
Theoretical Study of the Geometry of Dibenzoazepine Analogues

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
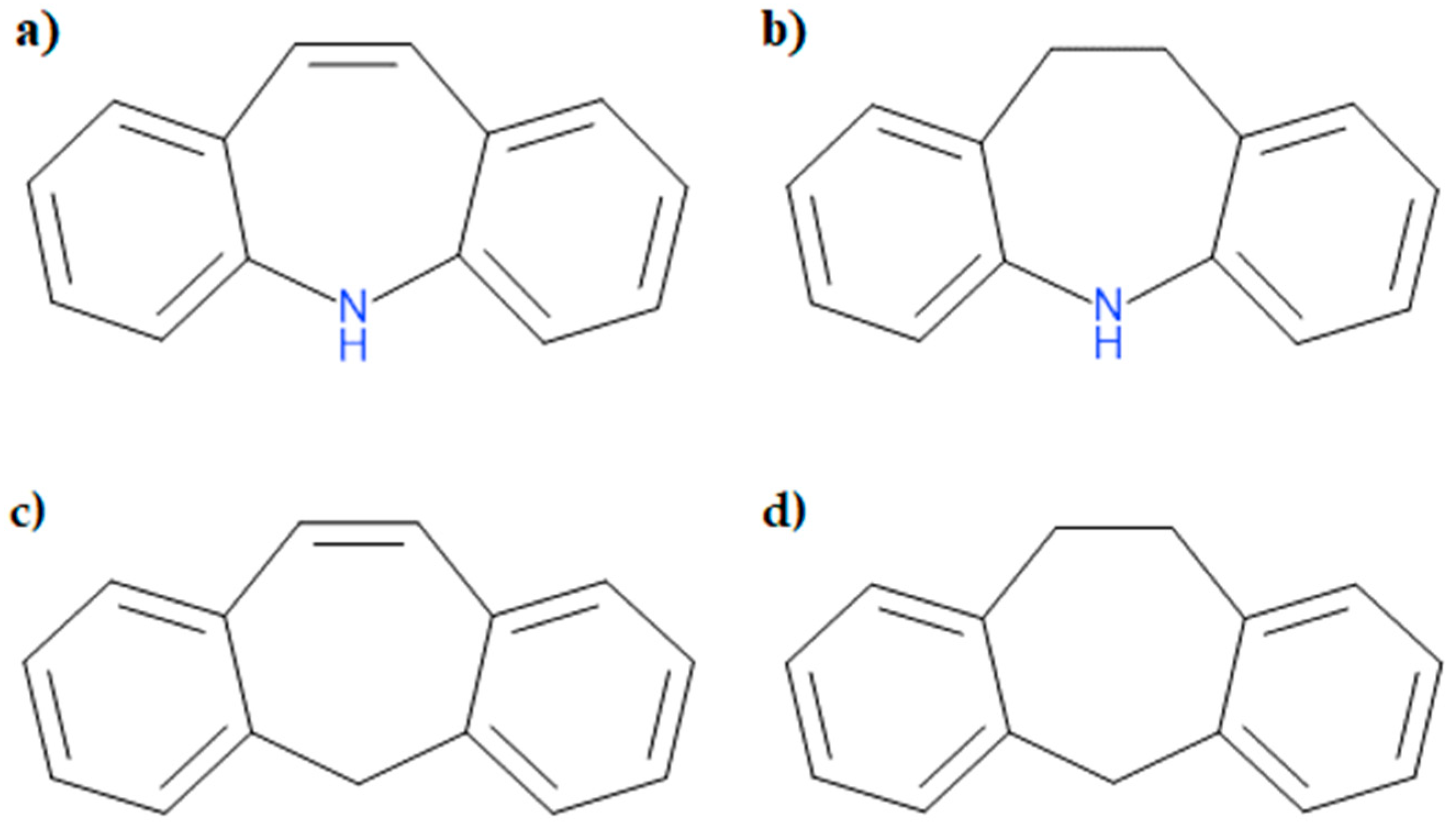
