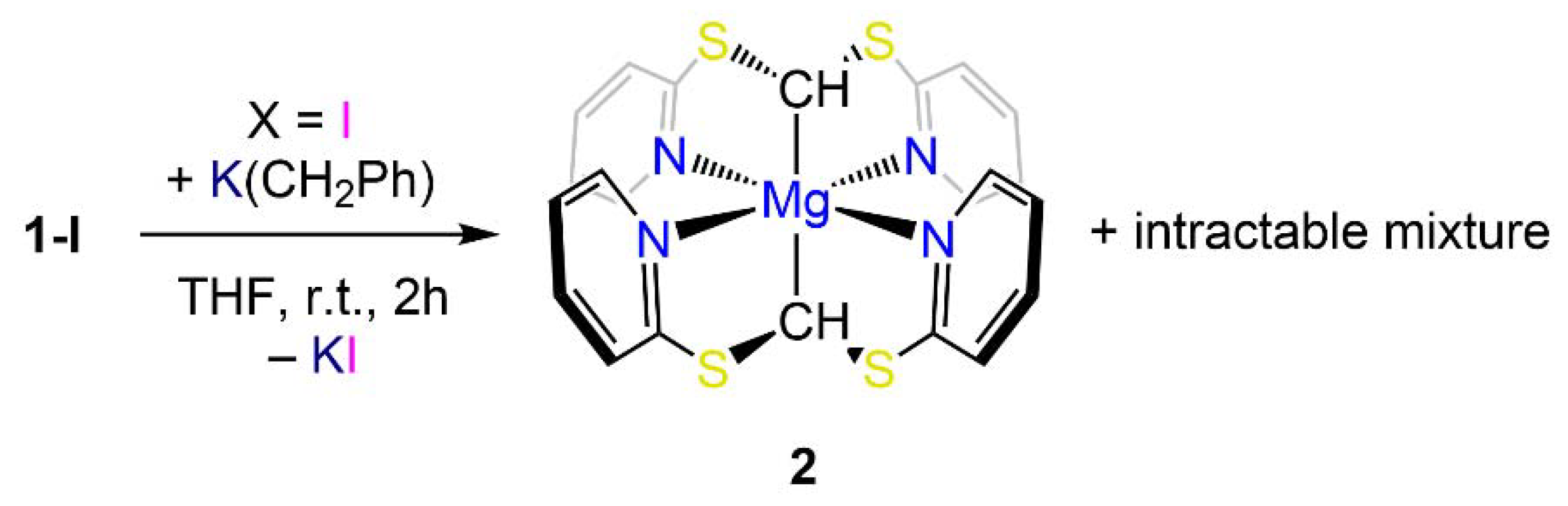
2. Results
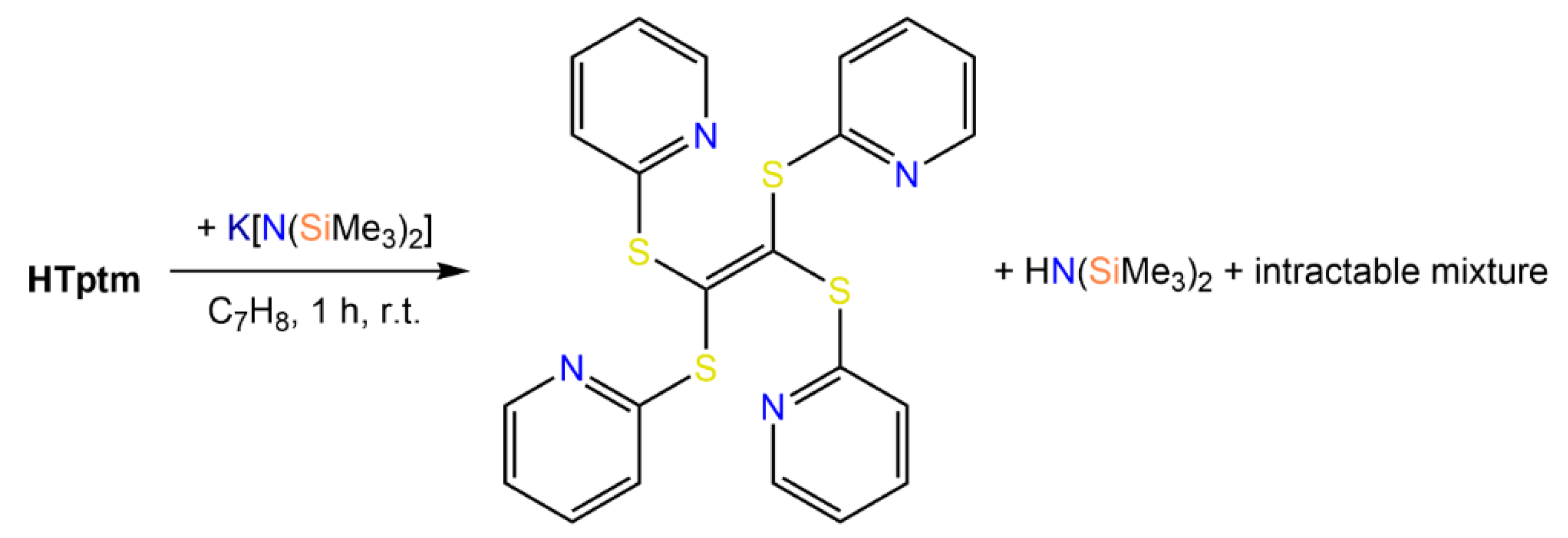
Synthesis and NMR Characterization
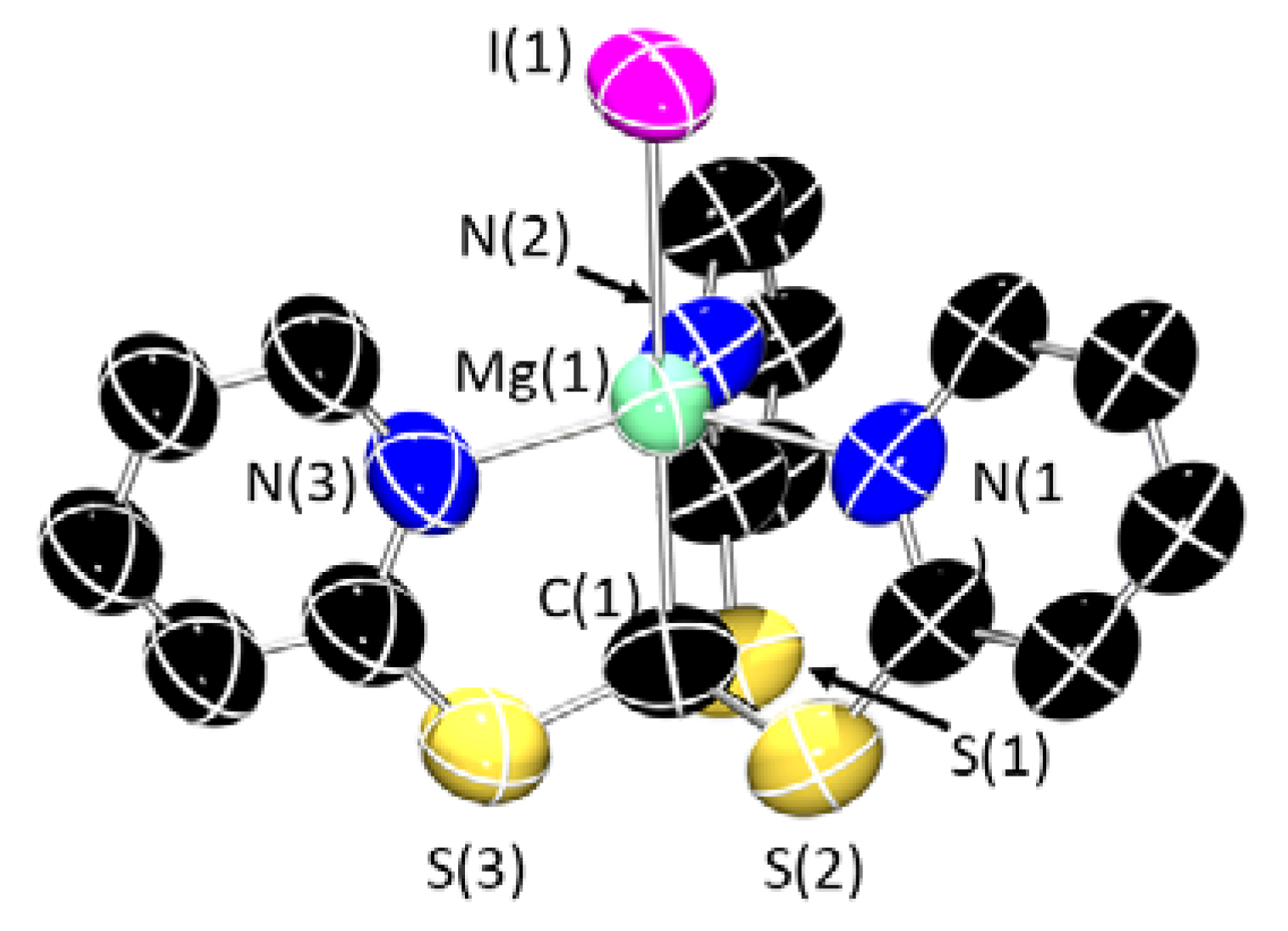
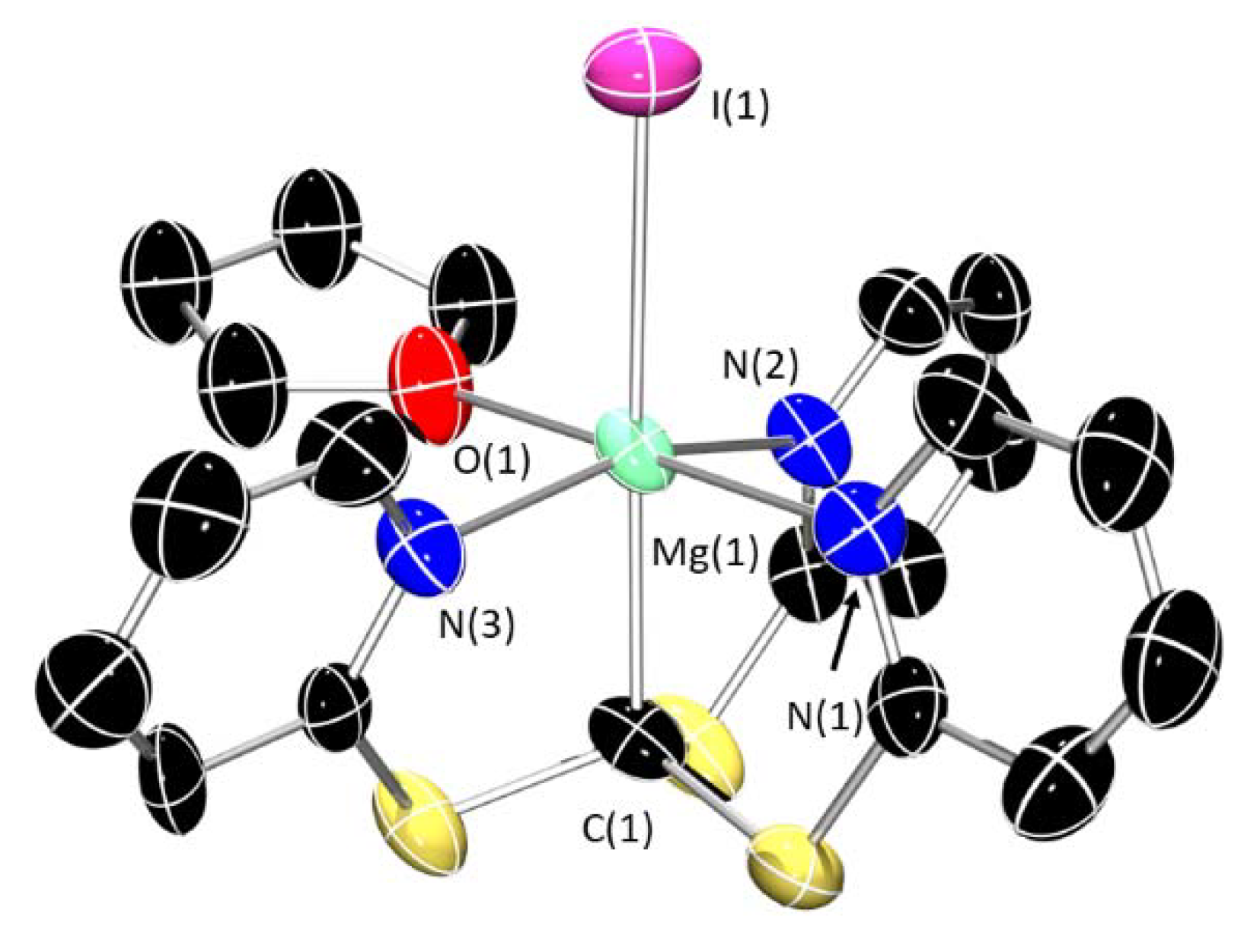
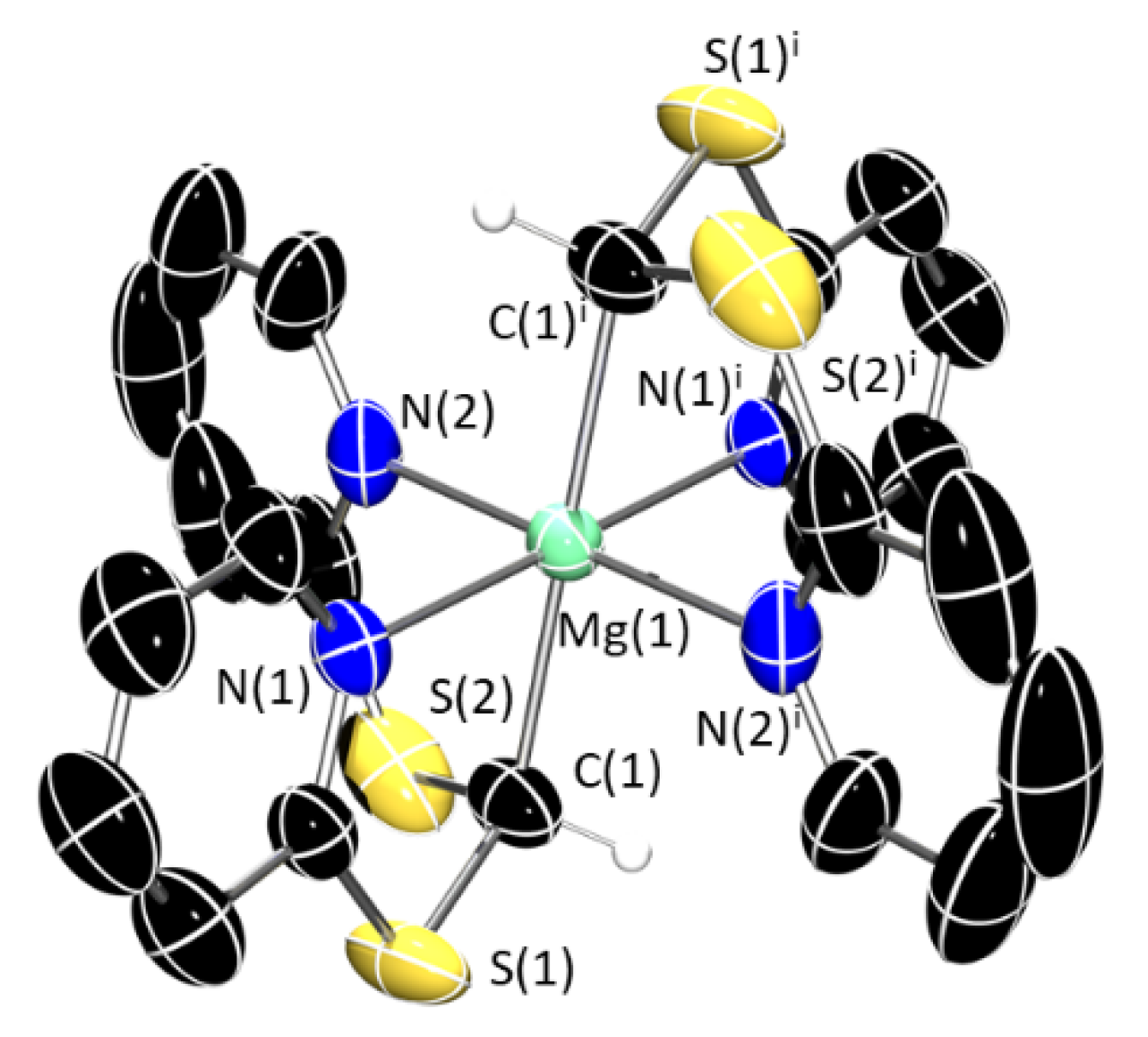
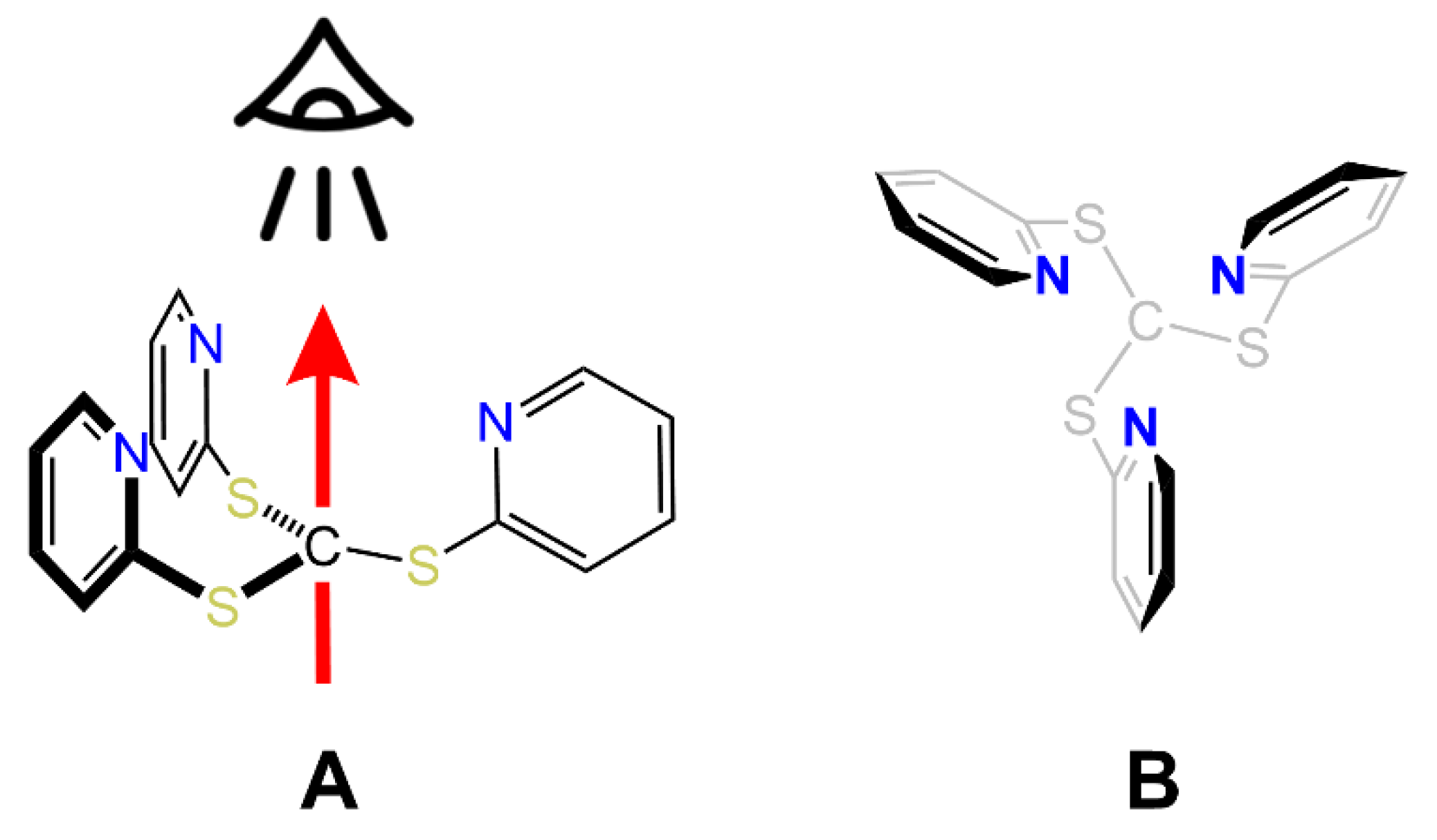
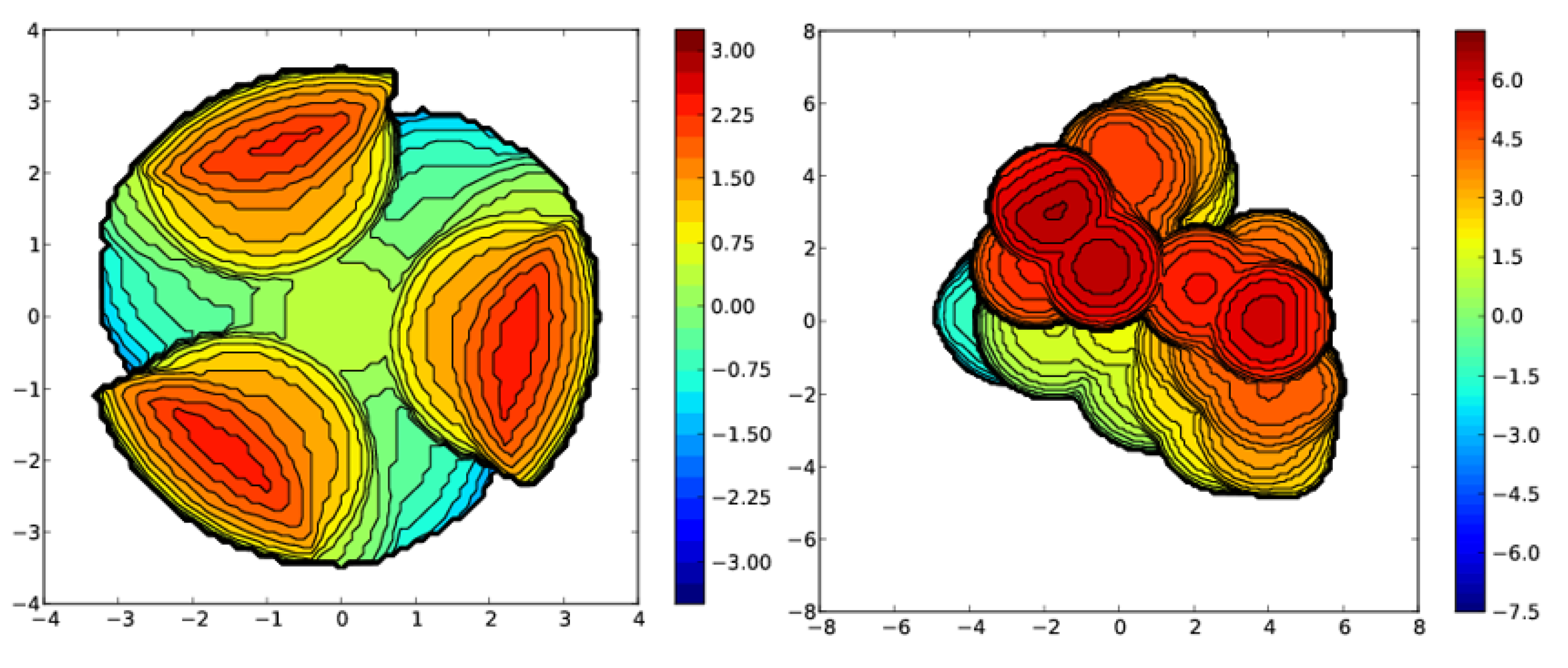
3. Structural Characterization and Buried Volume Calculations
4. Discussion
5. Conclusions
6. Materials and Methods
6.1. General Methods
6.2. Synthesis
6.3. Computational Methods
6.4. Crystallographic Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bourget-Merle, L.; Lappert, M.F.; Severn, J.R. The Chemistry of β-Diketiminatometal Complexes. Chem. Rev. 2002, 102, 3031–3065. [Google Scholar] [CrossRef] [PubMed]
- Lappert, M.F.; Protchenko, A.; Power, P.; Seeber, A. Metal Amides; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Kays, D.L. Extremely bulky amide ligands in main group chemistry. Chem. Soc. Rev. 2016, 45, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Bigmore, H.R.; Lawrence, S.C.; Mountford, P.; Tredget, C.S. Coordination, organometallic and related chemistry of tris(pyrazolyl)methane ligands. Dalton Trans. 