
Structure and Computational Studies of New Sulfonamide Compound: {(4-nitrophenyl)sulfonyl}tryptophan
 , , , , , , , , ,
, , , , , , , , ,
Abstract
:1. Introduction
2. Results
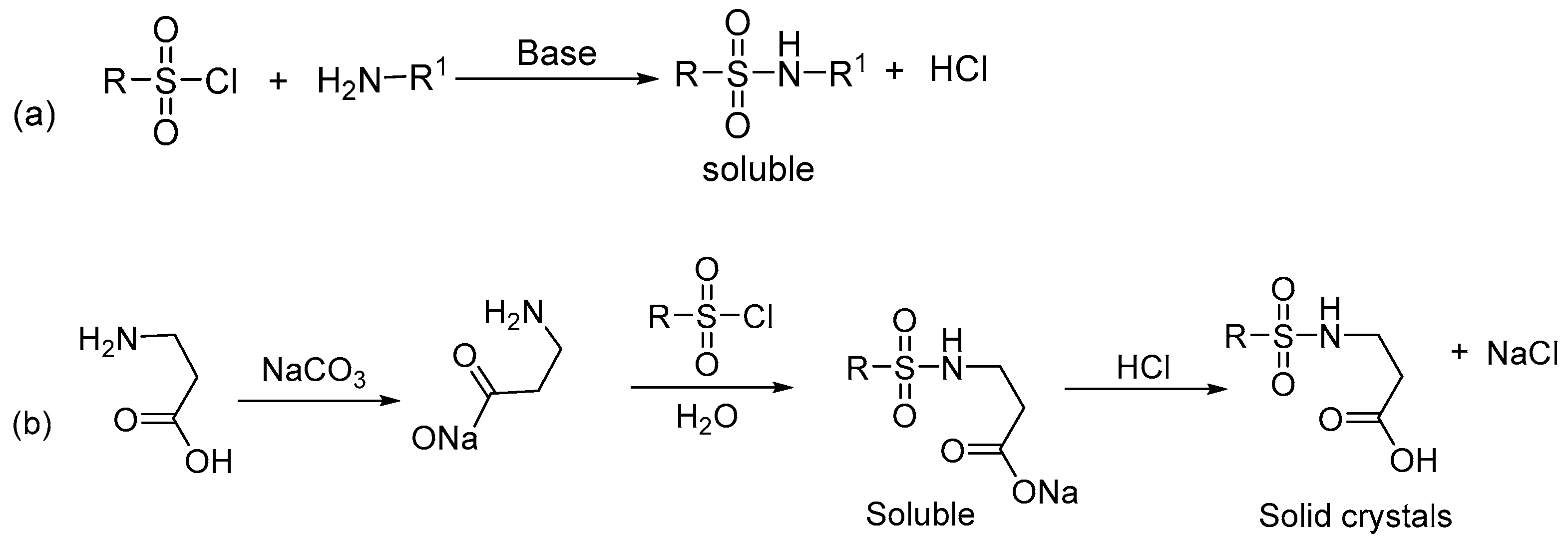
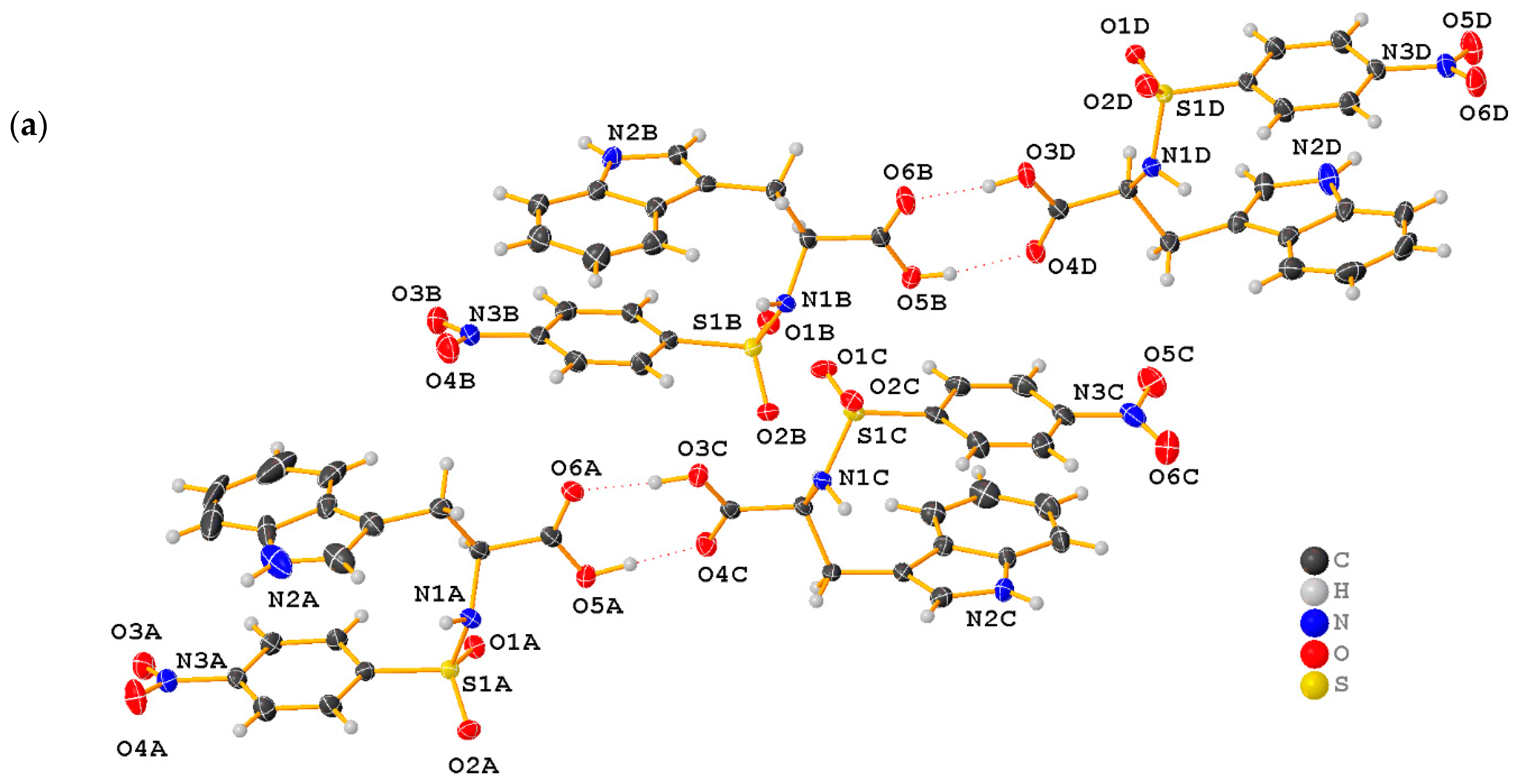
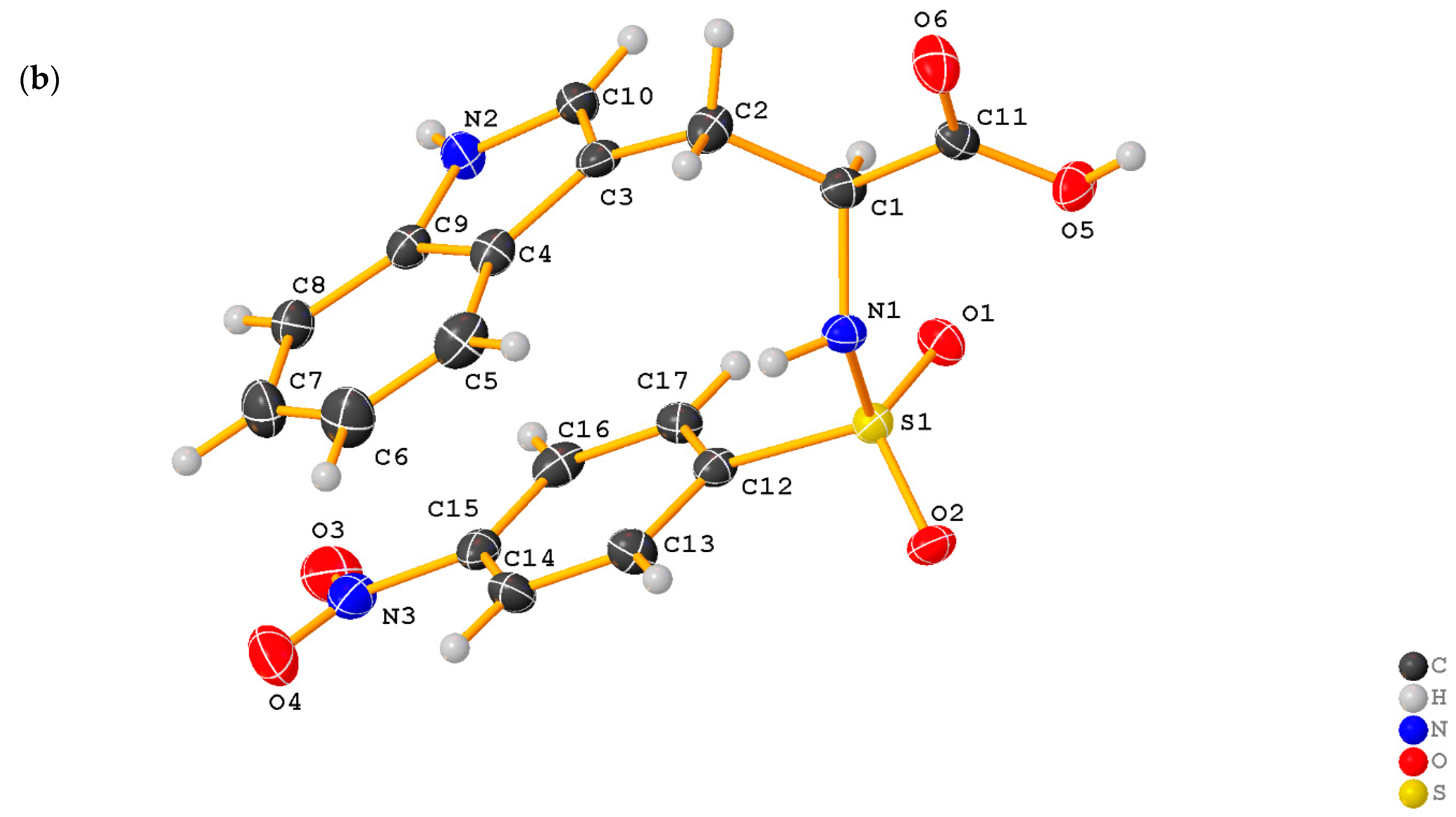
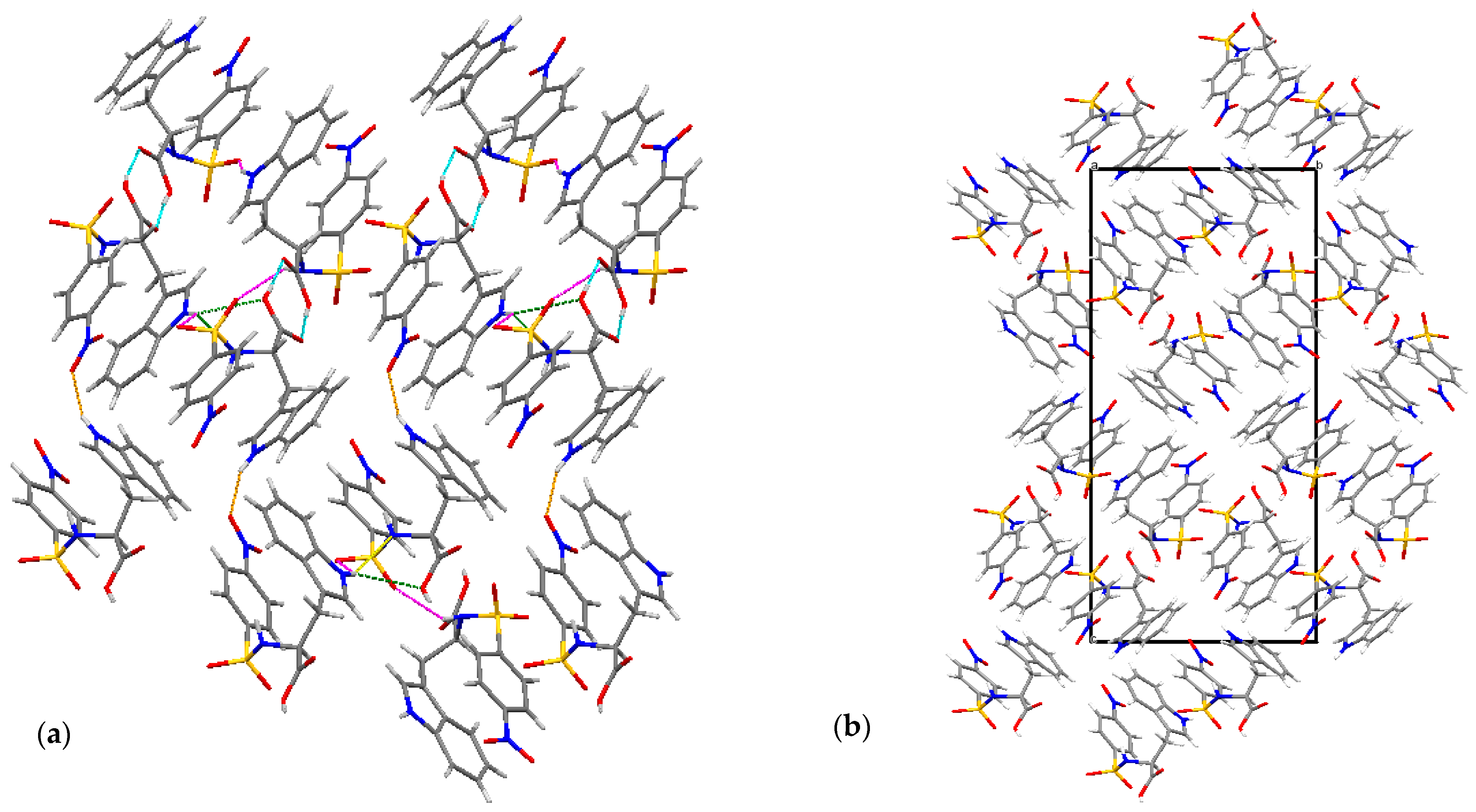
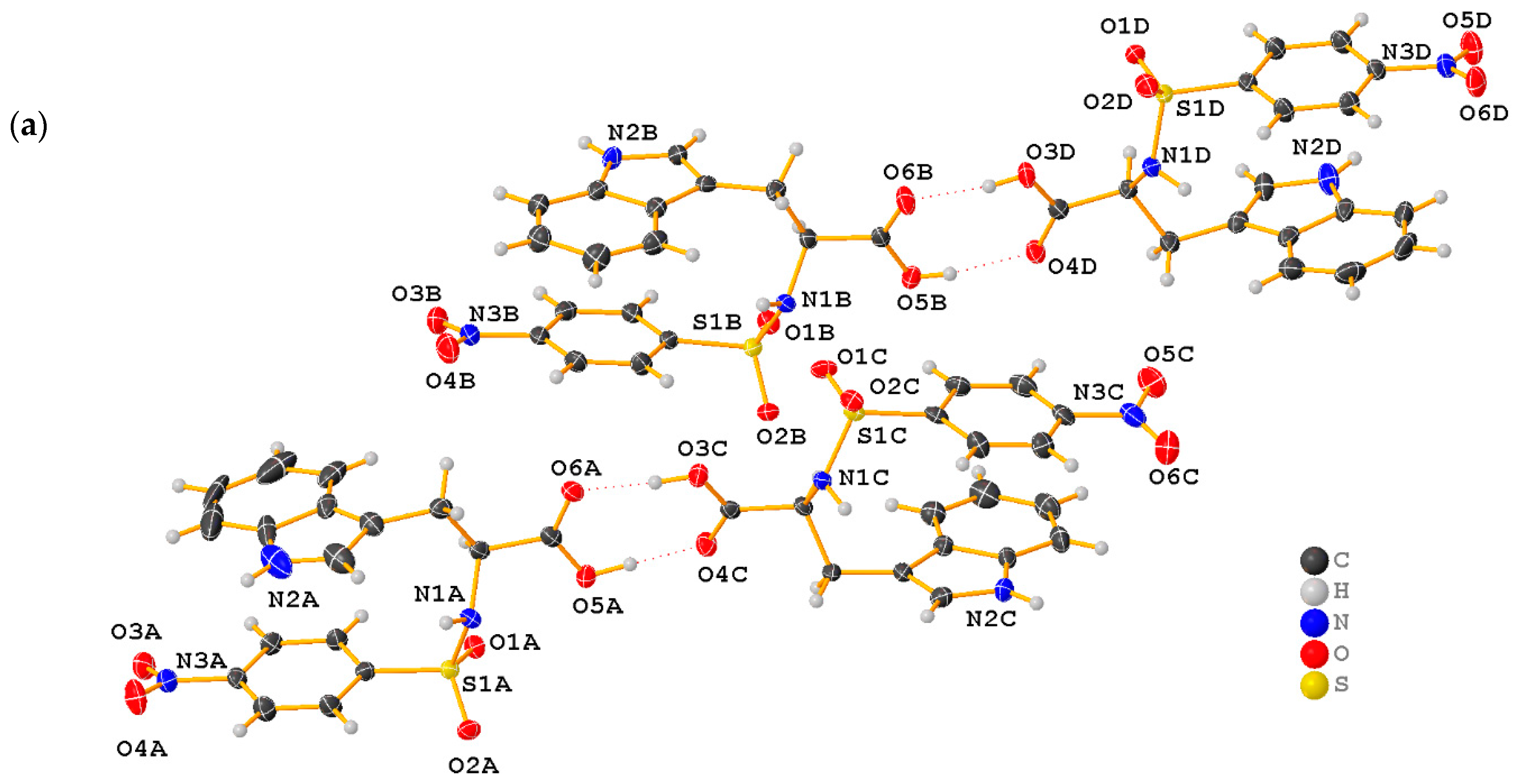
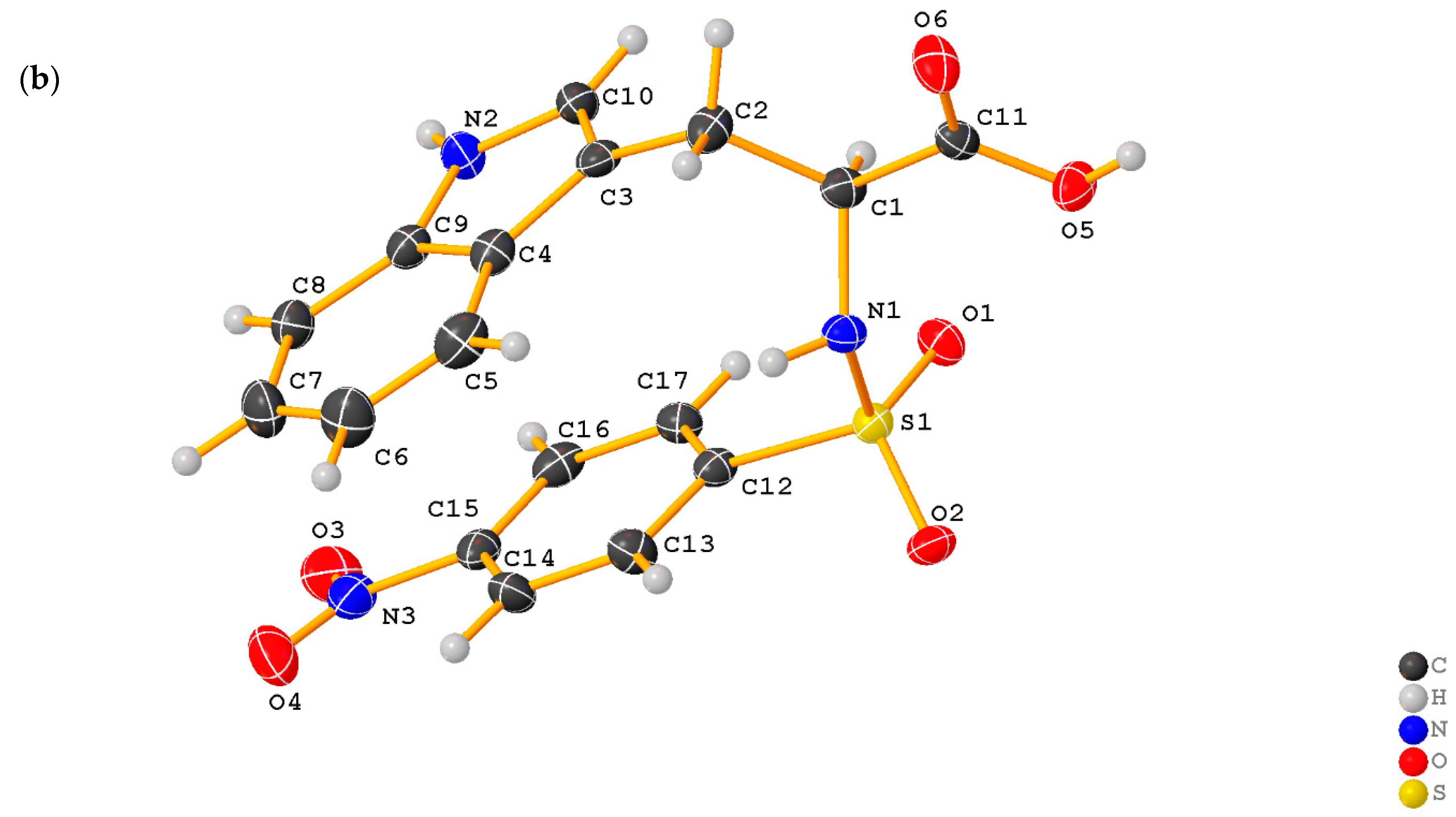
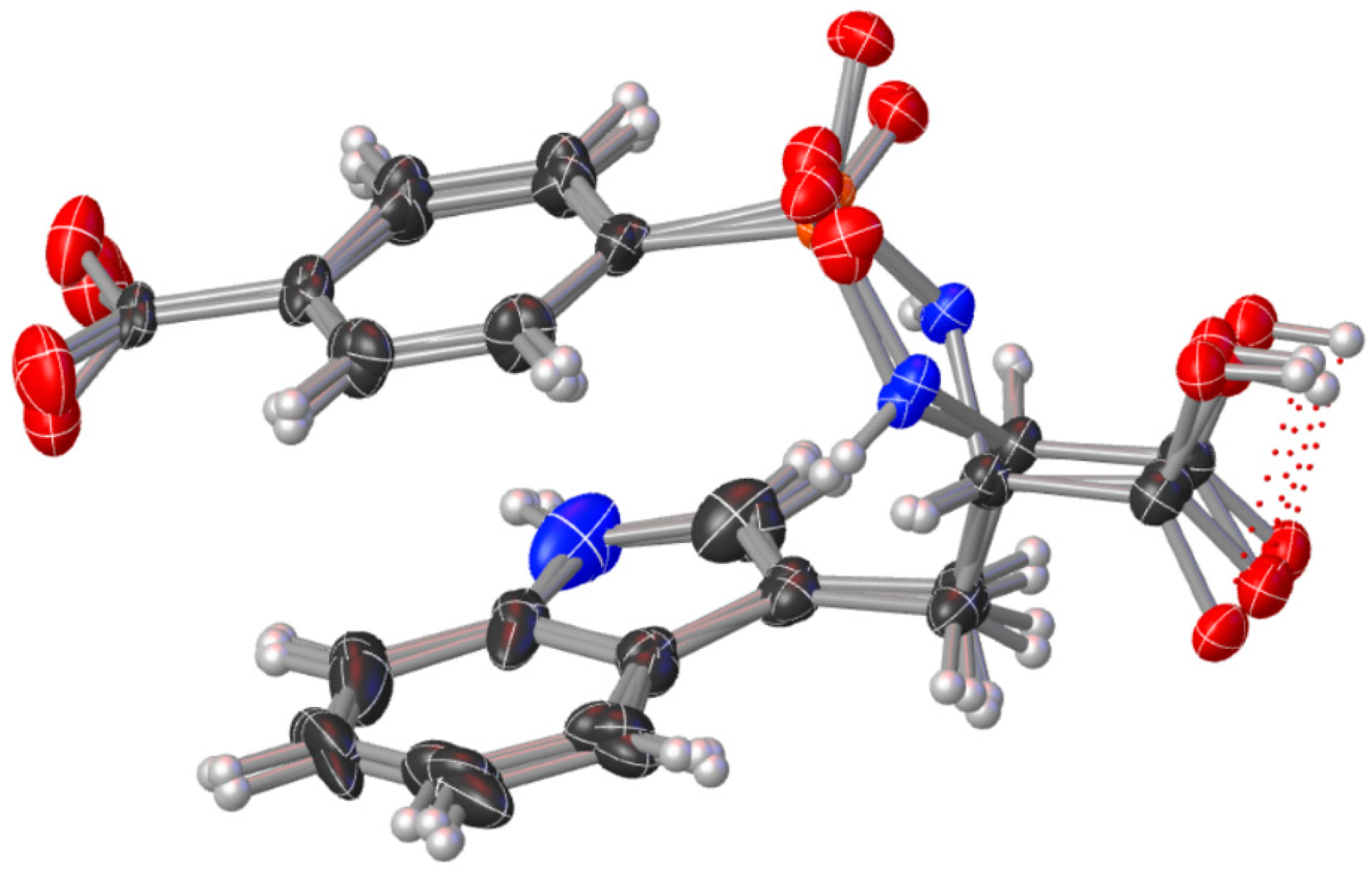
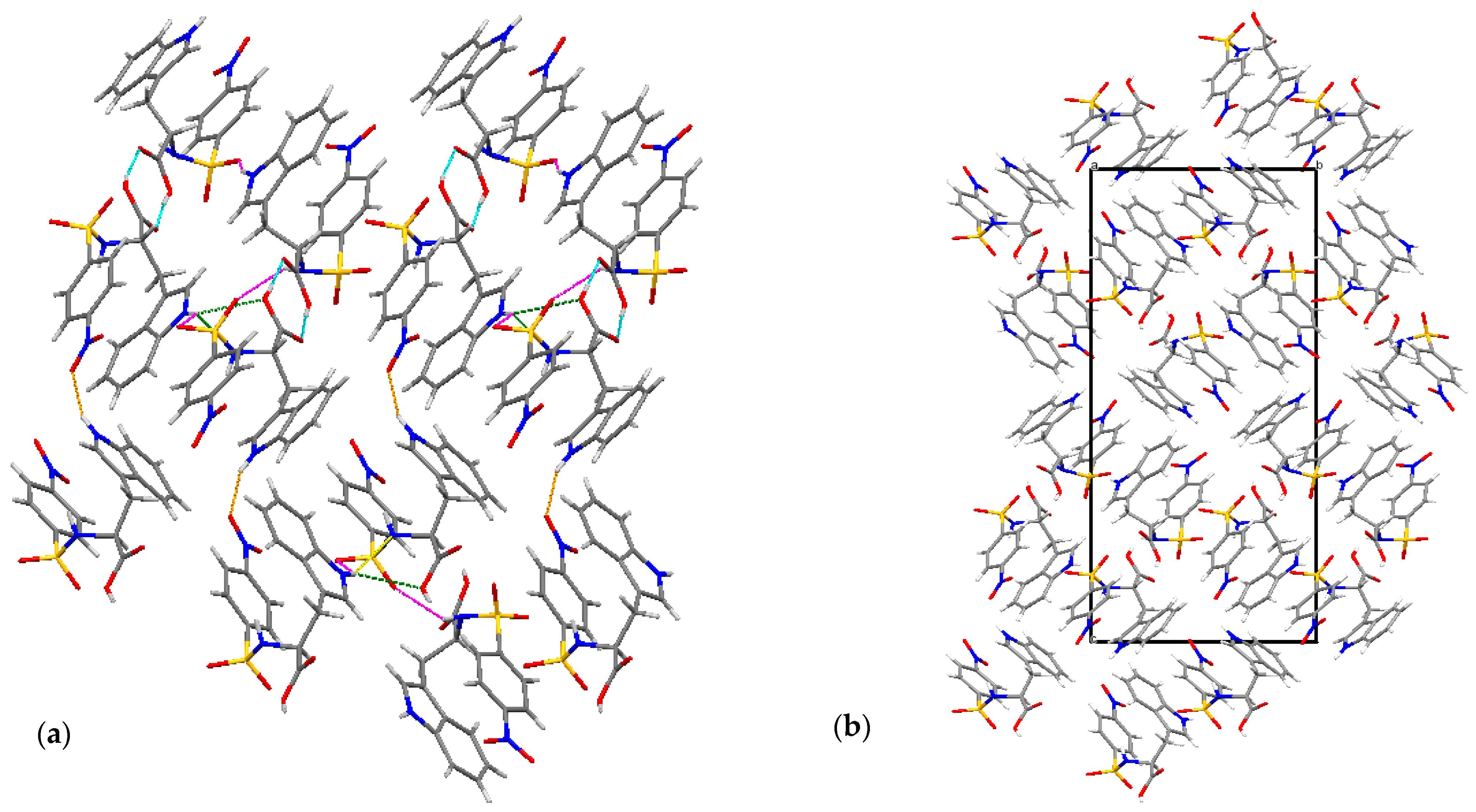
2.1. X-ray Crystallographic Structure Analysis
2.2. Electronic Spectra
2.3. Vibrational Analysis
2.4. 13C and 1H NMR Spectral Analysis
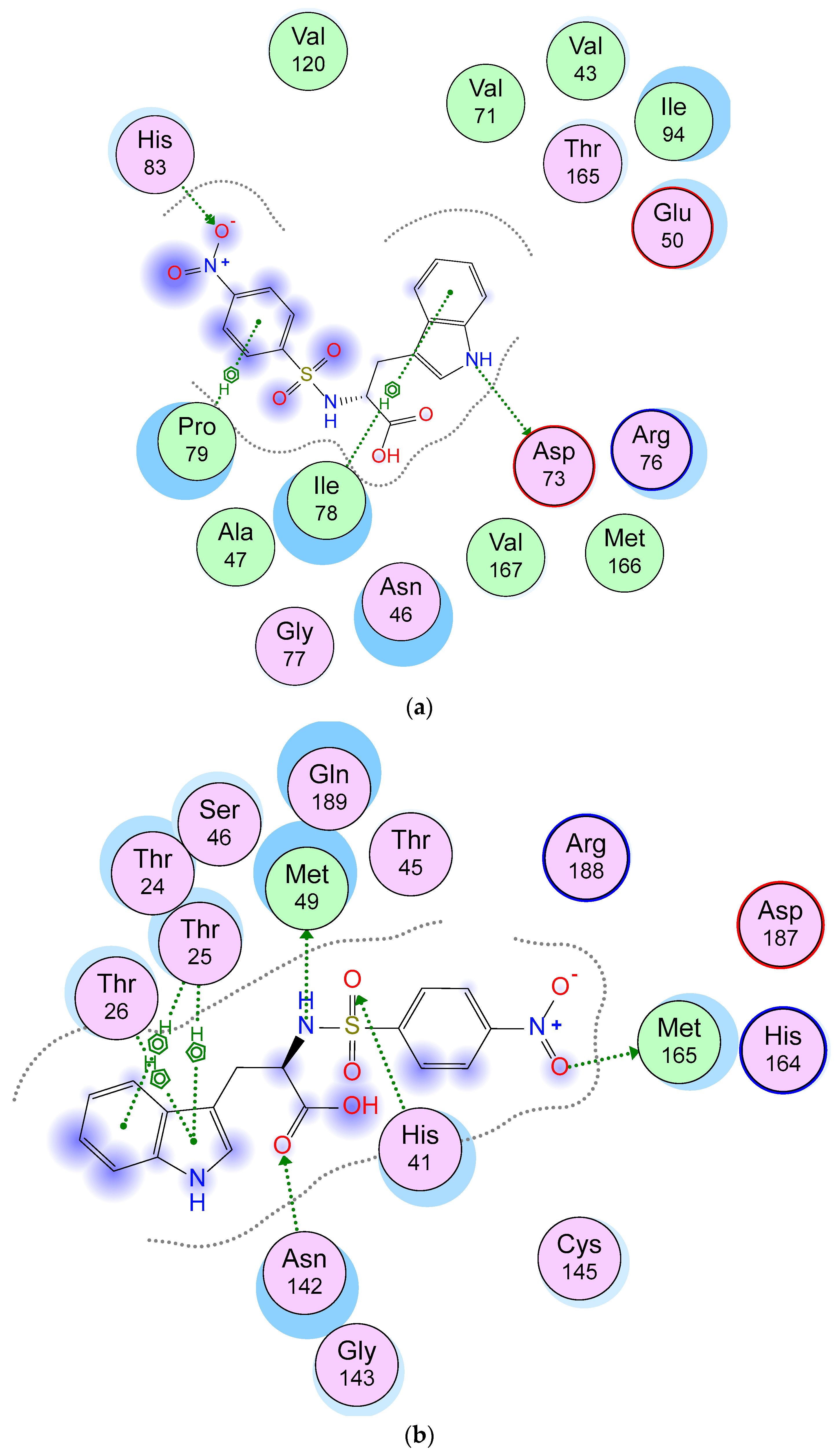
2.5. Molecular Docking Studies
3. Materials and Methods
3.1. Materials and Techniques
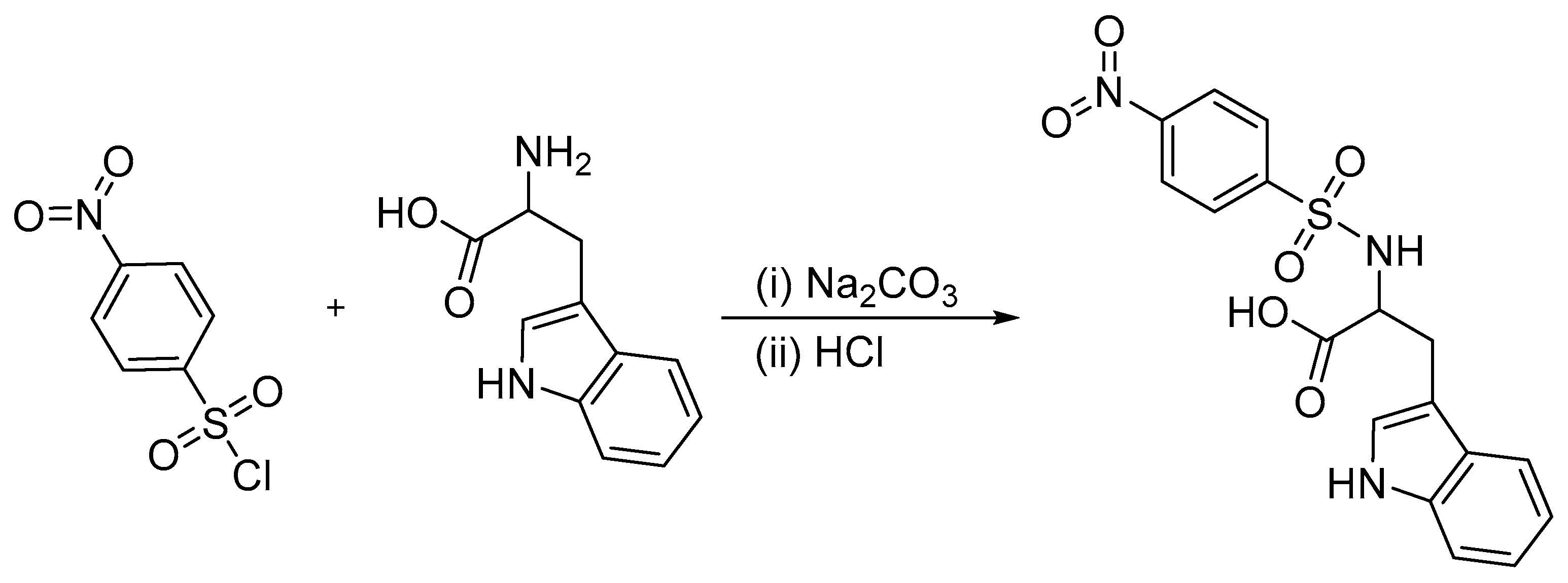
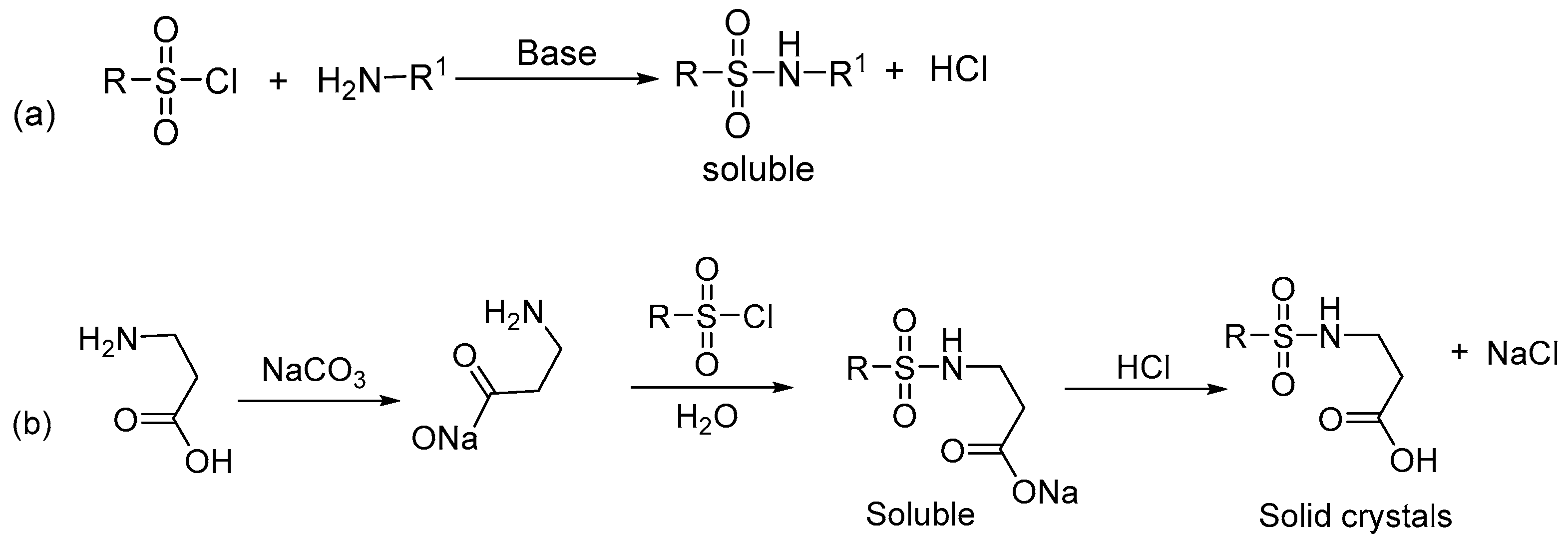
3.2. Synthesis of ((4-nitrophenyl)sulfonyl)tryptophan (DNSPA)
3.3. X-ray Determination of DNSPA
3.4. Computational Method
3.5. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Behmadi, H.; Saadati, S.M.; Roshani, M.; Ghaemy, M. Synthesis of new disulphonamides from different substituted diamino pyridines. Eclética Química 2009, 34, 27–31. [Google Scholar] [CrossRef]
- Hopper, D.W.; Vera, M.D.; Howa, D.; Sabatini, J.; Xiang, J.S.; Ipek, M.; Thomason, J.; Hub, Y.; Feyfant, E.; Wang, Q.; et al. Synthesis and biological evaluation of ((4-keto)-phenoxy)methyl biphenyl-4-sulphonamides: A class of potent aggrecanase-1 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2487–2491. [Google Scholar] [CrossRef] [PubMed]
- Cheong, M.S.; Seo, K.H.; Chohra, H.; Yoon, Y.E.; Choe, H.; Kantharaj, V.; Lee, Y.B. Influence of sulfonamide contamination derived from veterinary antibiotics on plant growth and development. Antibiotics 2020, 9, 456. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.N.M.; Yahaya, N.; Zain, N.N.M.; Lim, V.; Kamaruzaman, S.; Saad, B.; Nishiyama, N.; Yoshida, N.; Hirota, Y. Thiolfunctionalized magnetic carbon nanotubes for magnetic microsolid phase extraction of sulfonamide antibiotics from milks and commercial chicken meat products. Food Chem. 2019, 276, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Protease inhibitors of the sulfonamide type: Anticancer, anti-inflammatory, and antiviral agents. Med. Res. Rev. 2003, 5, 535–558. [Google Scholar] [CrossRef]
- Wright, S.W.; Hallstrom, K.N. A convenient preparation of heteroaryl sulfonamides and sulfonyl fluorides from heteroaryl thiols. J. Org. Chem. 2006, 71, 1080–1084. [Google Scholar] [CrossRef]