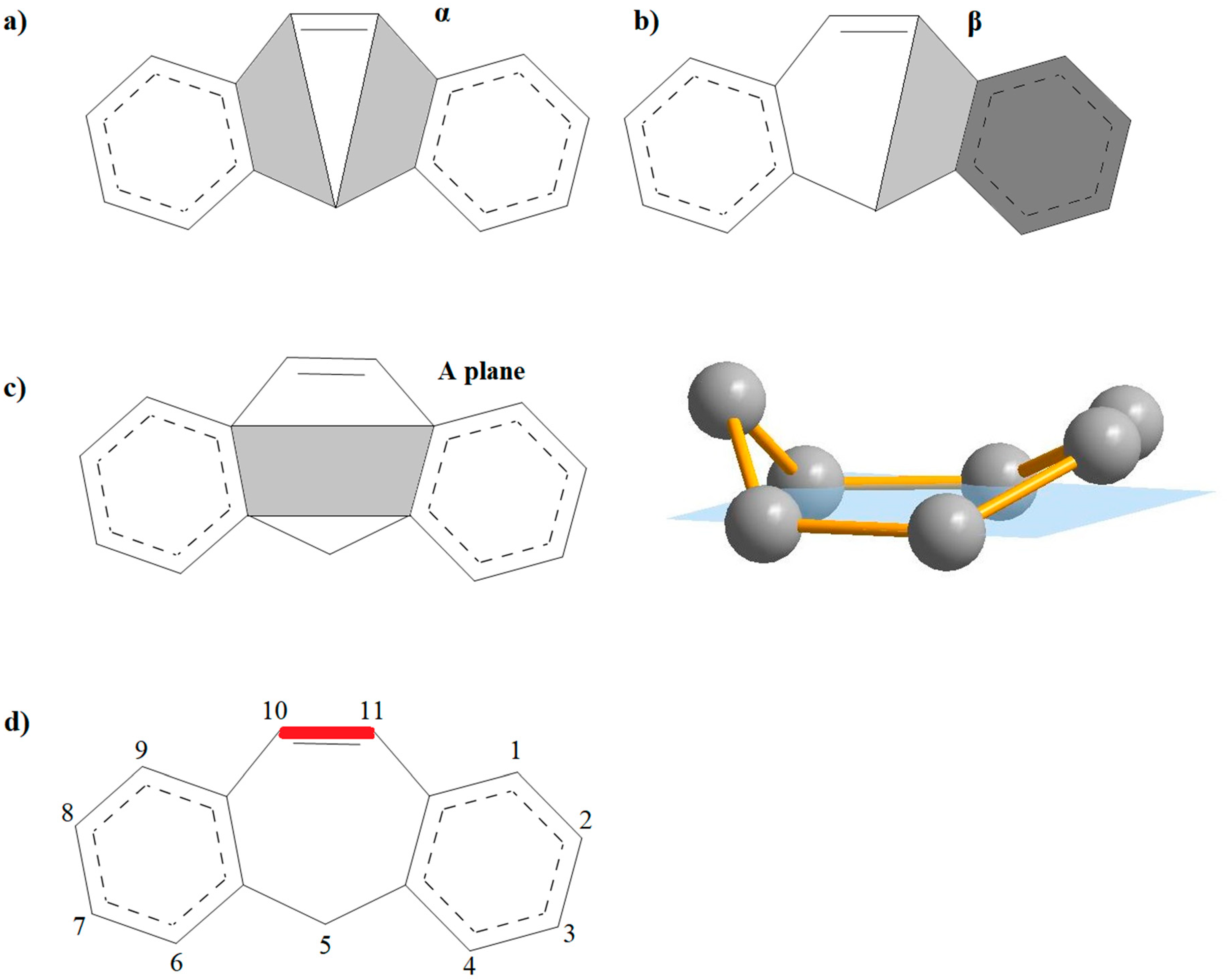
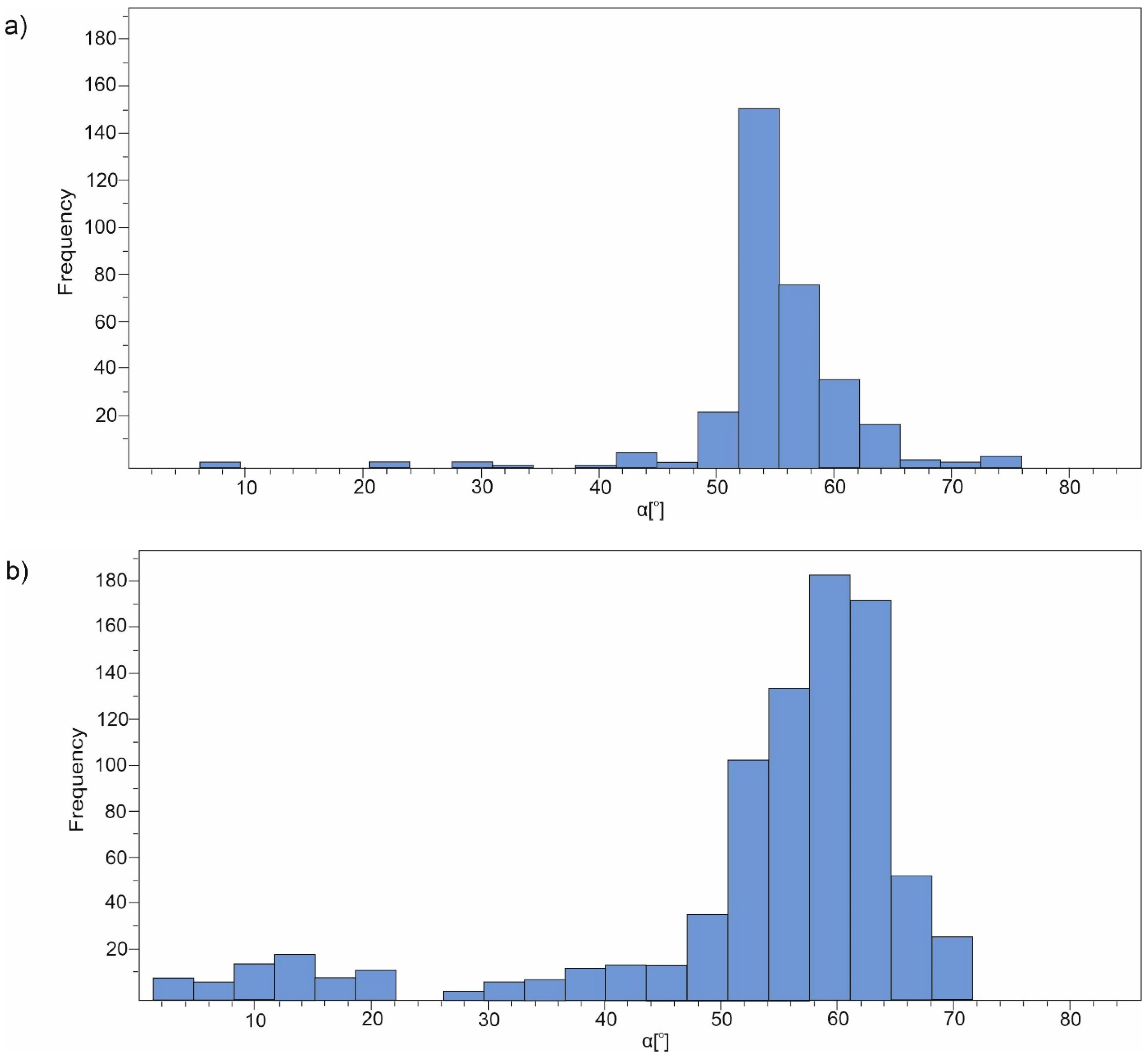
