
21 Fluorescent Protein-Based DNA Staining Dyes

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
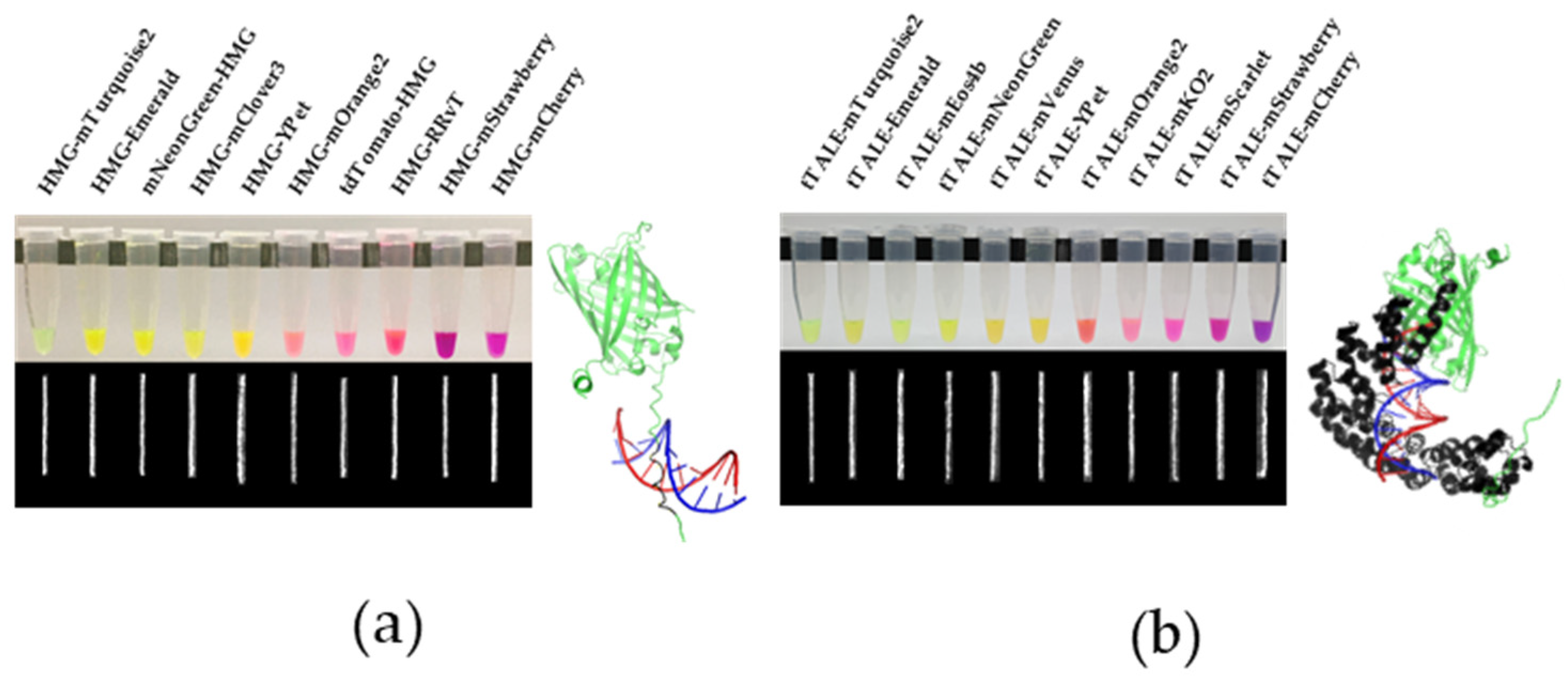
2.1. Construction and Expression of Diverse HMG-FPs and tTALE-FPs
2.2. Brightness Optimization of FP-DBPs
2.3. A/T-Rich Specific Staining by tTALE-FP
3. Materials and Methods
3.1. Chemicals
3.2. FP-DBP Recombinants
3.3. Molecular Cloning
3.4. Polydimethylsiloxane (PDMS) Microfluidic Devices
3.5. Positively-Charged Surface Preparation
3.6. Microscopy
3.7. DNA Imaging on a Positively Charged Surface and Fluorescence Intensity Measurement
3.8. DNA Imaging on a Positively Charged Surface with Microchannels
3.9. Fluorescence Intensity Measurement with the Fluorometer
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Hapsari, N.D.; Lee, S.; Jo, K. DNA binding fluorescent proteins as single-molecule probes. Analyst 2020, 145, 4079–4095. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M. Fluorescent Proteins 101: Photoactivatable Fluorescent Proteins; Addgene Blog: Watertown, NY, USA, 2017. [Google Scholar]
- Snapp, E. Design and use of fluorescent fusion proteins in cell biology. Curr. Protoc. Cell Biol. 2005, 21, 21 4 1–21 4 13. [Google Scholar] [CrossRef]
- Heim, R.; Prasher, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef]
- Lambert, T.J. FPbase: A community-editable fluorescent protein database. Nat. Methods. 2019, 16, 277–278. [Google Scholar] [CrossRef]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 1992, 111, 229–233. [Google Scholar] [CrossRef]
- Tomosugi, W.; Matsuda, T.; Tani, T.; Nemoto, T.; Kotera, I.; Saito, K.; Horikawa, K.; Nagai, T. An ultramarine fluorescent protein with increased photostability and pH insensitivity. Nat. Methods. 2009, 6, 351–353. [Google Scholar] [CrossRef]
- Nagel, G.; Szellas, T.; Huhn, W.; Kateriya, S.; Adeishvili, N.; Berthold, P.; Ollig, D.; Hegemann, P.; Bamberg, E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef]
- Hirano, M.; Ando, R.; Shimozono, S.; Sugiyama, M.; Takeda, N.; Kurokawa, H.; Deguchi, R.; Endo, K.; Haga, K.; Takai-Todaka, R.; et al. A highly photostable and bright green fluorescent protein. Nat. Biotechnol. 2022, 40, 1132–1142. [Google Scholar] [CrossRef]
- Merzlyak, E.M.; Goedhart, J.; Shcherbo, D.; Bulina, M.E.; Shcheglov, A.S.; Fradkov, A.F.; Gaintzeva, A.; Lukyanov, K.A.; Lukyanov, S.; Gadella, T.W.; et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods. 2007, 4, 555–557. [Google Scholar] [CrossRef]
- Patterson, G.H.K.; Susan, M.; Sharif, W.D.; Kain, S.R.; Piston, D.W. Use of the Green Fluorescent Protein and Its Mutants in Quantitative Fluorescence Microscopy. Biophys J. 1997, 73, 2782–2790. [Google Scholar] [CrossRef]