- Eze, F.U.; Onyeyilim, E.; Ugwu, D.I.; Ezeokonkwo, M.A.; Udaya, O.P.; Uzoewulu, C.P.; Eze, C.C.; Okonkwo, V.I. Synthesis, Characterization and Anti-Inflammatory Activities of N-Aryl Substituted Benzenesulphonamide Derivatives. Can. J. Pure Appl. Sci. 2020, 14, 5117–5123. [Google Scholar]
- Caddick, S.; Wilden, J.D.; Judd, D.B. Direct synthesis of sulfonamides and activated sulfonate esters from sulfonic acids. J. Am. Chem. Soc. 2004, 126, 1024–1025. [Google Scholar] [CrossRef]
- Frost, C.G.; Hartley, J.P.; Griffin, D. Catalytic arylation of sulfamoyl chlorides: A practical synthesis of sulfonamides. Synlett 2002, 11, 1928–1930. [Google Scholar] [CrossRef]
- Harmata, M.; Zheng, P.; Huang, C.; Gomes, M.G.; Jing, W.; Ranyanil, K.; Balan, G.; Calkins, N.L. Expedient synthesis of sulfinamides from sulfonyl chlorides. J. Org. Chem. 2007, 72, 683–685. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, R.; Srinivas, B.; Kumar, V.P.; Narender, M.; Rao, K.R. β-Cyclodextrin-catalyzed monosulfonylation of amines and amino acids in water. Adv. Synth. Catal. 2007, 349, 1873–1876. [Google Scholar] [CrossRef]
- Rad, M.N.S.; Khalafi-Nezhad, A.; Asrari, Z.; Behrouz, S.; Amini, Z.; Behrouz, M. One-pot synthesis of sulfonamides from primary and secondary amine derived sulfonate salts using cyanuric chloride. Synthesis 2009, 30, 3983–3988. [Google Scholar] [CrossRef]
- Deedar, A.; Sayyeda, T.A.; Zainab, S.; Muhammad, M.N.; Mariya, R.; Tayyaba, A.S.; Shafia, I.; Muhammad, R.S.; Abdul, H.; Jamshed, I. Utilization of transition metal fluoride-based solid support catalysts for the synthesis of sulfonamides: Carbonic anhydrase inhibitory activity and in silico study. RSC Adv. 2022, 12, 3165. [Google Scholar]
- Huang, X.; Wang, J.; Ni, Z.; Wang, S.; Pan, Y. Copper-mediated S–N formation via an oxygen-activated radical process: A new synthesis method for sulphonamides. Chem. Commun. 2014, 50, 4582. [Google Scholar] [CrossRef]
- Kumar, S.; Parumala, R.; Peddinti, R.K. Metal-free synthesis of sulfonamides via iodine-catalysed oxidative coupling of sulfonyl hydrazides and amines. Tetrahedron Lett. 2016, 57, 1232–1235. [Google Scholar]
- Jafarpour, M.; Rezaeifard, A.; Golshani, T. A green catalyst-free method for the synthesis of sulfonamides and sulfonylazides. Phosphorus Sulfur Silicon 2011, 186, 140–148. [Google Scholar] [CrossRef]
