
Gene Expression and Protein Abundance of Nuclear Receptors in Human Intestine and Liver: A New Application for Mass Spectrometry-Based Targeted Proteomics

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Assay Characteristics
2.2. Method Validation
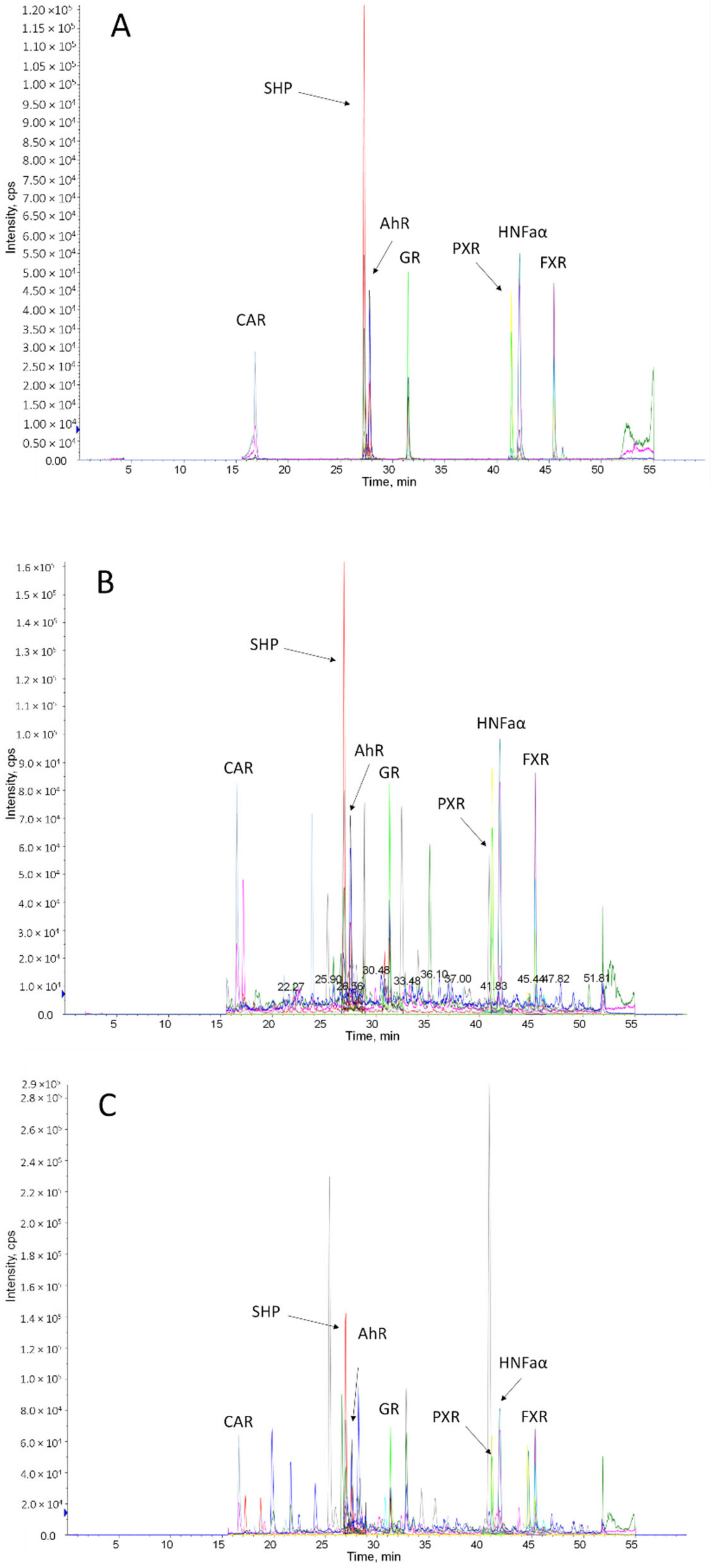
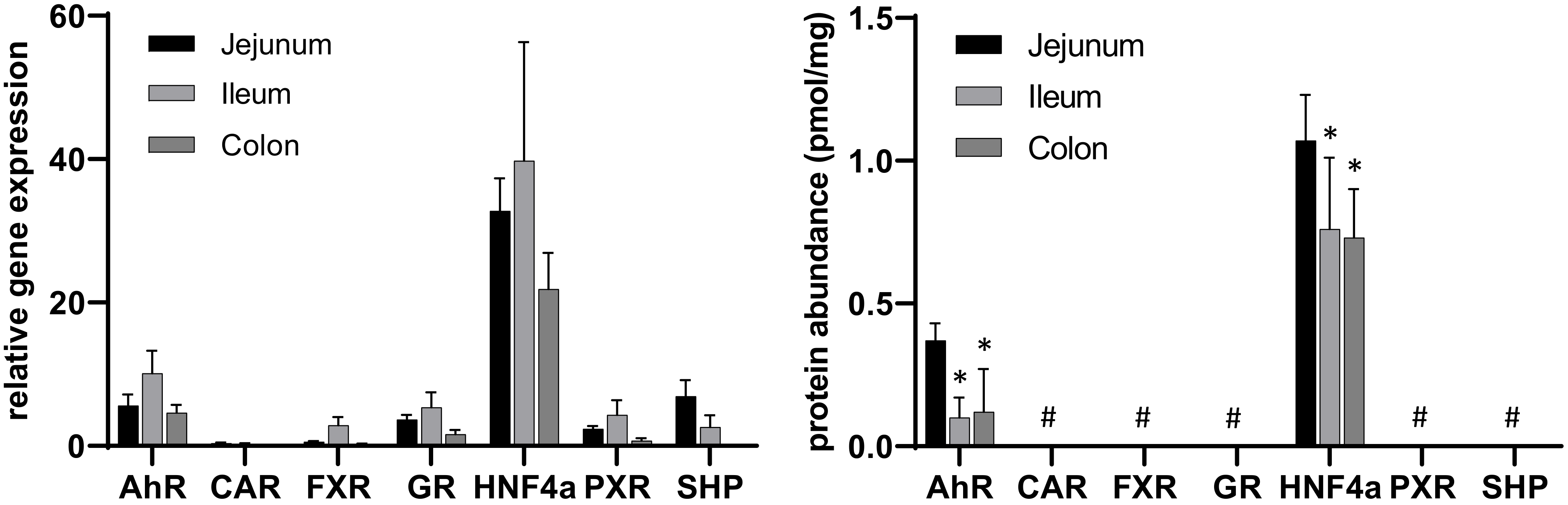
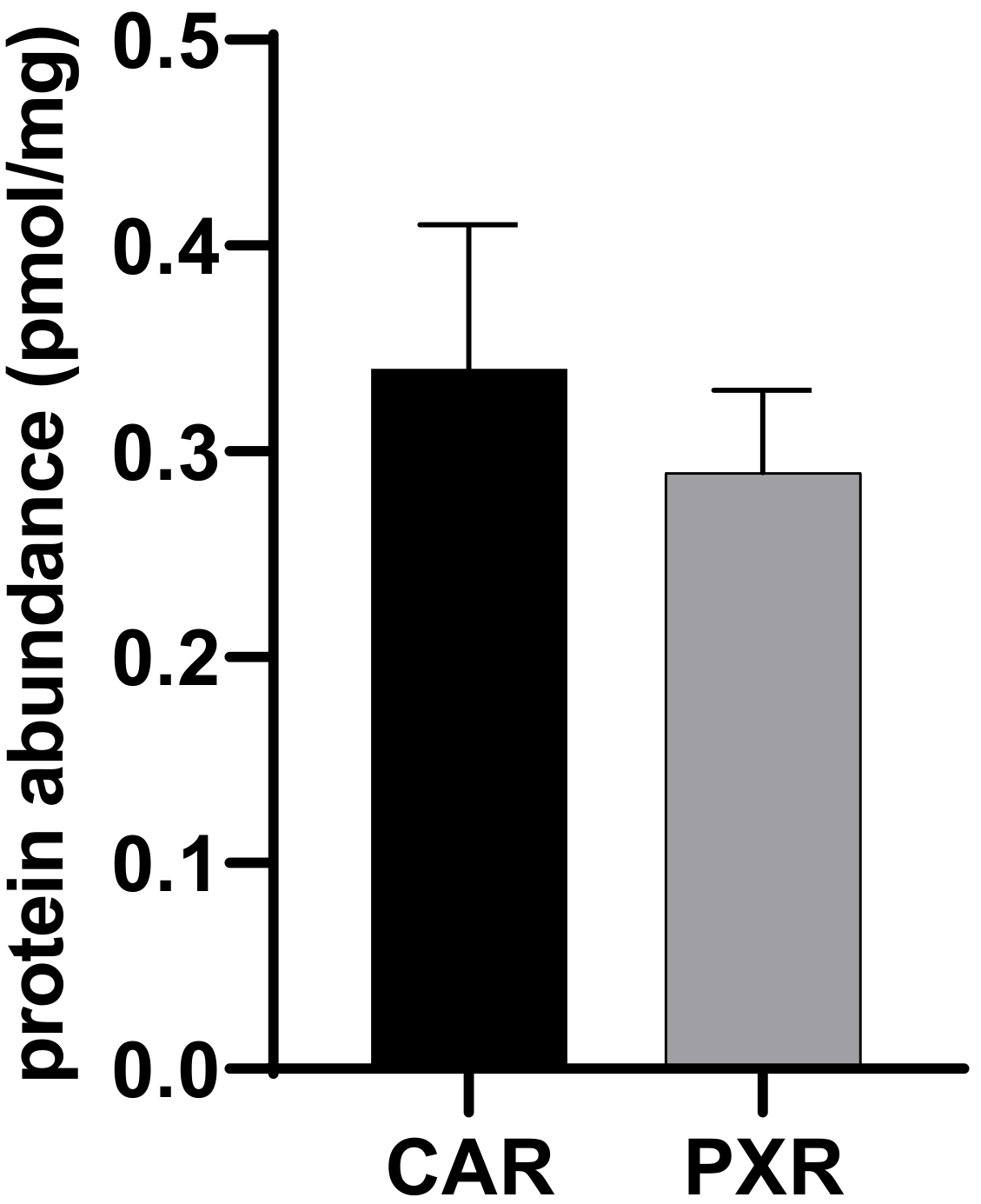
2.3. Application of the Method
3. Materials and Methods
3.1. Reagents and Consumables
3.2. Intestinal Tissue
3.3. Gene Expression Analysis
3.4. Protein Quantification by LC-MS/MS Analysis
3.4.1. Identification of Proteotypic Peptides
3.4.2. Sample Preparation and Digestion Procedure
3.4.3. LC-MS/MS Analysis
3.4.4. Preparation of Calibration Curves, Method Validation and Sample Measurements
3.5. Generation of MDCKII-CAR and -PXR
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Taub, M.E.; Chothe, P.P.; Chu, X.; Giacomini, K.M.; Kim, R.B.; Ray, A.S.; Stocker, S.L.; Unadkat, J.D.; Wittwer, M.B.; et al. Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance. Clin. Pharmacol. Ther. 2018, 104, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.W.; Brackman, D.J.; Ennis, E.A.; Sugiyama, Y.; Kamdem, L.K.; Blanchard, R.; Galetin, A.; Zhang, L.; Giacomini, K.M. Influence of Transporter Polymorphisms on Drug Disposition and Response: A Perspective from the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 803–817. [Google Scholar] [CrossRef]
- Hirota, T.; Tanaka, T.; Takesue, H.; Ieiri, I. Epigenetic regulation of drug transporter expression in human tissues. Expert Opin. Drug Metab. Toxicol. 2017, 13, 19–30. [Google Scholar] [CrossRef]
- Peng, L.; Zhong, X. Epigenetic regulation of drug metabolism and transport. Acta Pharm. Sin. B 2015, 5, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Zanger, U.M.; Klein, K.; Thomas, M.; Rieger, J.K.; Tremmel, R.; Kandel, B.A.; Klein, M.; Magdy, T. Genetics, epigenetics, and regulation of drug-metabolizing cytochrome p450 enzymes. Clin. Pharmacol. Ther. 2014, 95, 258–261. [Google Scholar] [CrossRef]