2005, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Stradiotto, M.; Lundgren, R.J. Ligand Design in Metal Chemistry: Reactivity and Catalysis; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Trofimenko, S. Recent advances in poly(pyrazolyl)borate (scorpionate) chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Wolf, B.M.; Stuhl, C.; Maichle-Mössmer, C.; Anwander, R. Dimethylcalcium. J. Am. Chem. Soc. 2018, 140, 2373–2383. [Google Scholar] [CrossRef]
- Westerhausen, M.; Koch, A.; Görls, H.; Krieck, S. Heavy Grignard Reagents: Synthesis, Physical and Structural Properties, Chemical Behavior, and Reactivity. Chem. Eur. J. 2017, 23, 1456–1483. [Google Scholar] [CrossRef]
- Rauch, M.; Ruccolo, S.; Parkin, G. Synthesis, Structure, and Reactivity of a Terminal Magnesium Hydride Compound with a Carbatrane Motif, [TismPriBenz]MgH: A Multifunctional Catalyst for Hydrosilylation and Hydroboration. J. Am. Chem. Soc. 2017, 139, 13264–13267. [Google Scholar] [CrossRef]
- Rauch, M.; Parkin, G. Zinc and Magnesium Catalysts for the Hydrosilylation of Carbon Dioxide. J. Am. Chem. Soc. 2017, 139, 18162–18165. [Google Scholar] [CrossRef]