- Vicente, D.A.; Galdino, D.; Navarro, M.; Menezes, P.H. Electrochemical synthesis of sulfonamides in graphite powder macro electrode. Green Chem. 2020, 22, 5262–5266. [Google Scholar] [CrossRef]
- Jiang, J.Y.; Wang, Q.Q.; Liang, S.; Hu, L.M.; Little, R.D.; Zeng, C.C. Electrochemical oxidative amination of sodium sulfinates: Synthesis of sulfonamides mediated by nh4i as a redox catalyst. J. Org. Chem. 2016, 81, 4713–4719. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Y.; Yuan, G. Electrosynthesis of arylsulfonamides from amines and sodium sulfinates using h2o-nai as the electrolyte solution at room temperature. Chin. J. Chem. 2016, 34, 1277–1282. [Google Scholar] [CrossRef]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palmad, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Gioiello, A.; Rosatelli, E.; Teofrasti, M.; Filipponi, P.; Pellicciari, R. Building a sulfonamide library by eco-friendly flow synthesis. ACS Comb. Sci. 2013, 15, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Shovan, M.; Suniti, M. Synthesis of sulfonamide and their synthetic and therapeutic applications: Recent advances. Tetrahedron 2020, 76, 131662. [Google Scholar]
- Sonntag, N.O.V. The reactions of aliphatic acid chlorides. Chem. Rev. 1953, 52, 237–476. [Google Scholar] [CrossRef]
- Medebielle, M.; Onomura, O.; Keirouz, R.; Okada, E.; Yano, H.; Terauchi, T. Indium-mediated reformatsky-type reaction of β-aminovinylchlorodifluoromethyl ketones with heteroaryl. Synthesis 2002, 17, 2601–2608. [Google Scholar] [CrossRef]
- Wydysh, E.A.; Medghalchi, S.M.; Vadlamudi, A.; Townsend, C.A. Design and synthesis of small molecule glycerol 3-phosphate acyltransferase inhibitors. J. Med. Chem. 2009, 52, 3317–3327. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, N.; Khan, I.U.; Yar, M.; Afzal, S.; Simpson, J.N. N-Bis(2-hydroxyethyl)-4-methylbenzenesulfonamide. Acta Cryst. 2012, E68, o2019. [Google Scholar] [CrossRef] [Green Version]
- Olayinka, O.A.; Oluwole, B.F.; Feipeng, W.; Johnbull, O.E.; Zheng, S. Room temperature synthesis and antibacterial activity of new sulfonamides containing n,n-diethyl-substituted amidomoieties. Int. J. Med. Chem. 2012, 2012, 367815. [Google Scholar]
- Galal, A.; Mahmoud, K. Antiproliferative Evaluation and molecular docking studies of some sulfonyl-α-l-amino acid derivatives coupled with anisamide scaffold. Egypt. J. Chem. 2021, 64, 3–4. [Google Scholar] [CrossRef]
