

N-Arylation of 3-Formylquinolin-2(1H)-ones Using Copper(II)-Catalyzed Chan–Lam Coupling

 , , and
, , and
Abstract
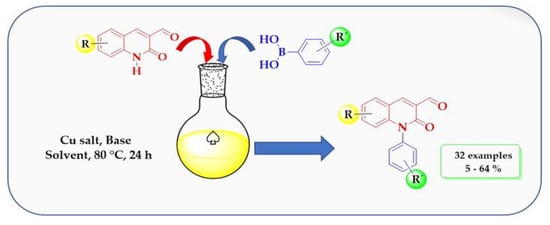
1. Introduction
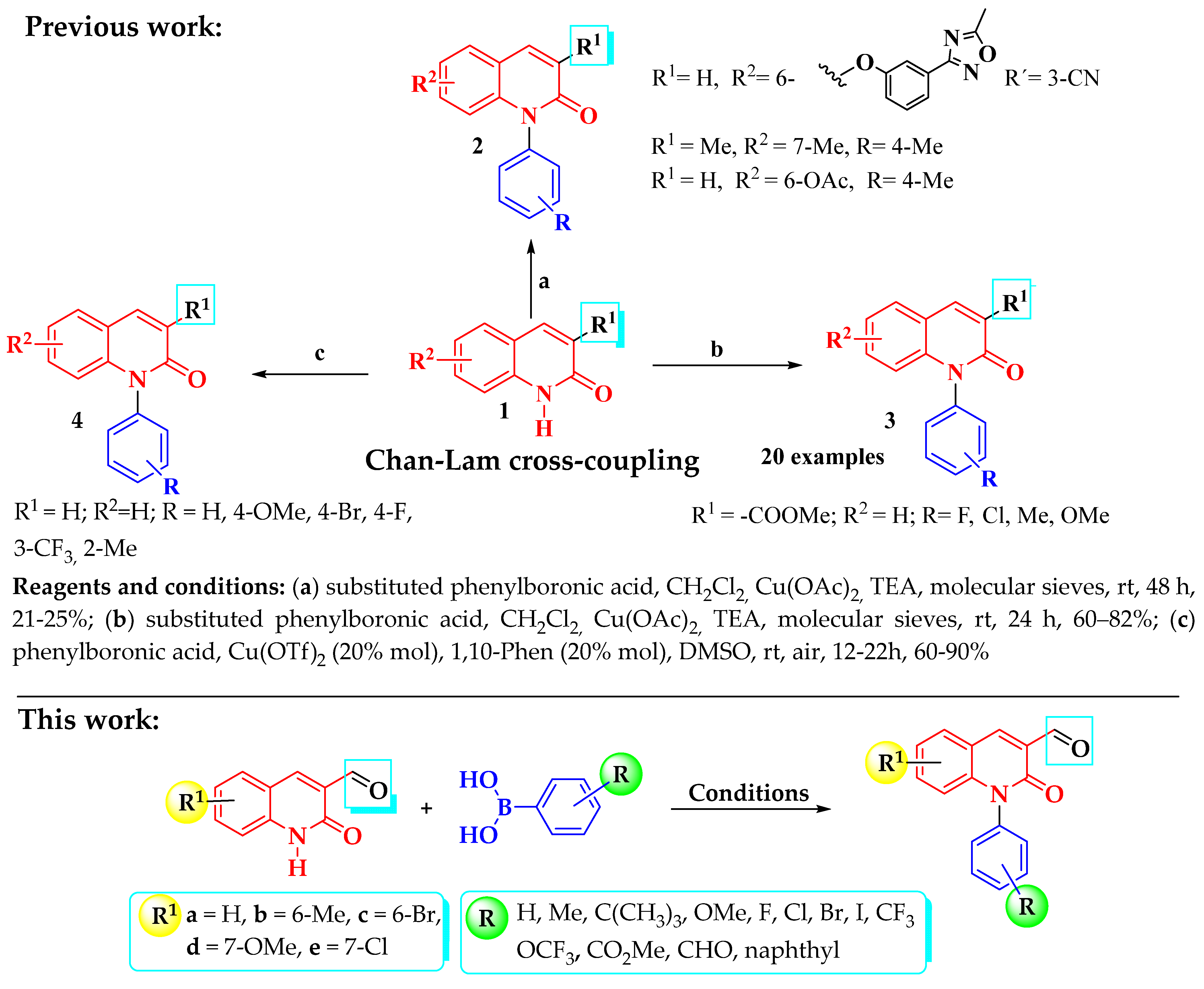
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedures and Characterization Data of Compounds
3.2.1. Synthesis of 2-Chloroquinoline-3-carbaldehydes (6a–e)
Synthesis of 2-Oxo-1,2-dihydroquinoline-3-carbaldehyde (3-formyl-2-quinolones 7a–e)
2-Oxo-1,2-dihydroquinoline-3-carbaldehyde (7a)
6-Methyl-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (7b)
6-Bromo-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (7c)
7-Chloro-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (7d)
7-Methoxy-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (7e)
3.2.2. Synthesis of N-Aryl-3-formylquinolone Derivatives (9a–x, 10a–h)
2-Oxo-1-(p-tolyl)-1,2-dihydroquinoline-3-carbaldehyde (9a)
2-Oxo-1-phenyl-1,2-dihydroquinoline-3-carbaldehyde (9b)
2-Oxo-1-(m-tolyl)-1,2-dihydroquinoline-3-carbaldehyde (9c)
1-(3-(tert-Butyl)phenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9d)
1-(3-Methoxyphenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9e)
1-(3-Fluorophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9f)
1-(3-Chlorophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9g)
1-(3-Bromophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9h)
1-(3-Iodophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9i)
2-Oxo-1-(3-(trifluoromethyl)phenyl)-1,2-dihydroquinoline-3-carbaldehyde (9j)
2-Oxo-1-(3-(trifluoromethoxy)phenyl)-1,2-dihydroquinoline-3-carbaldehyde (9k)
Methyl 3-(3-formyl-2-oxoquinolin-1(2H)-yl)benzoate (9l)
1-(3-Formylphenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9m)
1-(4-(tert-Butyl)phenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9n)
1-(4-Methoxyphenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9o)
1-(4-Fluorophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9p)
1-(4-Chlorophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9q)
1-(4-Bromophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9r)
1-(4-Iodophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9s)
2-Oxo-1-(4-(trifluoromethyl)phenyl)-1,2-dihydroquinoline-3-carbaldehyde (9t)
2-Oxo-1-(4-(trifluoromethoxy)phenyl)-1,2-dihydroquinoline-3-carbaldehyde (9u)
Methyl 4-(3-Formyl-2-oxoquinolin-1(2H)-yl)benzoate (9v)
1-(4-Formylphenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9w)
1-(Naphthalen-2-yl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (9x)
6-Methyl-2-oxo-1-(p-tolyl)-1,2-dihydroquinoline-3-carbaldehyde (10a)
1-(4-Fluorophenyl)-6-methyl-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (10b)