- Wang, L.; Jackson, W.C.; Steinbach, P.A.; Tsien, R.Y. Evolution of new nonantibody proteins via iterative somatic hypermutation. Proc. Natl. Acad. Sci. USA 2004, 101, 16745–16749. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tsien, R.Y. Evolving proteins in mammalian cells using somatic hypermutation. Nat. Protoc. 2006, 1, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.G.; Depernet, H.; Gotthard, G.; Schultz, D.T.; Navizet, I.; Lambert, T.; Adams, S.R.; Torreblanca-Zanca, A.; Chu, M.; Bindels, D.S.; et al. Aequorea’s secrets revealed: New fluorescent proteins with unique properties for bioimaging and biosensing. PLoS Biol. 2020, 18, e3000936. [Google Scholar] [CrossRef]
- Wiens, M.D.; Shen, Y.; Li, X.; Salem, M.A.; Smisdom, N.; Zhang, W.; Brown, A.; Campbell, R.E. A Tandem Green-Red Heterodimeric Fluorescent Protein with High FRET Efficiency. Chembiochem 2016, 17, 2361–2367. [Google Scholar] [CrossRef]
- Lee, S.; Oh, Y.; Lee, J.; Choe, S.; Lim, S.; Lee, H.S.; Jo, K.; Schwartz, D.C. DNA binding fluorescent proteins for the direct visualization of large DNA molecules. Nucleic Acids Res. 2016, 44, e6. [Google Scholar] [CrossRef]
- Jo, K.; Dhingra, D.M.; Odijk, T.; de Pablo, J.J.; Graham, M.D.; Runnheim, R.; Forrest, D.; Schwartz, D.C. A single-molecule barcoding system using nanoslits for DNA analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 2673–2678. [Google Scholar] [CrossRef]
- Teague, B.; Waterman, M.S.; Goldstein, S.; Potamousis, K.; Zhou, S.; Reslewic, S.; Sarkar, D.; Valouev, A.; Churas, C.; Kidd, J.M.; et al. High-resolution human genome structure by single-molecule analysis. Proc. Natl. Acad. Sci. USA 2010, 107, 10848–10853. [Google Scholar] [CrossRef]
- Kounovsky-Shafer, K.L.; Hernandez-Ortiz, J.P.; Potamousis, K.; Tsvid, G.; Place, M.; Ravindran, P.; Jo, K.; Zhou, S.; Odijk, T.; de Pablo, J.J.; et al. Electrostatic confinement and manipulation of DNA molecules for genome analysis. Proc. Natl. Acad. Sci. USA 2017, 114, 13400–13405. [Google Scholar] [CrossRef]
- Lam, E.T.; Hastie, A.; Lin, C.; Ehrlich, D.; Das, S.K.; Austin, M.D.; Deshpande, P.; Cao, H.; Nagarajan, N.; Xiao, M.; et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012, 30, 771–776. [Google Scholar] [CrossRef]
- Nyberg, L.K.; Persson, F.; Berg, J.; Bergstrom, J.; Fransson, E.; Olsson, L.; Persson, M.; Stalnacke, A.; Wigenius, J.; Tegenfeldt, J.O.; et al. A single-step competitive binding assay for mapping of single DNA molecules. Biochem. Biophys. Res. Commun. 2012, 417, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.N.; Emilsson, G.; Nyberg, L.K.; Noble, C.; Stadler, L.S.; Fritzsche, J.; Moore, E.R.; Tegenfeldt, J.O.; Ambjornsson, T.; Westerlund, F. Competitive binding-based optical DNA mapping for fast identification of bacteria—Multi-ligand transfer matrix theory and experimental applications on Escherichia coli. Nucleic Acids Res. 2014, 42, e118. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kawamoto, Y.; Vaijayanthi, T.; Park, J.; Bae, J.; Kim-Ha, J.; Sugiyama, H.; Jo, K. TAMRA-polypyrrole for A/T sequence visualization on DNA molecules. Nucleic Acids Res. 2018, 46, e108. [Google Scholar] [CrossRef]