- Taheri, E.; Mirjafary, Z.; Saeidian, H. Highly efficient regioselective synthesis, spectroscopic characterizations and DFT calculations of novel hydroxymethylated 1,4-disubstituted-1,2,3-triazole-based sulfonamides. J. Mol. Struct. 2018, 1157, 418–424. [Google Scholar] [CrossRef]
- Bonyad, S.R.; Mirjafary, Z.; Saeidian, H.; Rouhani, M. Efficient synthesis, spectroscopic characterization and DFT based studies of novel 1-amide 4-sulfonamide-1,2,3-triazole derivatives. J. Mol. Struct. 2019, 1197, 164–170. [Google Scholar] [CrossRef]
- Alizadeh, M.; Mirjafary, Z.; Saeidian, H. Straightforward synthesis, spectroscopic characterizations and comprehensive DFT calculations of novel 1-ester 4-sulfonamide-1,2,3-triazole scaffolds. J. Mol. Struct. 2020, 1203, 127405. [Google Scholar] [CrossRef]
- Verma, S.K.; Verma, R.; Xue, F.; Thakur, P.K.; Girish, Y.R.; Rakesh, K.P. Antibacterial activities of sulfonyl or sulfonamide containing heterocyclic derivatives and its structure-activity relationships (SAR) studies: A critical review. Bioorg. Chem. 2020, 105, 104400. [Google Scholar] [CrossRef] [PubMed]
- Ul Hassan, A.; Sumrra, S.H.; Imran, M.; Chohan, Z.H. New 3d multifunctional metal chelates of sulfonamide: Spectral, vibrational, molecular modeling, DFT, medicinal and in silico studies. J. Mol. Struct. 2022, 1254, 132305. [Google Scholar] [CrossRef]
- Okafor, S.N.; Angsantikul, P.; Ahmed, H. Discovery of Novel HIV Protease Inhibitors Using Modern Computational Techniques. Int. J. Mol. Sci. 2022, 23, 12149. [Google Scholar] [CrossRef]
- Obasi, L.N.; Oruma, U.S.; Al-Swaidan, I.A.; Ramasami, P.; Ezeorah, C.J.; Ochonogor, A.E. Synthesis, Characterization and Antibacterial Studies of N-(Benzothiazol-2-yl)-4-chlorobenzenesulphonamide and Its Neodymium(III) and Thallium(III) Complexes. Molecules 2017, 22, 153. [Google Scholar] [CrossRef]
- Oruma, U.S.; Ukoha, P.O.; Uzoewulu, C.P.; Ndefo, J.C.; Ugwuoke, S.C.; Ukwueze, N.N.; Eze, T.E.; Ekowo, L.C.; Eze, F.U.; Chinaegbomkpa, U.V.; et al. Synthesis, Biological and In Silico Studies of a Tripodal Schiff Base Derived from 2,4,6-Triamino-1,3,5-triazine and Its Trinuclear Dy(III), Er(III), and Gd(III) Salen Capped Complexes. Molecules 2021, 26, 4379. [Google Scholar] [CrossRef]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. Editorial: In silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612. [Google Scholar] [CrossRef]
- Dimasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inf. 2020, 39, e2000028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhu, H.; Niu, G.; Shi, E.; Chen, J.; Sun, B.; Chen, W.; Zhou, H.; Yang, C. Synthesis, modification and docking studies of 5-sulfonyl isatin derivatives as SARS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2014, 22, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.A.; Athar, F. Anti-HIV and Anti-HCV drugs are the putative inhibitors of RNA-dependent-RNA polymerase activity of NSP12 of the SARS CoV-2 (COVID-19). Pharm. Pharmacol. Int. J. 2020, 8, 163–172. [Google Scholar] [CrossRef]
- Ugwu, D.I.; Okoro, U.C.; Ukoha, P.O.; Okafor, S.; Ibezim, A.; Kumar, N.M. Synthesis, characterization, molecular docking and in vitro antimalarial properties of new carboxamides bearing sulphonamide. Eur. J. Med. Chem. 2017, 135, 349–369. [Google Scholar] [CrossRef]
- Ugwu, D.I.; Okoro, U.C.; Mishra, N.K. Synthesis, characterization and in vitro antitrypanosomal activities of new carboxamides bearing quinoline moiety. PLoS ONE 2018, 13, e0191234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugwu, D.I.; Eze, F.U.; Ogboo, B.C.; Okoro, V.N.; Ugwu, M.C.; Okafor, S.N.; Ayogu, J.I.; Attah, S.I. Synthesis of multi-target benzene-sulphonamide derivatives for the treatment of trypanosomiasis. Med. Chem. 2019, 9, 83–92. [Google Scholar]
- Ugwu, D.I.; Okoro, U.C.; Ukoha, P.O.; Gupta, A.; Okafor, S.N. Novel anti-inflammatory and analgesic agents: Synthesis, molecular docking and in vivo studies. J. Enzym. Inhib. Med. Chem. 2018, 33, 405–415. [Google Scholar] [CrossRef]
- Ugwu, D.I.; Okoro, U.C.; Ahmad, H. New carboxamide derivatives bearing benzenesulphonamide as a selective COX-II inhibitor: Design, synthesis and structureactivity relationship. PLoS ONE 2017, 12, e0183807. [Google Scholar] [CrossRef] [Green Version]
- Ugwu, D.I.; Okoro, U.C.; Mishra, N.K. Synthesis, characterization and anthelmintic activity evaluation of pyrimidine derivatives bearing carboxamide and sulphonamide moieties. J. Serb. Chem. Soc. 2018, 83, 401–409. [Google Scholar] [CrossRef]
- Murthy, P.K.; Suneetha, V.; Armakovi, S.; Armakovi, S.J.; Suchetan, P.A.; Giri, L.; Rao, R.S. Synthesis, characterization and computational study of the newly synthetised sulfonamide molecule. J. Mol. Struct. 2018, 1153, 212–229. [Google Scholar] [CrossRef]
- Ozochukwu, I.S.; Okpareke, O.C.; Izuogu, D.C.; Ibezim, A.; Ujam, O.T.; Asegbeloyin, J.N. N’-(Pyridin-3-ylmethylene) benzenesulfonohydrazide: Crystal structure, DFT, Hirshfeld surface and in silico anticancer studies. Eur. J. Chem. 2021, 12, 256–264. [Google Scholar] [CrossRef]