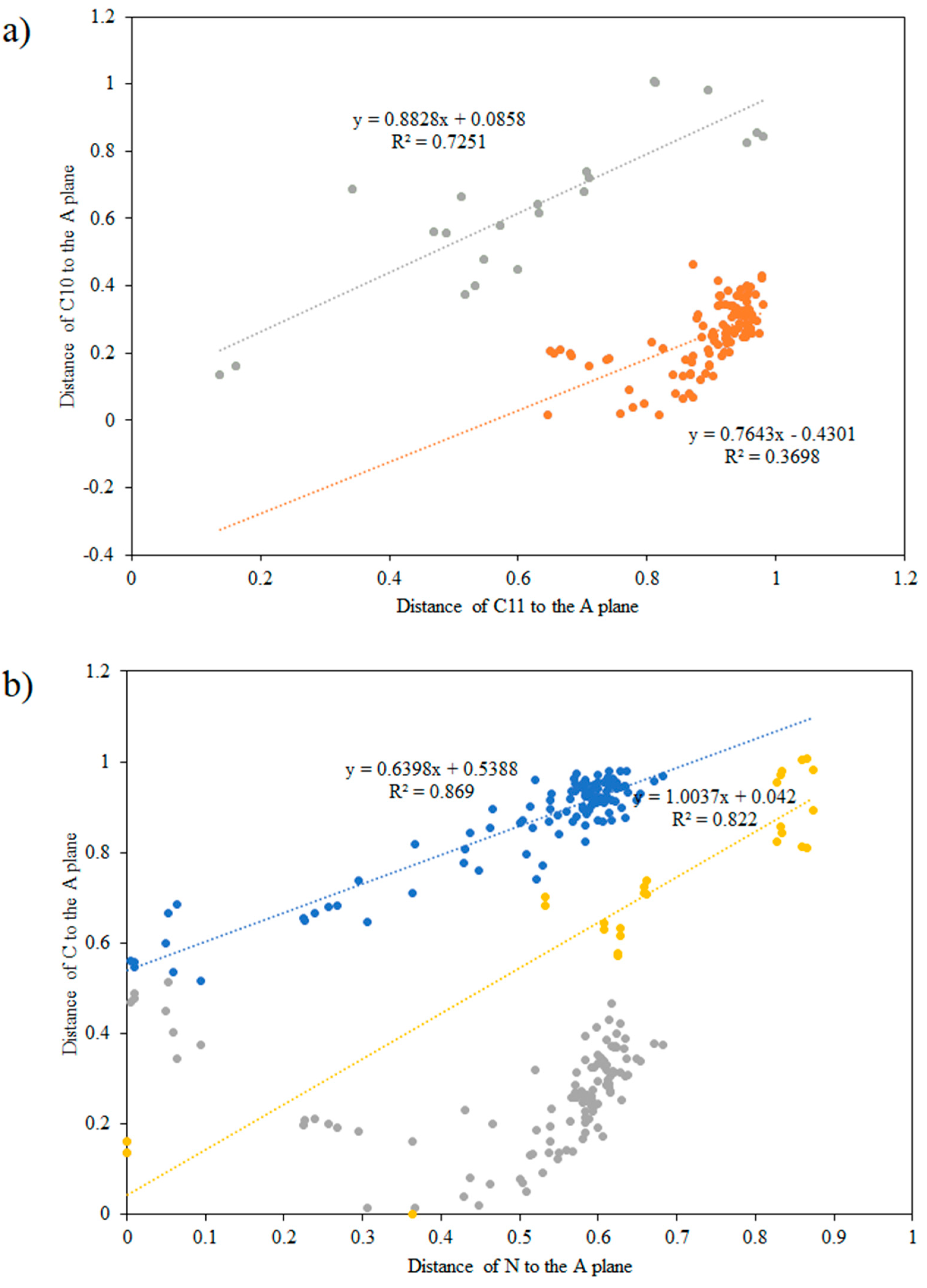
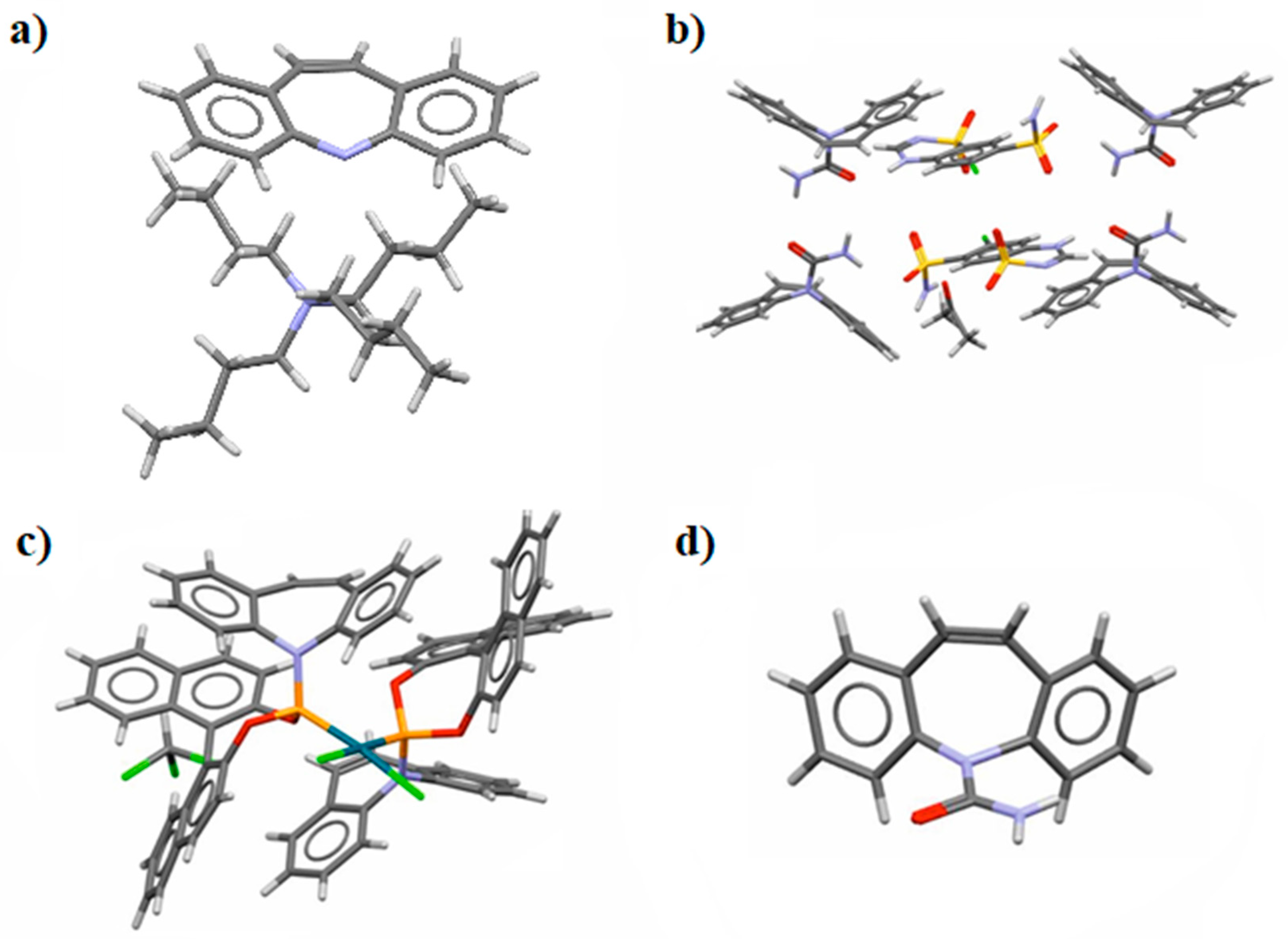