6-Bromo-2-oxo-1-(p-tolyl)-1,2-dihydroquinoline-3-carbaldehyde (10c)
6-Bromo-1-(4-fluorophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (10d)
7-Methoxy-2-oxo-1-(p-tolyl)-1,2-dihydroquinoline-3-carbaldehyde (10e)
1-(4-Fluorophenyl)-7-methoxy-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (10f)
7-Chloro-2-oxo-1-(p-tolyl)-1,2-dihydroquinoline-3-carbaldehyde (10g)
7-Chloro-1-(4-fluorophenyl)-2-oxo-1,2-dihydroquinoline-3-carbaldehyde (10h)
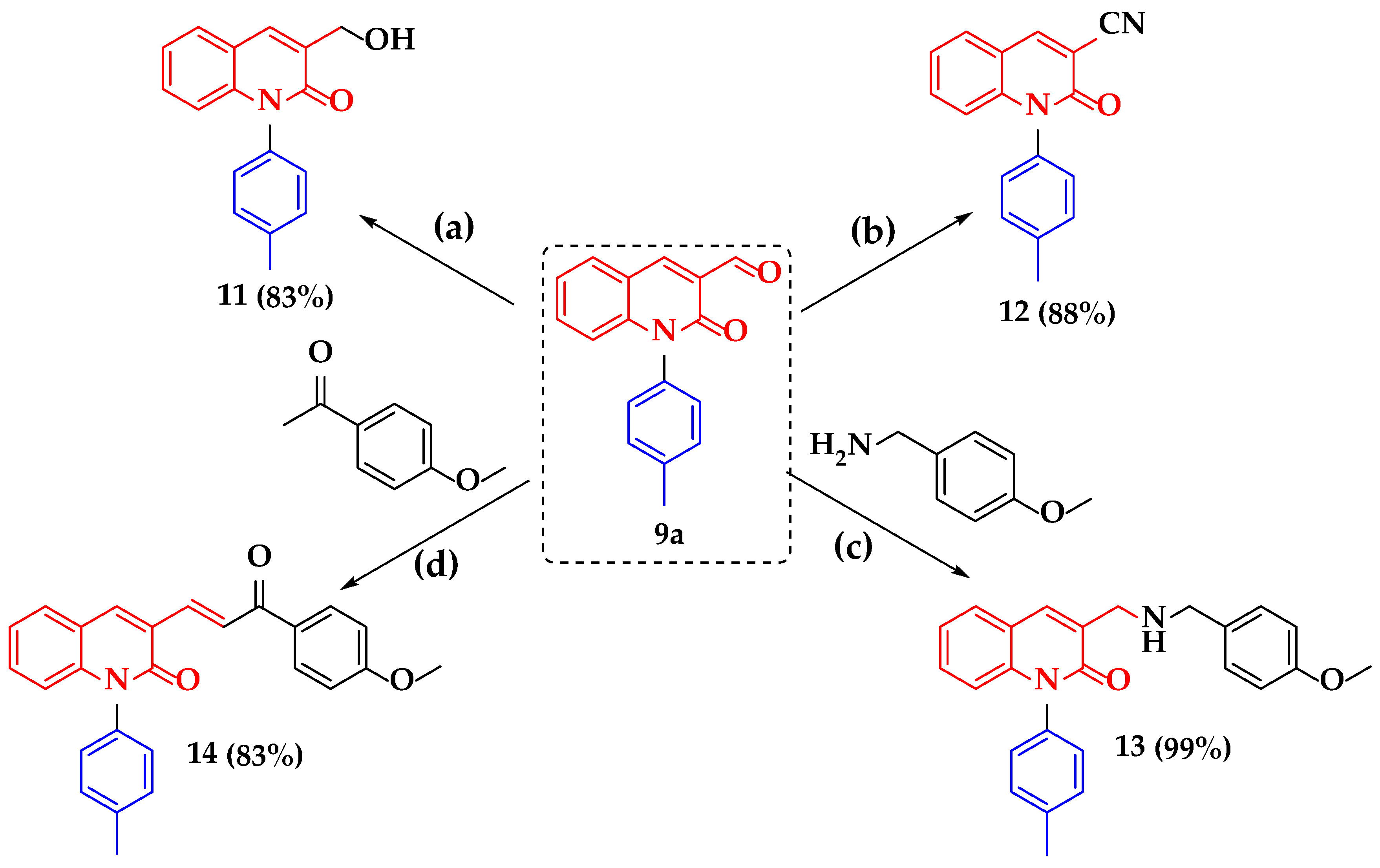
3.2.3. Synthesis of 3-(Hydroxymethyl)-1-(p-tolyl)quinolin-2(1H)-one (11)
3.2.4. Synthesis of 2-Oxo-1-(p-tolyl)-1,2-dihydroquinoline-3-carbonitrile (12)
3.2.5. Synthesis of 3-(((4-Methoxybenzyl)amino)methyl)-1-(p-tolyl)quinolin-2(1H)-one (13)
3.2.6. Synthesis of (E)-3-(3-(4-Methoxyphenyl)-3-oxoprop-1-en-1-yl)-1-(p-tolyl)quinolin-2(1H)-one (14)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shobeiri, N.; Rashedi, M.; Mosaffa, F.; Zarghi, A.; Ghandadi, M.; Ghasemi, A.; Ghodsi, R. Synthesis and Biological Evaluation of Quinoline Analogues of Flavones as Potential Anticancer Agents and Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2016, 114, 14–23. [Google Scholar] [CrossRef]
- Brown, C.E.; McNulty, J.; Bordón, C.; Yolken, R.; Jones-Brando, L. Enol Ethers as Carbonyl Surrogates in a Modification of the Povarov Synthesis of 3-Aryl Quinolines and Their Anti-Toxoplasma Activity. Org. Biomol. Chem. 2016, 14, 5951–5955. [Google Scholar] [CrossRef] [PubMed]
- Kappenberg, Y.G.; Ketzer, A.; Stefanello, F.S.; Salbego, P.R.S.; Acunha, T.V.; Abbadi, B.L.; Bizarro, C.V.; Basso, L.A.; Machado, P.; Martins, M.A.P.; et al. Synthesis and Photophysical, Thermal and Antimycobacterial Properties of Novel 6-Amino-2-Alkyl(Aryl/Heteroaryl)-4-(Trifluoromethyl) Quinolines. New J. Chem. 2019, 43, 12375–12384. [Google Scholar] [CrossRef]
- dos Santos, G.C.; de Andrade Bartolomeu, A.; Ximenes, V.F.; da Silva-Filho, L.C. Facile Synthesis and Photophysical Characterization of New Quinoline Dyes. J. Fluoresc. 2017, 27, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Bonacorso, H.G.; Rodrigues, M.B.; Iglesias, B.A.; da Silveira, C.H.; Feitosa, S.C.; Rosa, W.C.; Martins, M.A.P.; Frizzo, C.P.; Zanatta, N. New 2-(Aryl/Heteroaryl)-6-(Morpholin-4-Yl/Pyrrolidin-1-Yl)-(4-Trifluoromethyl)Quinolines: Synthesis via Buchwald–Hartwig Amination, Photophysics, and Biomolecular Binding Properties. New J. Chem. 2018, 42, 10024–10035. [Google Scholar] [CrossRef]
- Sukpattanacharoen, C.; Kungwan, N. Theoretical Insights of Solvent Effect on Excited-State Proton Transfers of 2-Aryl-3-Hydroxyquinolone. J. Mol. Liq. 2021, 325, 115035. [Google Scholar] [CrossRef]