- Gubu, A.; Li, L.; Ning, Y.; Zhang, X.; Lee, S.; Feng, M.; Li, Q.; Lei, X.; Jo, K.; Tang, X. Bioorthogonal Metabolic DNA Labelling using Vinyl Thioether-Modified Thymidine and o-Quinolinone Quinone Methide. Chemistry 2018, 24, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Neely, R.K.; Dedecker, P.; Hotta, J.-I.; Urbanavičiūtė, G.; Klimašauskas, S.; Hofkens, J. DNA fluorocode: A single molecule, optical map of DNA with nanometre resolution. Chem. Sci. 2010, 1, 453–460. [Google Scholar] [CrossRef]
- Gabrieli, T.; Michaeli, Y.; Avraham, S.; Torchinsky, D.; Margalit, S.; Schutz, L.; Juhasz, M.; Coruh, C.; Arbib, N.; Zhou, Z.S.; et al. Chemoenzymatic labeling of DNA methylation patterns for single-molecule epigenetic mapping. Nucleic Acids Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Cheon, N.Y.; Cha, J.; Kim, A.; Kim, H.I.; Lee, L.; Kim, K.O.; Jo, K.; Lee, J.Y. High-throughput single-molecule imaging system using nanofabricated trenches and fluorescent DNA-binding proteins. Biotechnol. Bioeng. 2020, 117, 1640–1648. [Google Scholar] [CrossRef]
- Kim, K.I.; Lee, S.; Jin, X.; Kim, S.J.; Jo, K.; Lee, J.H. DNA Binding Peptide Directed Synthesis of Continuous DNA Nanowires for Analysis of Large DNA Molecules by Scanning Electron Microscope. Small 2017, 13, 1601926. [Google Scholar] [CrossRef]
- Kim, K.I.; Yoon, S.; Chang, J.; Lee, S.; Cho, H.H.; Jeong, S.H.; Jo, K.; Lee, J.H. Multifunctional Heterogeneous Carbon Nanotube Nanocomposites Assembled by DNA-Binding Peptide Anchors. Small 2020, 16, e1905821. [Google Scholar] [CrossRef]
- Graneli, A.; Yeykal, C.C.; Robertson, R.B.; Greene, E.C. Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 1221–1226. [Google Scholar] [CrossRef]
- Giepmans, B.N.; Adams, S.R.; Ellisman, M.H.; Tsien, R.Y. The fluorescent toolbox for assessing protein location and function. Science 2006, 312, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Wang, C.; Song, J.; Kim, D.-G.; Oh, Y.; Ko, W.; Lee, J.; Park, J.; Lee, H.S.; Jo, K. Investigation of various fluorescent protein–DNA binding peptides for effectively visualizing large DNA molecules. RSC Adv. 2016, 6, 46291–46298. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.; Won, N.; Shin, E.; Kim, S.-H.; Chun, M.-Y.; Gu, J.; Jung, G.-Y.; Lim, K.-I.; Jo, K. Single-molecule DNA visualization using AT-specific red and non-specific green DNA-binding fluorescent proteins. Analyst 2019, 144, 921–927. [Google Scholar] [CrossRef]