- Santo, R.; Miyamoto, R.; Tanaka, R.; Nishioka, T.; Sato, K.; Toyota, K.; Obata, M.; Yano, S.; Kinoshita, I.; Ichimura, A.; et al. Diamagnetic–Paramagnetic Conversion of Tris(2-pyridylthio)methylcopper(III) through a Structural Change from Trigonal Bipyramidal to Octahedral. Angew. Chem. Int. Ed. 2006, 45, 7611–7614. [Google Scholar] [CrossRef]
- Kinoshita, I.; Wright, L.J.; Kubo, S.; Kimura, K.; Sakata, A.; Yano, T.; Miyamoto, R.; Nishioka, T.; Isobe, K. Design and synthesis of copper complexes of novel ligands based on the pyridine thiolate group. Dalton Trans. 2003, 10, 1993–2003. [Google Scholar] [CrossRef]
- Miyamoto, R.; Santo, R.; Matushita, T.; Nishioka, T.; Ichimura, A.; Teki, Y.; Kinoshita, I. A complete series of copper(II) halide complexes (X = F, Cl, Br, I) with a novel Cu(II)–C(sp3) bond. Dalton Trans. 2005, 19, 3179–31866. [Google Scholar] [CrossRef] [PubMed]
- Kitano, K.; Kuwamura, N.; Tanaka, R.; Santo, R.; Nishioka, T.; Ichimura, A.; Kinoshita, I. Synthesis and characterization of tris(2-pyridylthio)methanido Zn complex with a Zn–C bond and DFT calculation of its one-electron oxidized species. Chem. Commun. 2008, 11, 1314–1316. [Google Scholar] [CrossRef] [PubMed]