- Echeverry-Gonzalez, C.A.; Villamizar, M.C.O.; Kouznetsov, V.V. The Remarkable Selectivity of the 2-Arylquinoline-Based Acyl Hydrazones toward Copper Salts: Exploration of Their Catalytic Applications in the Copper Catalysed N -Arylation of Indole Derivatives and C1-Alkynylation of Tetrahydroisoquinolines via the A 3 Reaction. New J. Chem. 2021, 45, 243–250. [Google Scholar] [CrossRef]
- Heravi, M.M.; Kheilkordi, Z.; Zadsirjan, V.; Heydari, M.; Malmir, M. Buchwald-Hartwig Reaction: An Overview. J. Organomet. Chem. 2018, 861, 17–104. [Google Scholar] [CrossRef]
- Hardouin Duparc, V.; Bano, G.L.; Schaper, F. Chan–Evans–Lam Couplings with Copper Iminoarylsulfonate Complexes: Scope and Mechanism. ACS Catal. 2018, 8, 7308–7325. [Google Scholar] [CrossRef]
- Evano, G.; Theunissen, C.; Pradal, A. Impact of Copper-Catalyzed Cross-Coupling Reactions in Natural Product Synthesis: The Emergence of New Retrosynthetic Paradigms. Nat. Prod. Rep. 2013, 30, 1467. [Google Scholar] [CrossRef]
- Doyle, M.G.J.; Lundgren, R.J. Oxidative Cross-Coupling Processes Inspired by the Chan–Lam Reaction. Chem. Commun. 2021, 57, 2724–2731. [Google Scholar] [CrossRef]
- Chen, J.-Q.; Li, J.-H.; Dong, Z.-B. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311–3331. [Google Scholar] [CrossRef]
- Yu, S.; Saenz, J.; Srirangam, J.K. Facile Synthesis of N-Aryl Pyrroles via Cu(II)-Mediated Cross Coupling of Electron Deficient Pyrroles and Arylboronic Acids. J. Org. Chem. 2002, 67, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Bakkestuen, A.K.; Gundersen, L.-L. Regioselective N-9 Arylation of Purines Employing Arylboronic Acids in the Presence of Cu(II). Tetrahedron Lett. 2003, 44, 3359–3362. [Google Scholar] [CrossRef]
- Lasalle, M.; Hoguet, V.; Hennuyer, N.; Leroux, F.; Piveteau, C.; Belloy, L.; Lestavel, S.; Vallez, E.; Dorchies, E.; Duplan, I.; et al. Topical Intestinal Aminoimidazole Agonists of G-Protein-Coupled Bile Acid Receptor 1 Promote Glucagon Like Peptide-1 Secretion and Improve Glucose Tolerance. J. Med. Chem. 2017, 60, 4185–4211. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Velasco, O.A.; Saavedra-Olavarría, J.; Araya-Santelices, D.A.A.; Hermosilla-Ibáñez, P.; Cassels, B.K.; Pérez, E.G. Synthesis of N -Arylcytisine Derivatives Using the Copper-Catalyzed Chan-Lam Coupling. J. Nat. Prod. 2021, 84, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.; Jang, E.; Choi, S.; Hong, S. Visible-Light-Photocatalyzed Synthesis of Phenanthridinones and Quinolinones via Direct Oxidative C–H Amidation. Org. Lett. 2018, 20, 240–243. [Google Scholar] [CrossRef]