3.1. Geometry of the Investigated Compounds
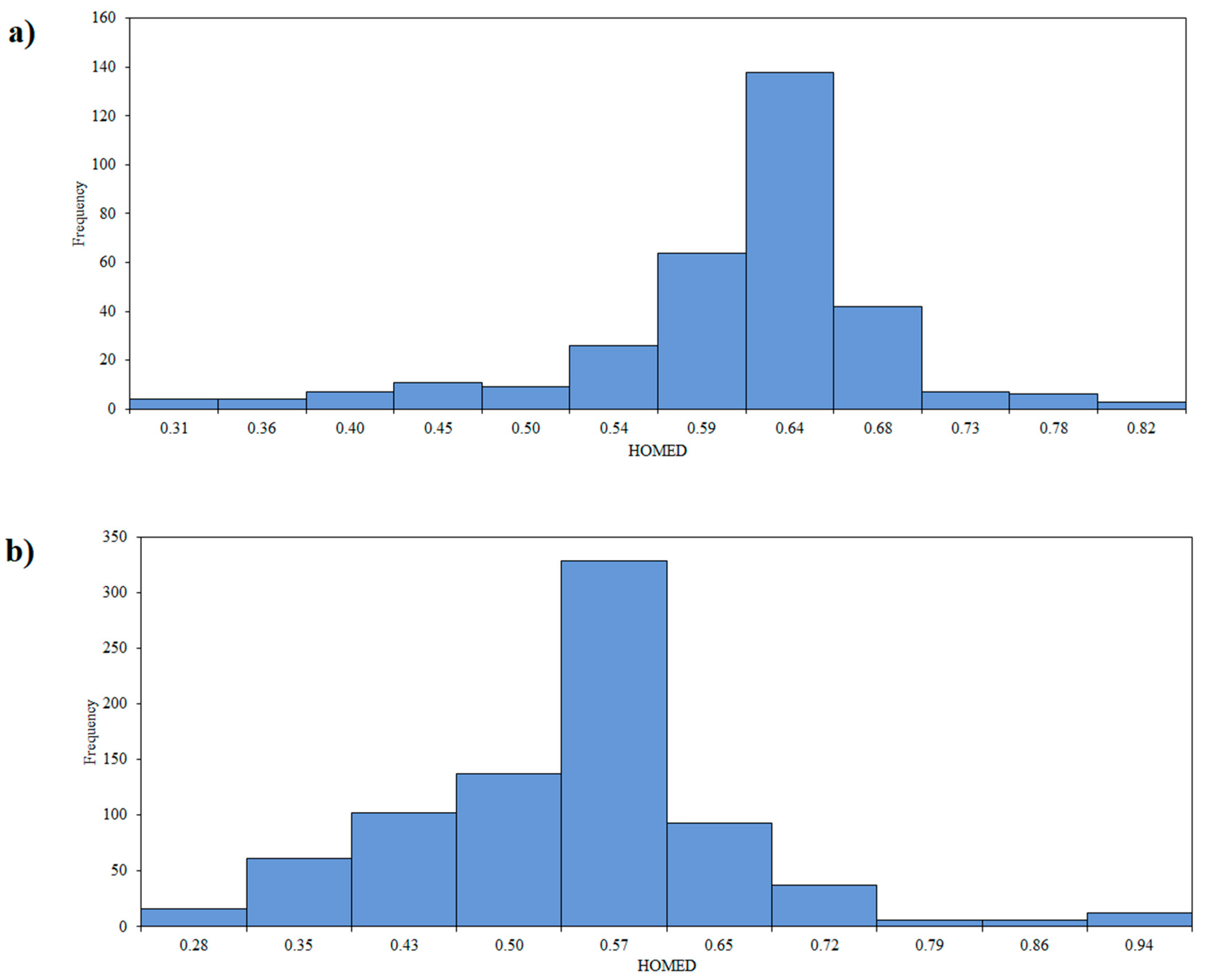