- Czuba, L.C.; Hillgren, K.M.; Swaan, P.W. Post-translational modifications of transporters. Pharmacol. Ther. 2018, 192, 88–99. [Google Scholar] [CrossRef]
- Evers, R.; Piquette-Miller, M.; Polli, J.W.; Russel, F.G.M.; Sprowl, J.A.; Tohyama, K.; Ware, J.A.; de Wildt, S.N.; Xie, W.; Brouwer, K.L.R. Disease-Associated Changes in Drug Transporters May Impact the Pharmacokinetics and/or Toxicity of Drugs: A White Paper from the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 900–915. [Google Scholar] [CrossRef]
- Dunvald, A.-C.D.; Järvinen, E.; Mortensen, C.; Stage, T.B. Clinical and Molecular Perspectives on Inflammation-Mediated Regulation of Drug Metabolism and Transport. Clin. Pharmacol. Ther. 2021, 112, 277–290. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Woody, S.; Sun, M.; Cui, W. Nuclear-receptor-mediated regulation of drug- and bile-acid-transporter proteins in gut and liver. Drug Metab. Rev. 2013, 45, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, K.L.R.; Evers, R.; Hayden, E.; Hu, S.; Li, C.Y.; Meyer Zu Schwabedissen, H.E.; Neuhoff, S.; Oswald, S.; Piquette-Miller, M.; Saran, C.; et al. Regulation of Drug Transport Proteins-From Mechanisms to Clinical Impact: A White Paper on Behalf of the International Transporter Consortium. Clin. Pharmacol. Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Gessner, A.; König, J.; Fromm, M.F. Clinical Aspects of Transporter-Mediated Drug-Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and drug metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: Implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef]
- Rodrigues, A.D.; Lai, Y.; Shen, H.; Varma, M.V.S.; Rowland, A.; Oswald, S. Induction of Human Intestinal and Hepatic Organic Anion Transporting Polypeptides: Where Is the Evidence for Its Relevance in Drug-Drug Interactions? Drug Metab. Dispos. 2020, 48, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.D.; Rowland, A. Profiling of Drug-Metabolizing Enzymes and Transporters in Human Tissue Biopsy Samples: A Review of the Literature. J. Pharmacol. Exp. Ther. 2020, 372, 308–319. [Google Scholar] [CrossRef]