- Raghavan, S.; Manogaran, P.; Gadepalli Narasimha, K.K.; Kalpattu Kuppusami, B.; Mariyappan, P.; Gopalakrishnan, A.; Venkatraman, G. Synthesis and Anticancer Activity of Novel Curcumin–Quinolone Hybrids. Bioorg. Med. Chem. Lett. 2015, 25, 3601–3605. [Google Scholar] [CrossRef]
- Charushin, V.N.; Mochulskaya, N.N.; Antipin, F.V.; Kotovskaya, S.K.; Nosova, E.V.; Ezhikova, M.A.; Kodess, M.I.; Kravchenko, M.A. Synthesis and Antimycobacterial Evaluation of New (2-Oxo-2H-Chromen-3-Yl) Substituted Fluoroquinolones. J. Fluor. Chem. 2018, 208, 15–23. [Google Scholar] [CrossRef]
- Xu, M.; Wagerle, T.; Long, J.K.; Lahm, G.P.; Barry, J.D.; Smith, R.M. Insecticidal Quinoline and Isoquinoline Isoxazolines. Bioorg. Med. Chem. Lett. 2014, 24, 4026–4030. [Google Scholar] [CrossRef]
- Kaur, P.; Anuradha; Chandra, A.; Tanwar, T.; Sahu, S.K.; Mittal, A. Emerging Quinoline- and Quinolone-Based Antibiotics in the Light of Epidemics. Chem. Biol. Drug Des. 2022, 100, 765–785. [Google Scholar] [CrossRef] [PubMed]
- Vil’, V.A.; Grishin, S.S.; Baberkina, E.P.; Alekseenko, A.L.; Glinushkin, A.P.; Kovalenko, A.E.; Terent’ev, A.O. Electrochemical Synthesis of Tetrahydroquinolines from Imines and Cyclic Ethers via Oxidation/Aza-Diels-Alder Cycloaddition. Adv. Synth. Catal. 2022, 364, 1098–1108. [Google Scholar] [CrossRef]
- Senerovic, L.; Opsenica, D.; Moric, I.; Aleksic, I.; Spasić, M.; Vasiljevic, B. Quinolines and Quinolones as Antibacterial, Antifungal, Anti-Virulence, Antiviral and Anti-Parasitic Agents. In Advances in Microbiology, Infectious Diseases and Public Health: Volume 14; Springer International Publishing: Cham, Switzerland, 2020; pp. 37–69. ISBN 978-3-030-53647-3. [Google Scholar]
- Ghosh, J.; Swarup, V.; Saxena, A.; Das, S.; Hazra, A.; Paira, P.; Banerjee, S.; Mondal, N.B.; Basu, A. Therapeutic Effect of a Novel Anilidoquinoline Derivative, 2-(2-Methyl-Quinoline-4ylamino)-N-(2-Chlorophenyl)-Acetamide, in Japanese Encephalitis: Correlation with in Vitro Neuroprotection. Int. J. Antimicrob. Agents 2008, 32, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Sureshkumar, B.; Mary, Y.S.; Panicker, C.Y.; Suma, S.; Armaković, S.; Armaković, S.J.; Van Alsenoy, C.; Narayana, B. Quinoline Derivatives as Possible Lead Compounds for Anti-Malarial Drugs: Spectroscopic, DFT and MD Study. Arab. J. Chem. 2020, 13, 632–648. [Google Scholar] [CrossRef]
- Full Article: Synthesis and Evaluation of Antioxidant Activity of New Quinoline-2-Carbaldehyde Hydrazone Derivatives: Bioisosteric Melatonin Analogues. Available online: https://www.tandfonline.com/doi/full/10.3109/14756366.2015.1005012 (accessed on 9 October 2022).