- Sattler, W.; Parkin, G. Synthesis, Structure, and Reactivity of a Mononuclear Organozinc Hydride Complex: Facile Insertion of CO2 into a Zn–H Bond and CO2-Promoted Displacement of Siloxide Ligands. J. Am. Chem. Soc. 2011, 133, 9708–9711. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Sattler, W.; Parkin, G. Structural characterization of zinc bicarbonate compounds relevant to the mechanism of action of carbonic anhydrase. Chem. Sci. 2012, 3, 2015–2019. [Google Scholar] [CrossRef]
- Sattler, W.; Ruccolo, S.; Parkin, G. Synthesis, Structure, and Reactivity of a Terminal Organozinc Fluoride Compound: Hydrogen Bonding, Halogen Bonding, and Donor–Acceptor Interactions. J. Am. Chem. Soc. 2013, 135, 18714–18717. [Google Scholar] [CrossRef] [PubMed]
- Stuhl, C.; Miachle-Mössmer, C.; Anwander, R. Magnesium Stung by Nonclassical Scorpionate Ligands: Synthesis and Cone-Angle Calculations. Chem. Eur. J. 2018, 24, 14254–14268. [Google Scholar] [CrossRef] [PubMed]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. [Google Scholar] [CrossRef]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [Green Version]