- Bats, J.W.; Coppens, P.; Koctzle, T.F. The experimental charge density in sulfur-containing molecules. A study of the deformation electron density in sulfamic acid at 78 K by X-ray and neutron diffraction. Acta Crystallogr. Sect. B 1977, 33, 37–45. [Google Scholar] [CrossRef]
- Khan, I.U.; Mubashar-ur-Rehman, H.; Aziza, S.; Harrison, W.T.A. 3-(1H-Indol-3-yl)-2-(2-nitro-benzene-sulfonamido)-propanoic acid including an unknown solvate. Acta Cryst. 2012, E68, o2019. [Google Scholar]
- Hehre, W.J. A Guide to Molecular Mechanisms and Quantum Chemical Calculations Wavefunction; Wavefunction Inc.: Irvine, CA, USA, 2003; pp. 153–181. [Google Scholar]
- Jamroz, M.H. Vibrational energy distribution analysis VEDA 4. Warsaw 2004. Available online: https://www.scienceopen.com/document?vid=c3aca357-33d7-4fa8-8fb3-6711575c8e32 (accessed on 4 May 2022).
- Roeges, N.P.G. Guide to the Interpretation of Infrared Spectra of Organic Structure; John Wiley & Sons Inc.: New York, NY, USA, 1994; pp. 1–6. [Google Scholar]
- Asegbeloyin, J.N.; Izuogu, D.C.; Oyeka, E.E.; Okpareke, O.C.; Ibezim, A. Crystal structure, non-covalent interaction and molecular docking studies of 2-{[2-phenylsulfonyl)hydrazinylidene] methyl} benzoic acid and its dysprosium catalysed cyclised product: 2-(phenyl-sulfonyl) phthalazin-1 (2H)-one. J. Mol. Struct. 2019, 1175, 219–229. [Google Scholar] [CrossRef]
- Bellamy, L.J. The Infrared Spectra of Complex Molecules; Chapman and Hall: London, UK, 1980; Volume 2, pp. 1–5. [Google Scholar]
- Pinlajer, K.; Kleinpeter, E. Carbon13 Chemical Shifts in Structure and Spectrochemical Analysis; VCH Publishers: Deerfield Beach, FL, USA, 1994; pp. 10–20. [Google Scholar]
- Arshad, M.N.; Faidallah, H.M.; Asiri, A.M.; Kosar, N.; Mahmood, T. Structural, spectroscopic and nonlinear optical properties of sulfonamide derivatives; experimental and theoretical study. J. Mol. Struct. 2020, 1202, 127393. [Google Scholar] [CrossRef]
- Panchaud, P.; Bruyère, T.; Blumstein, A.-C.; Bur, D.; Chambovey, A.; Ertel, E.A.; Gude, M.; Hubschwerlen, C.; Jacob, L.; Kimmerlin, T.; et al. Discovery and Optimization of Isoquinoline Ethyl Ureas as Antibacterial Agents. J. Med. Chem. 2017, 60, 3755–3775. [Google Scholar] [CrossRef]
- CrysAlisPro Software System. Rigaku Oxford Diffraction; CrysAlisPro Software System: Yarnton, UK, 2021. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement, and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, B37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I. Quantum Chemical and Statistical Theory of Solutions—A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Fox DJ Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-H∙∙∙A | d(D∙∙∙H) | d(H∙∙∙A) | d(D∙∙∙A) | <(DHA) |