- Shin, E.; Kim, W.; Lee, S.; Bae, J.; Kim, S.; Ko, W.; Seo, H.S.; Lim, S.; Lee, H.S.; Jo, K. Truncated TALE-FP as DNA Staining Dye in a High-salt Buffer. Sci. Rep. 2019, 9, 17197. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef]
- Beliveau, B.J.; Boettiger, A.N.; Avendano, M.S.; Jungmann, R.; McCole, R.B.; Joyce, E.F.; Kim-Kiselak, C.; Bantignies, F.; Fonseka, C.Y.; Erceg, J.; et al. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun. 2015, 6, 7147. [Google Scholar] [CrossRef]
- Nieves, D.J.; Gaus, K.; Baker, M.A.B. DNA-Based Super-Resolution Microscopy: DNA-PAINT. Genes 2018, 9, 621. [Google Scholar] [CrossRef]
- Pattanayak, V.; Guilinger, J.P.; Liu, D.R. Determining the specificities of TALENs, Cas9, and other genome-editing enzymes. Methods Enzym. 2014, 546, 47–78. [Google Scholar]
- Pathania, M.; Torres-Reveron, J.; Yan, L.; Kimura, T.; Lin, T.V.; Gordon, V.; Teng, Z.Q.; Zhao, X.; Fulga, T.A.; Van Vactor, D.; et al. miR-132 enhances dendritic morphogenesis, spine density, synaptic integration, and survival of newborn olfactory bulb neurons. PLoS ONE 2012, 7, e38174. [Google Scholar]
- Shaner, N.C.; Lin, M.Z.; McKeown, M.R.; Steinbach, P.A.; Hazelwood, K.L.; Davidson, M.W.; Tsien, R.Y. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 2008, 5, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.W.; Daugherty, P.S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 23, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Chertkova, A.O.; Marieke, M.; Postma, M.; van Bommel, N.; van der Niet, S.; Batenburg, K.L.; Joosen, L.; Gadella, T.W.J., Jr.; Okada, Y.; Goedhart, J. Robust and Bright Genetically Encoded Fluorescent Markers for Highlighting Structures and Compartments in Mammalian Cells. bioRxiv 2020, 160374. [Google Scholar] [CrossRef]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W., Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef]
- Balleza, E.; Kim, J.M.; Cluzel, P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat. Methods 2018, 15, 47–51. [Google Scholar] [CrossRef]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef]
- Koushik, S.V.; Chen, H.; Thaler, C.; Puhl, H.L., 3rd; Vogel, S.S. Cerulean, Venus, and VenusY67C FRET reference standards. Biophys J. 2006, 91, L99–L101. [Google Scholar] [CrossRef]
- Paez-Segala, M.G.; Sun, M.G.; Shtengel, G.; Viswanathan, S.; Baird, M.A.; Macklin, J.J.; Patel, R.; Allen, J.R.; Howe, E.S.; Piszczek, G.; et al. Fixation-resistant photoactivatable fluorescent proteins for CLEM. Nat. Methods 2015, 12, 215–218. [Google Scholar] [CrossRef]