- Barone, G.W.; Gurley, B.J.; Ketel, B.L.; Lightfoot, M.L.; Abul-Ezz, S.R. Drug interaction between St. John’s wort and cyclosporine. Ann. Pharmacother. 2000, 34, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Ruschitzka, F.; Meier, P.J.; Turina, M.; Lüscher, T.F.; Noll, G. Acute heart transplant rejection due to Saint John’s wort. Lancet 2000, 355, 548–549. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S.; Yokoi, T. Tissue-specific mRNA expression profiles of human nuclear receptor subfamilies. Drug Metab. Pharmacokinet. 2004, 19, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Brueck, S.; Bruckmueller, H.; Wegner, D.; Busch, D.; Martin, P.; Oswald, S.; Cascorbi, I.; Siegmund, W. Transcriptional and Post-Transcriptional Regulation of Duodenal P-Glycoprotein and MRP2 in Healthy Human Subjects after Chronic Treatment with Rifampin and Carbamazepine. Mol. Pharm. 2019, 16, 3823–3830. [Google Scholar] [CrossRef] [PubMed]
- Fritz, A.; Busch, D.; Lapczuk, J.; Ostrowski, M.; Drozdzik, M.; Oswald, S. Expression of clinically relevant drug-metabolizing enzymes along the human intestine and their correlation to drug transporters and nuclear receptors: An intra-subject analysis. Basic Clin. Pharmacol. Toxicol. 2019, 124, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Greiner, B.; Eichelbaum, M.; Fritz, P.; Kreichgauer, H.P.; von Richter, O.; Zundler, J.; Kroemer, H.K. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Investig. 1999, 104, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtbecker, N.; Fromm, M.F.; Kroemer, H.K.; Ohnhaus, E.E.; Heidemann, H. The nifedipine-rifampin interaction. Evid. Induction Gut Wall Metab. Drug Metab. Dispos. 1996, 24, 1121–1123. [Google Scholar]