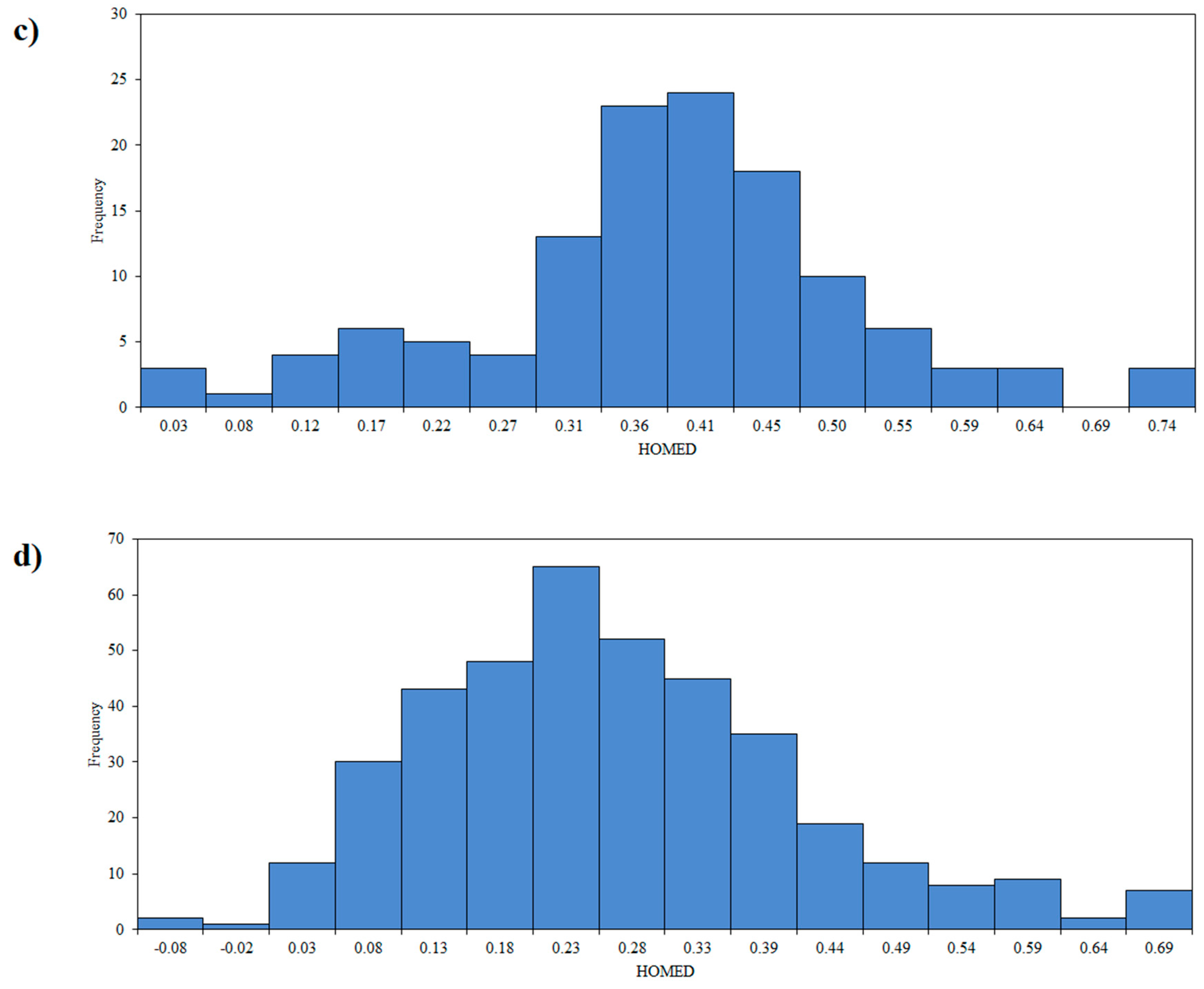
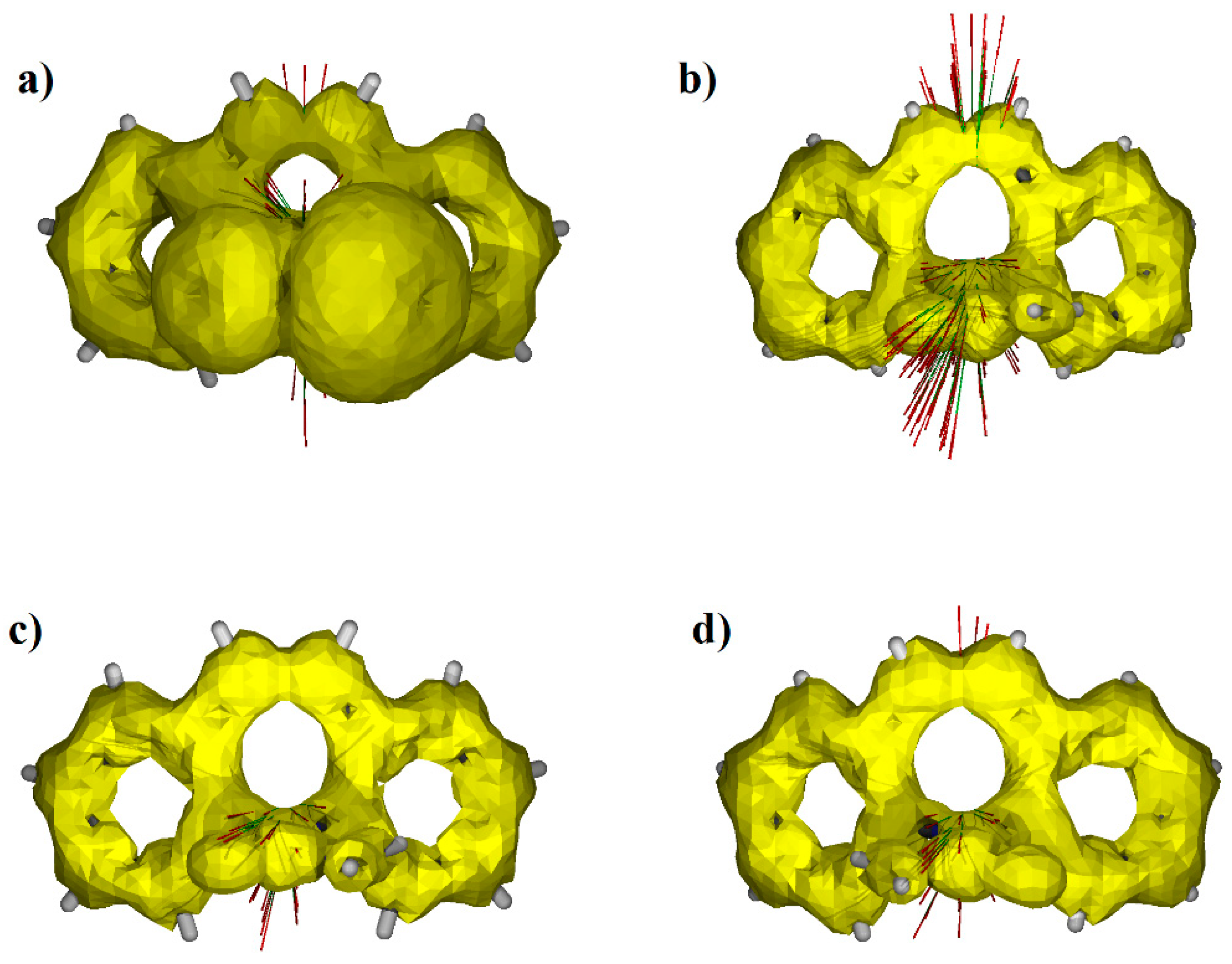