- Guzei, I.A.; Wendt, M. An improved method for the computation of ligand steric effects based on solid angles. Dalton Trans. 2006, 33, 3991–3999. [Google Scholar] [CrossRef]
- Clavier, H.; Nolan, S.P. Percent buried volume for phosphine and N-heterocyclic carbene ligands: Steric properties in organometallic chemistry. Chem. Commun. 2010, 46, 841–861. [Google Scholar] [CrossRef] [PubMed]
- Ortu, F.; Moxey, G.J.; Blake, A.J.; Lewis, W.; Kays, D.L. Tuning Coordination in s-Block Carbazol-9-yl Complexes. Chem. Eur. J. 2015, 21, 6949–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, S.; Causero, A.; Elsen, H.; Pahl, J.; Langer, J.; Harder, S. Ligand Effects in Calcium Catalyzed Ketone Hydroboration. Eur. J. Inorg. Chem. 2020, 2020, 1728–1735. [Google Scholar] [CrossRef]
- De Castro, V.D.; de Lima, G.M.; Filgueiras, C.A.L.; Gambardella, M.T.P. The molecular structure and spectral studies of mercaptopyridyl-based ligands. J. Mol. Struct. 2002, 609, 199–203. [Google Scholar] [CrossRef]
- Chakrabarti, N.; Sattler, W.; Parkin, G. Structural characterization of tris(pyrazolyl)hydroborato and tris(2-pyridylthio)methyl lithium compounds: Lithium in uncommon trigonal pyramidal and trigonal monopyramidal coordination environments. Polyhedron 2013, 58, 235–246. [Google Scholar] [CrossRef]