- Westphal, K.; Weinbrenner, A.; Zschiesche, M.; Franke, G.; Knoke, M.; Oertel, R.; Fritz, P.; von Richter, O.; Warzok, R.; Hachenberg, T.; et al. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: A new type of drug/drug interaction. Clin. Pharmacol. Ther. 2000, 68, 345–355. [Google Scholar] [CrossRef]
- Gorski, J. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin. Pharmacol. Ther. 2003, 74, 275–287. [Google Scholar] [CrossRef]
- Fromm, M.F.; Busse, D.; Kroemer, H.K.; Eichelbaum, M. Differential induction of prehepatic and hepatic metabolism of verapamil by rifampin. Hepatology 1996, 24, 796–801. [Google Scholar] [CrossRef]
- Oswald, S.; Meyer zu Schwabedissen, H.E.; Nassif, A.; Modess, C.; Desta, Z.; Ogburn, E.T.; Mostertz, J.; Keiser, M.; Jia, J.; Hubeny, A.; et al. Impact of efavirenz on intestinal metabolism and transport: Insights from an interaction study with ezetimibe in healthy volunteers. Clin. Pharmacol. Ther. 2012, 91, 506–513. [Google Scholar] [CrossRef]
- Mouly, S.; Lown, K.S.; Kornhauser, D.; Joseph, J.L.; Fiske, W.D.; Benedek, I.H.; Watkins, P.B. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin. Pharmacol. Ther. 2002, 72, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kaza, M.; Karaźniewicz-Łada, M.; Kosicka, K.; Siemiątkowska, A.; Rudzki, P.J. Bioanalytical method validation: New FDA guidance vs. EMA Guideline. Better Or Worse? J. Pharm. Biomed. Anal. 2019, 165, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Gröer, C.; Brück, S.; Lai, Y.; Paulick, A.; Busemann, A.; Heidecke, C.D.; Siegmund, W.; Oswald, S. LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J. Pharm. Biomed. Anal. 2013, 85, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Fallon, J.K.; Neubert, H.; Hyland, R.; Goosen, T.C.; Smith, P.C. Targeted quantitative proteomics for the analysis of 14 UGT1As and -2Bs in human liver using NanoUPLC-MS/MS with selected reaction monitoring. J. Proteome Res. 2013, 12, 4402–4413. [Google Scholar] [CrossRef]