|---|---|---|---|---|
| N1B-H1B∙∙∙O1C | 0.97(4) | 1.89(4) | 2.859(4) | 171(4) |
| O3C-H3C∙∙∙O6A | 0.88(6) | 1.80(6) | 2.678(3) | 173(7) |
| O3D-H3D∙∙∙O6B | 0.82 | 1.77(6) | 2.590(3) | 175(3) |
| O5A-H5A∙∙∙O4C | 0.90(6) | 1.71(6) | 2.610(3) | 173(5) |
| O5B-H5B∙∙∙O4D | 0.82 | 1.86(4) | 2.661(3) | 169(4) |
| Wavelength (nm) | Oscillator Strength (f) | Orbital Transition (%) |
|---|---|---|
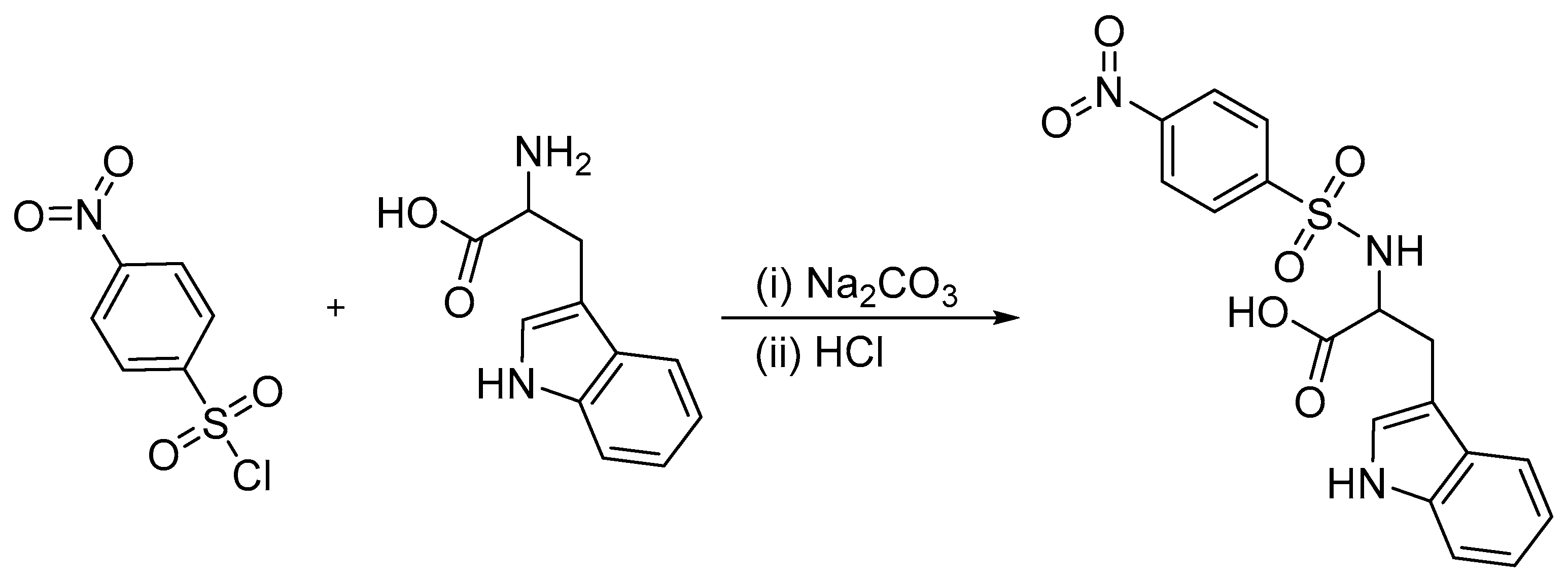
| 186 | 0.275 | HOMO-5 → LUMO + 2 (57%)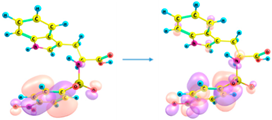 |
| 217 | 0.458 | HOMO → LUMO + 7 (36%)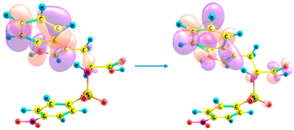 |
| 295 | 0.169 | HOMO-4 → LUMO (50%)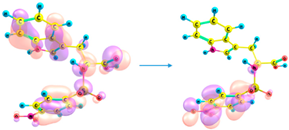 |
HOMO-3 → LUMO (46%)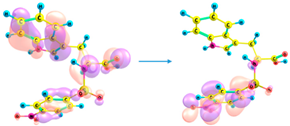 |
| Ligand | Receptor | Interaction | Distance (Å) | E (kcal/mol) |
|---|---|---|---|---|
| N 18 | OD2 ASP 73 | H-donor | 2.81 | −7.2 |
| O 3 | NE1 HIS 83 | H-acceptor | 3.69 | −0.3 |
| 6-ring | CD1 ILE 78 | pi-H | 3.67 | −0.3 |
| 6-ring | CG PRO 79 | pi-H | 4.38 | −0.3 |
| Ligand | Receptor | Interaction | Distance (Å) | E (kcal/mol) |
|---|---|---|---|---|
| O 1 | SD MET 165 | H-donor | 3.55 | 0.1 |
| N 13 | SD MET 49 | H-donor | 4.08 | −2.4 |
| O11 | NE2 HIS 41 | H-acceptor | 3.25 | −0.7 |
| O 26 | CB ASN 142 | H-acceptor | 3.38 | −0.3 |
| 5-ring | CA THR 25 | pi-H | 4.22 | −1.0 |
| 6-ring | CA THR 25 | pi-H | 4.32 | −0.6 |
| 5-ring | CG2 THR 25 | pi-H | 4.42 | −0.6 |
| 5-ring | N THR 26 | pi-H | 4.48 | −0.9 |
| Scheme. | Sulfonamide Analogue | Carbonyl Analogue |
|---|---|---|
| 1 | 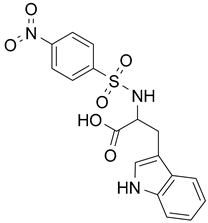 DNSPA |  DNSPA CO- analogue |
| ΔG = −6.35 kcal/mol | ΔG = −6.81 kcal/mol | |
| 2 | 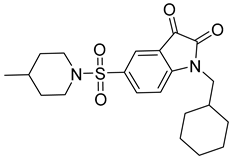 Compound 3 [6] | 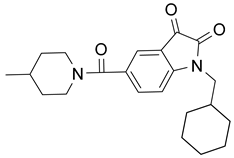 |
| ΔG = −7.07 kcal/mol | ΔG = −7.18 kcal/mol | |
| 3 | 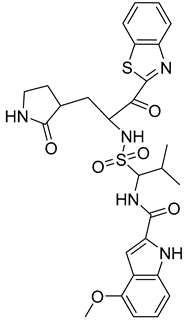 Compound 4 [6] |  |
| ΔG = −7.82 kcal/mol | ΔG = −8.31 kcal/mol | |
| 4 | 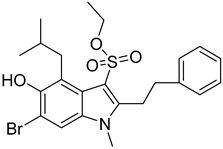 Compound 7 [6] | 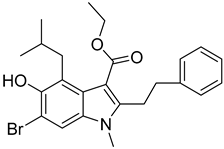 |
| ΔG = −7.49 kcal/mol | ΔG = −7.77 kcal/mol | |
| 5 |  Compound 8 [6] | 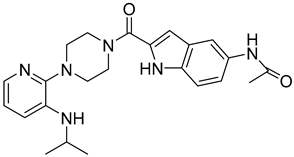 |
| ΔG = −7.43 kcal/mol | ΔG = −7.34 kcal/mol | |
| 6 | 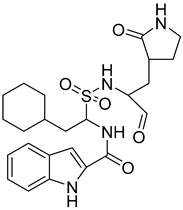 Compound 9 [6] |  |