- Abadi, A.H.; Hegazy, G.H.; El-Zaher, A.A. Synthesis of Novel 4-Substituted-7-Trifluoromethylquinoline Derivatives with Nitric Oxide Releasing Properties and Their Evaluation as Analgesic and Anti-Inflammatory Agents. Bioorg. Med. Chem. 2005, 13, 5759–5765. [Google Scholar] [CrossRef]
- Horta, P.; Secrieru, A.; Coninckxa, A.; Cristiano, M.L.S. Quinolones for Applications in Medicinal Chemistry: Synthesis and Structure. Targets Heterocycl. Syst. 2019, 22, 260. [Google Scholar] [CrossRef]
- Shiro, T.; Fukaya, T.; Tobe, M. The Chemistry and Biological Activity of Heterocycle-Fused Quinolinone Derivatives: A Review. Eur. J. Med. Chem. 2015, 97, 397–408. [Google Scholar] [CrossRef]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A Comprehensive Review on the Biological Interest of Quinoline and Its Derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef]
- El-Desoky, E.I.; El-Sayed, M.A.; Abd-ElGhani, G.E. Synthesis and Antimicrobial Evaluation of Some New Fused Quinolones Heterocyclic Compounds. Int. J. Mod. Org. Chem. 2018, 5, 21–35. [Google Scholar]
- Berger, Markus; Rehwinkel, Hartmut; May, Ekkehard; Schäcke, Heike WO2010009814 5-[(3,3,3-Trifluoro-2-Hydroxy-1-Arylpropyl) Amino]-1-Arylquinoline-2-Ones, Leur Procédé de Production et Leur Utilisation Comme Agents Anti-Inflammatoires. Available online: https://patentscope.wipo.int/search/fr/detail.jsf?docId=WO2010009814 (accessed on 8 September 2022).
- Weiss, M.; Boezio, A.; Boezio, C.; Butler, J.R.; Chu-Moyer, M.Y.; Dimauro, E.F.; Dineen, T.; Graceffa, R.; Guzman-Perez, A.; Huang, H.; et al. Composés De Sulfonamides Bicycliques Utilisés En Tant Qu’inhibiteurs Du Canal Sodique. Patent WO/2014/201206, 18 December 2014. [Google Scholar]
- Chen, M.-H.; Fitzgerald, P.; Singh, S.B.; O’Neill, E.A.; Schwartz, C.D.; Thompson, C.M.; O’Keefe, S.J.; Zaller, D.M.; Doherty, J.B. Synthesis and Biological Activity of Quinolinone and Dihydroquinolinone P38 MAP Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 2222–2226. [Google Scholar] [CrossRef]
- Tang, Q.; Zhai, X.; Tu, Y.; Wang, P.; Wang, L.; Wu, C.; Wang, W.; Xie, H.; Gong, P.; Zheng, P. Synthesis and Antiproliferative Activity of 6,7-Disubstituted-4-Phenoxyquinoline Derivatives Bearing the 2-Oxo-4-Chloro-1,2-Dihydroquinoline-3-Carboxamide Moiety. Bioorg. Med. Chem. Lett. 2016, 26, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Tao, K.; Peng, X.; Hu, C.; Lu, Y.; Wang, H. Synthesis of N-Aryl 2-Quinolinones via Intramolecular C(Sp 2 )–H Amidation of Knoevenagel Products. RSC Adv. 2016, 6, 104463–104466. [Google Scholar] [CrossRef]
- Mederski, W.W.K.R.; Lefort, M.; Germann, M.; Kux, D. N-Aryl Heterocycles via Coupling Reactions with Arylboronic Acids. Tetrahedron 1999, 55, 12757–12770. [Google Scholar] [CrossRef]
- Cui, H.; Peng, X.; Liu, J.; Ma, C.; Ji, Y.; Zhang, W.; Geng, M.; Li, Y. Design, Synthesis and Biological Evaluation of c-Met Kinase Inhibitors Bearing 2-Oxo-1,2-Dihydroquinoline Scaffold. Bioorg. Med. Chem. Lett. 2016, 26, 4483–4486. [Google Scholar] [CrossRef]
- Ikegai, K.; Nagata, Y.; Mukaiyama, T. N-Arylation of Pyridin-2(1H)-Ones with Pentavalent Organobismuth Reagents under Copper-Free Conditions. Bull. Chem. Soc. Jpn. 2006, 79, 761–767. [Google Scholar] [CrossRef]
- Ikegai, K.; Mukaiyama, T. Synthesis of N-Aryl Pyridin-2-Ones via Ligand Coupling Reactions Using Pentavalent Organobismuth Reagents. Chem. Lett. 2005, 34, 1496–1497. [Google Scholar] [CrossRef]
- López-Alvarado, P.; Avendaño, C.; Menéndez, J.C. 1,2-Dihydroquinolin-2-One (Carbostyril) Anions as Bidentate Nucleophiles in Their Reactions with Aryllead Triacetates: Synthesis of 1-Aryl- and 3-Aryl-Tetrahydroquinoline-2,5,8-Triones. J. Chem. Soc. Perkin 1 1997, 3, 229–234. [Google Scholar] [CrossRef]
- Wawzonek, S.; Van Truong, T. Preparation and Proton Spectra of 1-Aryl-1,2-Dihydro-2-Quinolones. J. Heterocycl. Chem. 1988, 25, 381–382. [Google Scholar] [CrossRef]