- Gröer, C.; Busch, D.; Patrzyk, M.; Beyer, K.; Busemann, A.; Heidecke, C.D.; Drozdzik, M.; Siegmund, W.; Oswald, S. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP-glucuronosyltransferases by mass spectrometry-based targeted proteomics. J. Pharm. Biomed. Anal. 2014, 100, 393–401. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Brown, P.W.; Ozer, J.S.; Lai, Y. Liquid chromatography/tandem mass spectrometry based targeted proteomics quantification of P-glycoprotein in various biological samples. Rapid Commun. Mass Spectrom. 2011, 25, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Ohtsuki, S.; Kamiie, J.; Suzuki, T.; Abe, T.; Terasaki, T. Simultaneous absolute quantification of 11 cytochrome P450 isoforms in human liver microsomes by liquid chromatography tandem mass spectrometry with in silico target peptide selection. J. Pharm. Sci. 2011, 100, 341–352. [Google Scholar] [CrossRef]
- Oswald, S.; Gröer, C.; Drozdzik, M.; Siegmund, W. Mass spectrometry-based targeted proteomics as a tool to elucidate the expression and function of intestinal drug transporters. AAPS J. 2013, 15, 1128–1140. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Peptide | Mass Transitions (m/z) | CE [V] | DP [V] | |
|---|---|---|---|---|---|
| Q1 | Q3 | ||||
| AhR | NDFSGEVDFR | 593.4 | 322.2 | 39 | 150 |
| 809.5 | 27 | 150 | |||
| 722.6 | 24 | 150 | |||
| AhR * | NDFSGEVDF[R(13C6;15N4)] | 598.2 | 332.2 | 39 | 150 |
| 819.4 | 27 | 150 | |||
| 732.4 | 24 | 150 | |||
| CAR | AQQTPVQLSK | 550.2 | 671.3 | 27 | 120 |
| 900.5 | 23 | 120 | |||
| 429.4 | 23 | 120 | |||
| CAR * | AQQTPVQLS[K(13C6;15N2)] | 554.1 | 679.4 | 27 | 120 |
| 908.5 | 23 | 120 | |||
| 429.1 | 23 | 120 | |||
| FXR | LQEPLLDVLQK | 648.5 | 371.3 | 27 | 190 |
| 925.6 | 27 | 190 | |||
| 388.4 | 39 | 190 | |||
| FXR * | LQEPLLDVLQ[K(13C6;15N2)] | 652.0 | 371.2 | 27 | 190 |
| 933.6 | 27 | 190 | |||
| 396.3 | 39 | 190 | |||
| GR | LLEESIANLNR | 635.9 | 787.4 | 31 | 170 |
| 356.4 | 28 | 170 | |||
| 485.2 | 26 | 170 | |||