- Kuwamura, N.; Kato, R.; Kitano, K.; Hirotsu, M.; Nishioka, T.; Hashimoto, H.; Kinoshita, I. Carbene–carbanion equilibrium for tris(2-pyridylthio)methanido Fe(II) complexes. Dalton Trans. 2010, 39, 9988–9993. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.A.P.; Smith, A.; Ortu, F.; Vitorica-Yrezabal, I.J.; Mills, D.P. Salt metathesis versus protonolysis routes for the synthesis of silylamide Hauser base (R2NMgX.; X = halogen) and amido-Grignard (R2NMgR) complexes. Dalton Trans. 2016, 45, 6004–6014. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.P.; Spray, E.; Vitorica-Yrezabal, I.J.; Singh, K.; Timmermann, V.M.; Sotorrios, L.; Macgregor, S.A.; Ortu, F. Synthesis, Characterisation and Reactivity of Group 2 Complexes with a Thiopyridyl Scorpionate Ligand. Dalton Trans. 2022. accepted manuscript. [Google Scholar] [CrossRef]
- Kloubert, T.; Müller, C.; Krieck, S.; Schlotthauer, T.; Görls, H.; Westerhausen, W. 3-(2-Pyridyl)-5-(2-thienyl)pyrazole and Complexes of Its Anion with Lithium, Magnesium, Calcium, and Zinc Ions. Eur. J. Inorg. Chem. 2012, 2012, 5991–6001. [Google Scholar] [CrossRef]
- Kanischchev, O.S.; Dolbier, W.R. Synthesis and Characterization of 2-Pyridylsulfur Pentafluorides. Angew. Chem. Int. Ed. 2015, 54, 280–284. [Google Scholar] [CrossRef]
- Schlosser, M.; Hartmann, J. Transmetalation and Double Metal Exchange: A Convenient Route to Organolithium Compounds of the Benzyl and Allyl Type. Angew. Chem. Int. Ed. 1973, 12, 508–509. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Höllwarth, A.; Böhme, M.; Dapprich, S.; Ehlers, A.W.; Gobbi, A.; Jonas, V.; Köhler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al-Bi and f-type polarization functions for Zn, Cd, Hg. Chem. Phys. Lett. 1993, 208, 237–240. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F.; Köhn, A.; Hättig, C. Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations. J. Chem. Phys. 2002, 116, 3175–3183. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SADABS: Program for Absorption Correction Using Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- CrysAlisPRO, Version 39.27b; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2017.