- Jung, S.-H.; Sung, D.-B.; Park, C.-H.; Kim, W.-S. Copper-Catalyzed N -Arylation of 2-Pyridones Employing Diaryliodonium Salts at Room Temperature. J. Org. Chem. 2016, 81, 7717–7724. [Google Scholar] [CrossRef]
- Lakshmi Narayana Sharma, K.; Suresh Kumar, C.; Kumaraswamy, S.; Krishna Reddy, V.; Kameswara Rao, N.S.; Raghu Babu, K.; Ramakrishna, G. Palladium-Catalyzed Domino Sequence for the Synthesis of N-Aryl Quinolinone-3-Carboxylate Derivatives and Their Anti-Proliferative Activity. Tetrahedron Lett. 2017, 58, 1127–1131. [Google Scholar] [CrossRef]
- Liu, J.; Ba, D.; Lv, W.; Chen, Y.; Zhao, Z.; Cheng, G. Base-Promoted Michael Addition/Smiles Rearrangement/ N -Arylation Cascade: One-Step Synthesis of 1,2,3-Trisubstituted 4-Quinolones from Ynones and Sulfonamides. Adv. Synth. Catal. 2020, 362, 213–223. [Google Scholar] [CrossRef]
- Mandewale, M.C.; Kokate, S.; Thorat, B.; Sawant, S.; Yamgar, R. Zinc Complexes of Hydrazone Derivatives Bearing 3,4-Dihydroquinolin-2(1H)-One Nucleus as New Anti-Tubercular Agents. Arab. J. Chem. 2019, 12, 4479–4489. [Google Scholar] [CrossRef]
- Bazine, I.; Cheraiet, Z.; Bensegueni, R.; Bensouici, C.; Boukhari, A. Synthesis, Antioxidant and Anticholinesterase Activities of Novel Quinoline-Aminophosphonate Derivatives. J. Heterocycl. Chem. 2020, 57, 2139–2149. [Google Scholar] [CrossRef]
- Raja, D.S.; Bhuvanesh, N.S.P.; Natarajan, K. A Novel Water Soluble Ligand Bridged Cobalt(II) Coordination Polymer of 2-Oxo-1,2-Dihydroquinoline-3-Carbaldehyde (Isonicotinic) Hydrazone: Evaluation of the DNA Binding, Protein Interaction, Radical Scavenging and Anticancer Activity. Dalton Trans. 2012, 41, 4365–4377. [Google Scholar] [CrossRef] [PubMed]
- Radini, I.A.M.; Elsheikh, T.M.Y.; El-Telbani, E.M.; Khidre, R.E. New Potential Antimalarial Agents: Design, Synthesis and Biological Evaluation of Some Novel Quinoline Derivatives as Antimalarial Agents. Molecules 2016, 21, 909. [Google Scholar] [CrossRef]
- Ghareeb, E.A.; Mahmoud, N.F.H.; El-Bordany, E.A.; El-Helw, E.A.E. Synthesis, DFT, and Eco-Friendly Insecticidal Activity of Some N-Heterocycles Derived from 4-((2-Oxo-1,2-Dihydroquinolin-3-Yl)Methylene)-2-Phenyloxazol-5(4H)-One. Bioorganic Chem. 2021, 112, 104945. [Google Scholar] [CrossRef]
- Govender, H.; Mocktar, C.; Kumalo, H.M.; Koorbanally, N.A. Synthesis, Antibacterial Activity and Docking Studies of Substituted Quinolone Thiosemicarbazones. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 1074–1081. [Google Scholar] [CrossRef]
- Conn, P.J.; Lindsley, C.W.; Hopkins, C.R.; Chauder, B.A.; Gogliotti, R.D.; Wood, M.R. Analogues de 1H-pyrrolo[3,2-c]quinoléine-4(5h)-one substitués utilisés comme modulateurs allostériques positifs du récepteur muscarinique à l’acétylcholine m4. Patent WO2012154731A1, 15 November 2012. [Google Scholar]
- Kolińska, J.; Grzelakowska, A.; Sokołowska, J. Dyes Based on the 2(1H)-quinolone Skeleton as Potential Colorimetric and Fluorescent Sensors for Cyanide Anions. Color. Technol. 2019, 135, 501–509. [Google Scholar] [CrossRef]
- Senthil Raja, D.; Ramachandran, E.; Bhuvanesh, N.S.P.; Natarajan, K. Synthesis, Structure and in Vitro Pharmacological Evaluation of a Novel 2-Oxo-1,2-Dihydroquinoline-3-Carbaldehyde (2′-Methylbenzoyl) Hydrazone Bridged Copper(II) Coordination Polymer. Eur. J. Med. Chem. 2013, 64, 148–159. [Google Scholar] [CrossRef]
- Sumana, T. Pushpa Iyengar Synthesis, Characterization And Antimicrobial Activity of Pharmaceutically Important 1,2-Dihydroquinoline Derivatives. J. Appl. Chem. 2015, 2, 2348–7968. [Google Scholar]
- SG52698—Benzoxazinones as Inhibitors of Hiv Reverse Transcriptase. Available online: https://patentscope.wipo.int/search/es/detail.jsf;jsessionid=7EA5F5D6EC4F4826BA1851ACA754AC31.wapp1nB?docId=SG1305690&_cid=P11-JYJURC-31566-35 (accessed on 8 September 2022).