| GR * | LLEESIANLN[R(13C6;15N4)] | 641.0 | 797.4 | 31 | 170 |
| 356.2 | 28 | 170 | |||
| 485.2 | 26 | 170 | |||
| HNF4α | DVLLLGNDYIVPR | 743.6 | 554.4 | 25 | 180 |
| 647.5 | 45 | 180 | |||
| 876.4 | 36 | 180 | |||
| HNF4α * | DVLLLGNDYIVP[R(13C6;15N4)] | 748.6 | 554.4 | 25 | 180 |
| 657.3 | 45 | 180 | |||
| 886.5 | 36 | 180 | |||
| PXR | VVDQLQEQFAITLK | 816.2 | 361.2 | 33 | 170 |
| 474.4 | 35 | 170 | |||
| 1077.4 | 36 | 170 | |||
| PXR * | VVDQLQEQFAITL[K(13C6;15N2)] | 820.3 | 369.2 | 33 | 170 |
| 482.4 | 35 | 170 | |||
| 1085.4 | 36 | 170 | |||
| SHP | VLLTASTLK | 472.8 | 620.4 | 20 | 130 |
| 326.1 | 17 | 130 | |||
| 448.2 | 32 | 130 | |||
| SHP * | VLLTASTL[K(13C6;15N2)] | 476.7 | 628.3 | 20 | 130 |
| 326.2 | 17 | 130 | |||
| 456.2 | 32 | 130 | |||
| Accuracy [%] | Precision [%] | Correlation Coefficient r | |||
|---|---|---|---|---|---|
| Within-Day | Between-Day | Within-Day | Between-Day | ||
| AhR | −1.7–13.2 | −1.2–0.3 | 2.3–4.5 | 3.2–9.3 | 0.9984–0.9999 |
| CAR | 0.5–12.0 | −2.3–(−0.8) | 4.4–7.6 | 2.7–7.6 | 0.9976–0.9999 |
| FXR | −1.8–6.3 | −3.0–2.3 | 0.9–3.5 | 3.3–5.4 | 0.9992–0.9999 |
| GR | −0.2–11.5 | −2.1–3.6 | 1.4–4.1 | 3.0–5.8 | 0.9992–0.9999 |
| HNF4α | −2.1–10.2 | −2.3–3.2 | 0.8–3.7 | 3.8–7.3 | 0.9995–0.9998 |
| PXR | −3.5–7.7 | −2.1–6.5 | 2.1–6.8 | 4.3–5.5 | 0.9988–0.9999 |
| SHP | −0.9–0.7 | −2.5–1.2 | 0.9–5.9 | 3.7–5.9 | 0.9992–0.9998 |
| Matrix Effect [%] | Rack Stability 24 h @ 4 °C [%] | Freeze–Thaw Stability [%] | |||
|---|---|---|---|---|---|
| 1st Cycle | 2nd Cycle | 3rd Cycle | |||
| AhR | 95.9–102.0 | 95.6–100.7 | 85.8–105.8 | 91.5–100.2 | 90.9–97.4 |
| CAR | 100.3–101.8 | 93.6–98.3 | 90.6–105.1 | 90.3–103.2 | 86.9–89.4 |
| FXR | 96.3–113.8 | 100.1–104.7 | 88.9–104.4 | 93.2–99.9 | 90.8–100.1 |
| GR | 91.2–101.5 | 97.1–102.9 | 90.8–106.6 | 92.9–102.1 | 95.1–99.5 |
| HNF4α | 88.5–95.8 | 98.2–104.3 | 87.6–104.9 | 91.7–100.7 | 89.3–99.1 |
| PXR | 82.8–91.3 | 99.8–104.6 | 86.6–98.5 | 91.0–95.8 | 93.3–93.6 |