- Dimalanta, E.T.; Lim, A.; Runnheim, R.; Lamers, C.; Churas, C.; Forrest, D.K.; de Pablo, J.J.; Graham, M.D.; Coppersmith, S.N.; Goldstein, S.; et al. A microfluidic system for large DNA molecule arrays. Anal. Chem. 2004, 76, 5293–5301. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorescent Protein | λex | λem | Brightness (εϕ) | Oligomerization |
|---|---|---|---|---|
| AausFP1 | 504 | 510 | 164.9 | Dimer |
| RRvT | 556 | 583 | 117.9 | Tandem dimer |
| tdTomato | 554 | 581 | 95.2 | Tandem dimer |
| mNeonGreen | 506 | 517 | 92.8 | Monomer |
| mClover3 | 506 | 518 | 84.8 | Monomer |
| YPet | 517 | 530 | 80.1 | Weak dimer |
| mScarlet | 569 | 595 | 70.0 | Monomer |
| mVenus | 515 | 527 | 66.6 | Monomer |
| mEos4b | 505 | 516 | 65.7 | Monomer |
| mKO2 | 551 | 565 | 39.56 | Monomer |
| Emerald | 487 | 509 | 39.1 | Monomer |
| mOrange2 | 549 | 565 | 34.8 | Monomer |
| mTurquoise2 | 434 | 474 | 27.9 | Monomer |
| mStrawberry | 549 | 565 | 26.1 | Monomer |
| eGFP | 488 | 507 | 33.5 | Weak dimer |
| avGFP | 395 | 509 | 19.8 | Dimer |
| mCherry | 574 | 596 | 15.8 | Monomer |
| Set | Excitation Filter | Mirror | Emission Filter |
|---|---|---|---|
| 1 | BrightLine Fluorescence filter 635/18 | FF652-DI01 | BrightLine Fluorescence filter 680/42 |
| 2 | RPB550-580 235297 | XF2086 580DRLP | BrightLine Fluorescence filter 641/75 |
| 3 | BrightLine Fluorescence filter 578/21 | FF596-DI01 | BrightLine Fluorescence filter 641/75 |
| 4 | BrightLine Fluorescence filter 531/40 | FF562-DI03 | BrightLine Fluorescence filter 593/40 |
| 5 | BrightLine Fluorescence filter 509/22 | FF526-DI01 | BrightLine Fluorescence filter 544/24 |
| 6 | BrightLine Fluorescence filter 472/30 | XF2443 | BrightLine Fluorescence filter 525/45 |
| 7 | BrightLine Fluorescence filter 474/27 | XF2443 | BrightLine Fluorescence filter 520/35 |
| 8 | BrightLine Basic Fluorescence filter 434/17 | MD-453 | BrightLine Basic Fluorescence filter 479/40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.T.; Oh, H.; Seo, M.J.; Lee, D.H.; Shin, J.; Bong, S.; Heo, S.; Hapsari, N.D.; Jo, K. 21 Fluorescent Protein-Based DNA Staining Dyes. Molecules 2022, 27, 5248. https://doi.org/10.3390/molecules27165248
Kim YT, Oh H, Seo MJ, Lee DH, Shin J, Bong S, Heo S, Hapsari ND, Jo K. 21 Fluorescent Protein-Based DNA Staining Dyes. Molecules. 2022; 27(16):5248. https://doi.org/10.3390/molecules27165248
Chicago/Turabian StyleKim, Yurie Tehee, Hyesoo Oh, Myung Jun Seo, Dong Hyeun Lee, Jieun Shin, Serang Bong, Sujeong Heo, Natalia Diyah Hapsari, and Kyubong Jo. 2022. "21 Fluorescent Protein-Based DNA Staining Dyes" Molecules 27, no. 16: 5248. https://doi.org/10.3390/molecules27165248
APA StyleKim, Y. T., Oh, H., Seo, M. J., Lee, D. H., Shin, J., Bong, S., Heo, S., Hapsari, N. D., & Jo, K. (2022). 21 Fluorescent Protein-Based DNA Staining Dyes. Molecules, 27(16), 5248. https://doi.org/10.3390/molecules27165248