| ΔG = −8.22 kcal/mol | ΔG = −8.87 kcal/mol |
| Identification Code | DNSPA |
|---|---|
| Empirical formula | C17H15N3O6S |
| Formula weight | 389.38 |
| Temperature/K | 110.2(5) |
| Crystal system | monoclinic |
| Space group | P21 |
| a/Å | 9.61560(10) |
| b/Å | 11.82720(10) |
| c/Å | 29.8731(3) |
| α/° | 90 |
| β/° | 94.9100(10) |
| γ/° | 90 |
| Volume/Å3 | 3384.87(6) |
| Z | 8 |
| ρcalc/g/cm3 | 1.528 |
| μ/mm−1 | 2.092 |
| F(000) | 1616.0 |
| Crystal size/mm3 | 0.26 × 0.26 × 0.2 |
| Radiation | CuKα (λ = 1.54184) |
| 2Θ range for data collection/° | 5.938 to 149.56 |
| Index ranges | −11 ≤ h ≤ 10, −14 ≤ k ≤ 13, −37 ≤ l ≤ 36 |
| Reflections collected | 27,634 |
| Independent reflections | 12167 [Rint = 0.0409, Rsigma = 0.0479] |
| Data/restraints/parameters | 12155/28/1007 |
| Goodness-of-fit on F2 | 1.045 |
| Final R indexes [I >= 2σ (I)] | R1 = 0.0341, wR2 = 0.0865 |
| Final R indexes [all data] | R1 = 0.0359, wR2 = 0.0880 |
| Largest diff. peak/hole/e Å−3 | 0.41/−0.39 |
| Flack parameter | −0.012(7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eze, F.U.; Ezeorah, C.J.; Ogboo, B.C.; Okpareke, O.C.; Rhyman, L.; Ramasami, P.; Okafor, S.N.; Tania, G.; Atiga, S.; Ejiyi, T.U.; et al. Structure and Computational Studies of New Sulfonamide Compound: {(4-nitrophenyl)sulfonyl}tryptophan. Molecules 2022, 27, 7400. https://doi.org/10.3390/molecules27217400
Eze FU, Ezeorah CJ, Ogboo BC, Okpareke OC, Rhyman L, Ramasami P, Okafor SN, Tania G, Atiga S, Ejiyi TU, et al. Structure and Computational Studies of New Sulfonamide Compound: {(4-nitrophenyl)sulfonyl}tryptophan. Molecules. 2022; 27(21):7400. https://doi.org/10.3390/molecules27217400
Chicago/Turabian StyleEze, Florence Uchenna, Chigozie Julius Ezeorah, Blessing Chinweotito Ogboo, Obinna Chibueze Okpareke, Lydia Rhyman, Ponnadurai Ramasami, Sunday Nwankwo Okafor, Groutso Tania, Simeon Atiga, Thomas Ugochukwu Ejiyi, and et al. 2022. "Structure and Computational Studies of New Sulfonamide Compound: {(4-nitrophenyl)sulfonyl}tryptophan" Molecules 27, no. 21: 7400. https://doi.org/10.3390/molecules27217400
APA StyleEze, F. U., Ezeorah, C. J., Ogboo, B. C., Okpareke, O. C., Rhyman, L., Ramasami, P., Okafor, S. N., Tania, G., Atiga, S., Ejiyi, T. U., Ugwu, M. C., Uzoewulu, C. P., Ayogu, J. I., Ekoh, O. C., & Ugwu, D. I. (2022). Structure and Computational Studies of New Sulfonamide Compound: {(4-nitrophenyl)sulfonyl}tryptophan. Molecules, 27(21), 7400. https://doi.org/10.3390/molecules27217400