| SHP | 97.2–99.3 | 93.4–100.8 | 92.3–104.8 | 93.7–99.5 | 93.3–98.5 |
| Protein (Alias) | Gene Name | TaqMan© Assay I.D. | Peptide |
|---|---|---|---|
| AhR (BHLHE76) | AhR | Hs00169233_m1 | NDFSGEVDFR |
| CAR | NR1I3 | Hs00901571_m1 | AQQTPVQLSK |
| FXR (BAR) | NR1H4 | Hs01026590_m1 | LQEPLLDVLQK |
| GR (GCR) | NR3C1 | Hs00353740_m1 | LLEESIANLNR |
| HNF4α (HNF4, NR2A1) | HNF4A | Hs00230853_m1 | DVLLLGNDYIVPR |
| PXR (BXR) | NR1I2 | Hs01114267_m1 | VVDQLQEQFAITLK |
| SHP (SHP1) | NR0B2 | Hs00222677_m1 | VLLTASTLK |
| Reference gene(s) | |||
| 18S | 18S | Hs99999901_s1 | - |
| GAPDH | GAPDH | Hs02758991_g1 | - |
| PGK1 | PGK1 | Hs00943178_g1 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wenzel, C.; Gödtke, L.; Reichstein, A.; Keiser, M.; Busch, D.; Drozdzik, M.; Oswald, S. Gene Expression and Protein Abundance of Nuclear Receptors in Human Intestine and Liver: A New Application for Mass Spectrometry-Based Targeted Proteomics. Molecules 2022, 27, 4629. https://doi.org/10.3390/molecules27144629
Wenzel C, Gödtke L, Reichstein A, Keiser M, Busch D, Drozdzik M, Oswald S. Gene Expression and Protein Abundance of Nuclear Receptors in Human Intestine and Liver: A New Application for Mass Spectrometry-Based Targeted Proteomics. Molecules. 2022; 27(14):4629. https://doi.org/10.3390/molecules27144629
Chicago/Turabian StyleWenzel, Christoph, Lisa Gödtke, Anne Reichstein, Markus Keiser, Diana Busch, Marek Drozdzik, and Stefan Oswald. 2022. "Gene Expression and Protein Abundance of Nuclear Receptors in Human Intestine and Liver: A New Application for Mass Spectrometry-Based Targeted Proteomics" Molecules 27, no. 14: 4629. https://doi.org/10.3390/molecules27144629
APA StyleWenzel, C., Gödtke, L., Reichstein, A., Keiser, M., Busch, D., Drozdzik, M., & Oswald, S. (2022). Gene Expression and Protein Abundance of Nuclear Receptors in Human Intestine and Liver: A New Application for Mass Spectrometry-Based Targeted Proteomics. Molecules, 27(14), 4629. https://doi.org/10.3390/molecules27144629