- Sheldrick, G. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POV-Ray Persistence of Vision Raytracer, Version 3.7; Pty. Ltd.: Williamston, Australia, 2013.
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
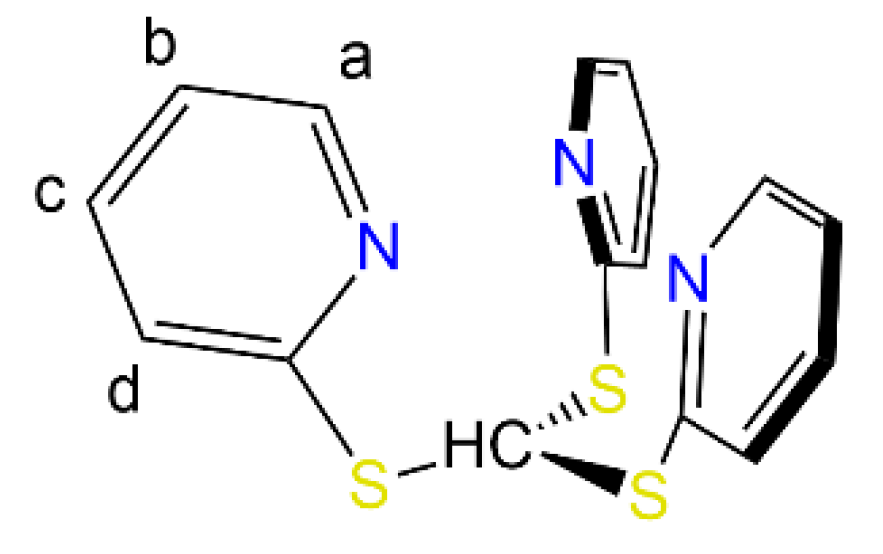
| Compound | Ha | Hc,d | Hb |
|---|---|---|---|
| 1-Cl | 9.61 | 6.45–6.48 | 6.19 |
| 1-Br | 8.22 | 6.18, 6.52 | 5.99 |
| I-I | 9.88 | 6.15–6.19 | 6.42 |
| Compound | %Vbur |
|---|---|
| HTptm | 66.7 |
| HTptmCF3 | 75.7 |
| 1-Cl/Br | 65.7 |
| 1-I (dimeric) | 65.7 |
| 1-I (monomeric) | 75.3 |
| 1-I·THF | 64.3 |
| 2 | 48.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, M.P.; Spray, E.; Vitorica-Yrezabal, I.J.; Singh, K.; Timmermann, V.M.; Sotorrios, L.; Ortu, F. Structural Investigation of Magnesium Complexes Supported by a Thiopyridyl Scorpionate Ligand. Molecules 2022, 27, 4564. https://doi.org/10.3390/molecules27144564
Stevens MP, Spray E, Vitorica-Yrezabal IJ, Singh K, Timmermann VM, Sotorrios L, Ortu F. Structural Investigation of Magnesium Complexes Supported by a Thiopyridyl Scorpionate Ligand. Molecules. 2022; 27(14):4564. https://doi.org/10.3390/molecules27144564
Chicago/Turabian StyleStevens, Matthew P., Emily Spray, Iñigo J. Vitorica-Yrezabal, Kuldip Singh, Vanessa M. Timmermann, Lia Sotorrios, and Fabrizio Ortu. 2022. "Structural Investigation of Magnesium Complexes Supported by a Thiopyridyl Scorpionate Ligand" Molecules 27, no. 14: 4564. https://doi.org/10.3390/molecules27144564
APA StyleStevens, M. P., Spray, E., Vitorica-Yrezabal, I. J., Singh, K., Timmermann, V. M., Sotorrios, L., & Ortu, F. (2022). Structural Investigation of Magnesium Complexes Supported by a Thiopyridyl Scorpionate Ligand. Molecules, 27(14), 4564. https://doi.org/10.3390/molecules27144564