- Abass, M.; Mostafa, B.B. Synthesis and Evaluation of Molluscicidal and Larvicidal Activities of Some Novel Enaminones Derived from 4-Hydroxyquinolinones: Part IX. Bioorg. Med. Chem. 2005, 13, 6133–6144. [Google Scholar] [CrossRef] [PubMed]
- Meth-Cohn, O.; Narine, B.; Tarnowski, B. A Versatile New Synthesis of Quinolines and Related Fused Pyridines, Part 5. The Synthesis of 2-Chloroquinoline-3-Carbaldehydes. J. Chem. Soc. Perkin 1 1981, 1520–1530. [Google Scholar] [CrossRef]
- Lam, P.Y.S. Chan–Lam Coupling Reaction: Copper-Promoted C–Element Bond Oxidative Coupling Reaction with Boronic Acids. Synth. Methods Drug Discov. 2016, 1, 242–273. [Google Scholar] [CrossRef]
- Janíková, K.; Jedinák, L.; Volná, T.; Cankař, P. Chan-Lam Cross-Coupling Reaction Based on the Cu2S/TMEDA System. Tetrahedron 2018, 74, 606–617. [Google Scholar] [CrossRef]
- Barraza, S.J.; Delekta, P.C.; Sindac, J.A.; Dobry, C.J.; Xiang, J.; Keep, R.F.; Miller, D.J.; Larsen, S.D. Discovery of Anthranilamides as a Novel Class of Inhibitors of Neurotropic Alphavirus Replication. Bioorg. Med. Chem. 2015, 23, 1569–1587. [Google Scholar] [CrossRef]
- West, M.J.; Fyfe, J.W.B.; Vantourout, J.C.; Watson, A.J.B. Mechanistic Development and Recent Applications of the Chan–Lam Amination. Chem. Rev. 2019, 119, 12491–12523. [Google Scholar] [CrossRef]
- Kumar, K.A.; Kannaboina, P.; Jaladanki, C.K.; Bharatam, P.V.; Das, P. Copper-Catalyzed N -Arylation of Tautomerizable Heterocycles with Boronic Acids and Its Application to Synthesis of Oxygenated Carbazoles. ChemistrySelect 2016, 1, 601–607. [Google Scholar] [CrossRef]
- Li, X.-H.; Ye, A.-H.; Liang, C.; Mo, D.-L. Substituent Effects of 2-Pyridones on Selective O-Arylation with Diaryliodonium Salts: Synthesis of 2-Aryloxypyridines under Transition -Metal-Free Conditions. Synthesis 2018, 50, 1699–1710. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Sporzyński, A. The Influence of Ortho-Substituents on the Properties of Phenylboronic Acids. J. Organomet. Chem. 2020, 913, 121202. [Google Scholar] [CrossRef]
- Munir, I.; Zahoor, A.F.; Rasool, N.; Naqvi, S.A.R.; Zia, K.M.; Ahmad, R. Synthetic Applications and Methodology Development of Chan–Lam Coupling: A Review. Mol. Divers. 2019, 23, 215–259. [Google Scholar] [CrossRef]
- Kurnia, K.A.; Setyaningsih, W.; Darmawan, N.; Yuliarto, B. A Comprehensive Study on the Impact of the Substituent on PKa of Phenylboronic Acid in Aqueous and Non-Aqueous Solutions: A Computational Approach. J. Mol. Liq. 2021, 326, 115321. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, S.; Zou, L.-H. Recent Advances in Direct Functionalization of Quinones: Recent Advances in Direct Functionalization of Quinones. Eur. J. Org. Chem. 2019, 2019, 2179–2201. [Google Scholar] [CrossRef]
- Zhang, R.; Luo, S. Bio-Inspired Quinone Catalysis. Chin. Chem. Lett. 2018, 29, 1193–1200. [Google Scholar] [CrossRef]
- Titi, A.; Al-Noaimi, M.; Kaddouri, Y.; El Ati, R.; Yousfi, E.B.; El Kodadi, M.; Touzani, R. Study of the Catecholase Catalytic Properties of Copper (II) Complexes Prepared in-Situ with Monodentate Ligands. Mater. Today Proc. 2019, 13, 1134–1142. [Google Scholar] [CrossRef]
- Cox, P.A.; Leach, A.G.; Campbell, A.D.; Lloyd-Jones, G.C. Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: PH–Rate Profiles, Autocatalysis, and Disproportionation. J. Am. Chem. Soc. 2016, 138, 9145–9157. [Google Scholar] [CrossRef]
- Gozdalik, J.T.; Adamczyk-Woźniak, A.; Sporzyński, A. Influence of Fluorine Substituents on the Properties of Phenylboronic Compounds. Pure Appl. Chem. 2018, 90, 677–702. [Google Scholar] [CrossRef]
- Hönel, M.; Vierhapper, F.W. Selectivity of Hydrogenations. Part 4 6- and 8-Substituted Quinaldines Yield of Tetrahydroderivatives and Basicities of Quinolines. Monatshefte Für Chem. Chem. Mon. 1984, 115, 1219–1228. [Google Scholar] [CrossRef]
- Hosmane, R.S.; Liebman, J.F. Paradoxes and Paradigms: Why Is Quinoline Less Basic than Pyridine or Isoquinoline? A Classical Organic Chemical Perspective. Struct. Chem. 2009, 20, 693–697. [Google Scholar] [CrossRef]
- Augustine, J.; Bombrun, A.; Atta, R. A Practical and Cost-Efficient, One-Pot Conversion of Aldehydes into Nitriles Mediated by ‘Activated DMSO’. Synlett 2011, 2011, 2223–2227. [Google Scholar] [CrossRef]
- Laali, K.K.; Insuasty, D.; Abonia, R.; Insuasty, B.; Bunge, S.D. Novel Quinoline–Imidazolium Adducts via the Reaction of 2-Oxoquinoline-3-Carbaldehyde and Quinoline-3-Carbaldehydes with 1-Butyl-3-Methylimidazolium Chloride [BMIM][Cl]. Tetrahedron Lett. 2014, 55, 4395–4399. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Insuasty, D.; Lima, S.; Gutiérrez, L.; Nogueras, M.; Marchal, A.; Abonia, R.; Andújar, S.; Spiegel, S.; Cobo, J.; et al. Design of New Quinolin-2-One-Pyrimidine Hybrids as Sphingosine Kinases Inhibitors. Bioorganic Chem. 2020, 94, 103414. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F.; Carson, K.G.; Harris, B.D.; Maryanoff, C.A.; Shah, R.D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures 1. J. Org. Chem. 1996, 61, 3849–3862. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, S.; Ahmad, Z.; Akhtar, T. Optical and Thermal Studies of Modified Terephthaldehyde–Acetone Polymer. Polym. Polym. Compos. 2020, 28, 572–578. [Google Scholar] [CrossRef]
- Abdou, W.M.; Khidre, R.E.; Kamel, A.A. Elaborating on Efficient Anti-Proliferation Agents of Cancer Cells and Anti-Inflammatory-Based N-Bisphosphonic Acids. Arch. Pharm. 2012, 345, 123–136. [Google Scholar] [CrossRef]
- Abonia, R.; Insuasty, D.; Castillo, J.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. Synthesis of Novel Quinoline-2-One Based Chalcones of Potential Anti-Tumor Activity. Eur. J. Med. Chem. 2012, 57, 29–40. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Solvent | Ligand (mol %) | Base | Yield (%) b |
| 1 | Cu(OAc)2 (10%) | MeCN | - | TEA | 0 |
| 2 | Cu(OAc)2 (10%) | MeCN | - | pyridine | 0 |
| 3 | Cu(OAc)2 (10%) | DMSO | - | TEA | 0 |
| 4 | Cu(OAc)2 (10%) | DMSO | - | pyridine | 15 |
| 5 | Cu(OAc)2 (10%) | DMF | - | TEA | 58 |
| 6 | Cu(OAc)2 (10%) | DMF | - | DMAP | 58 |
| 7 | Cu(OAc)2 (10%) | DMF | - | DIPEA | 53 |
| 8 | Cu(OAc)2 (10%) | DMF | - | Na2CO3.1.5 H2O2 | 52 |
| 9 | Cu(OAc)2 (10%) | DMF | - | Na2CO3 | 50 |
| 10 | Cu(OAc)2 (10%) | DMF | - | K2CO3 | 51 |
| 11 | Cu(OAc)2 (10%) | DMF | - | Cs2CO3 | 0 |
| 12 | Cu(OAc)2 (10%) | DMF | - | DBU | 0 |
| 13 | Cu(OAc)2 (10%) | DMF | - | quinoline | 41 |
| 14 | Cu(OAc)2 (10%) | DMF | - | pyridine | 60 |
| 15 c | Cu(OAc)2 (10%) | DMF | - | pyridine | 64 |
| 16 d | Cu(OAc)2 (10%) | DMF | - | pyridine | 64 |
| 17 | Cu(OAc)2 (20%) | DMF | - | pyridine | 47 |
| 18 | Cu(OAc)2 (10%) | DMF | Bipy (10%) | pyridine | 0 |
| 19 | Cu(OAc)2 (10%) | DMF | TMDA (10%) | pyridine | 21 |
| 20 | Cu(OTf)2 (10%) | DMF | - | pyridine | 27 |
| 21 | CuBr2 (10%) | DMF | - | pyridine | 23 |
| 22 | CuCl2 (10%) | DMF | - | pyridine | 15 |
 |
 |
 |
 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valencia, J.; Sánchez-Velasco, O.A.; Saavedra-Olavarría, J.; Hermosilla-Ibáñez, P.; Pérez, E.G.; Insuasty, D. N-Arylation of 3-Formylquinolin-2(1H)-ones Using Copper(II)-Catalyzed Chan–Lam Coupling. Molecules 2022, 27, 8345. https://doi.org/10.3390/molecules27238345
Valencia J, Sánchez-Velasco OA, Saavedra-Olavarría J, Hermosilla-Ibáñez P, Pérez EG, Insuasty D. N-Arylation of 3-Formylquinolin-2(1H)-ones Using Copper(II)-Catalyzed Chan–Lam Coupling. Molecules. 2022; 27(23):8345. https://doi.org/10.3390/molecules27238345
Chicago/Turabian StyleValencia, Jhesua, Oriel A. Sánchez-Velasco, Jorge Saavedra-Olavarría, Patricio Hermosilla-Ibáñez, Edwin G. Pérez, and Daniel Insuasty. 2022. "N-Arylation of 3-Formylquinolin-2(1H)-ones Using Copper(II)-Catalyzed Chan–Lam Coupling" Molecules 27, no. 23: 8345. https://doi.org/10.3390/molecules27238345
APA StyleValencia, J., Sánchez-Velasco, O. A., Saavedra-Olavarría, J., Hermosilla-Ibáñez, P., Pérez, E. G., & Insuasty, D. (2022). N-Arylation of 3-Formylquinolin-2(1H)-ones Using Copper(II)-Catalyzed Chan–Lam Coupling. Molecules, 27(23), 8345. https://doi.org/10.3390/molecules27238345