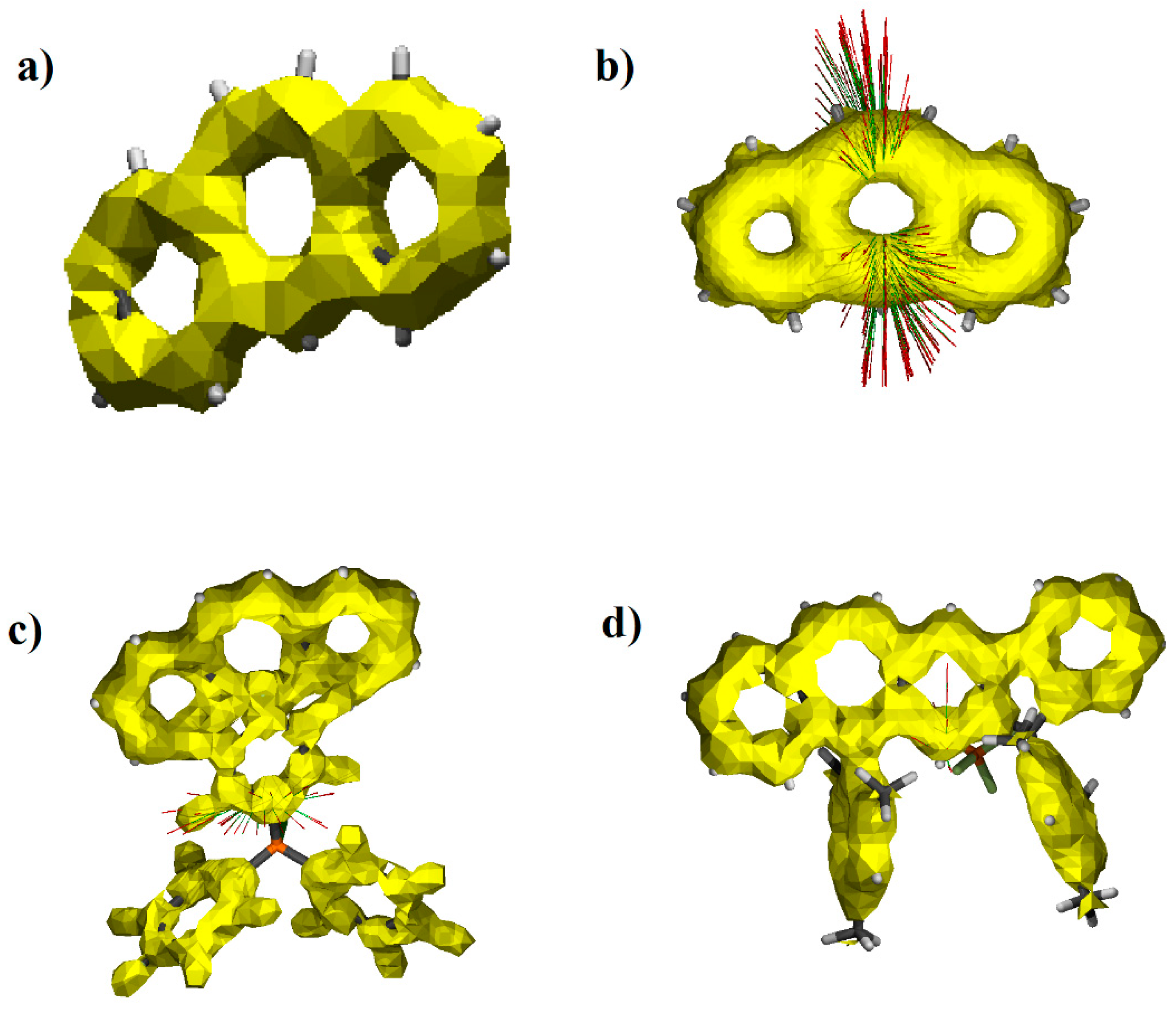
3.2. Aromaticity of the Central Ring of Investigated Compounds
3.3. Delocalization of Electrons
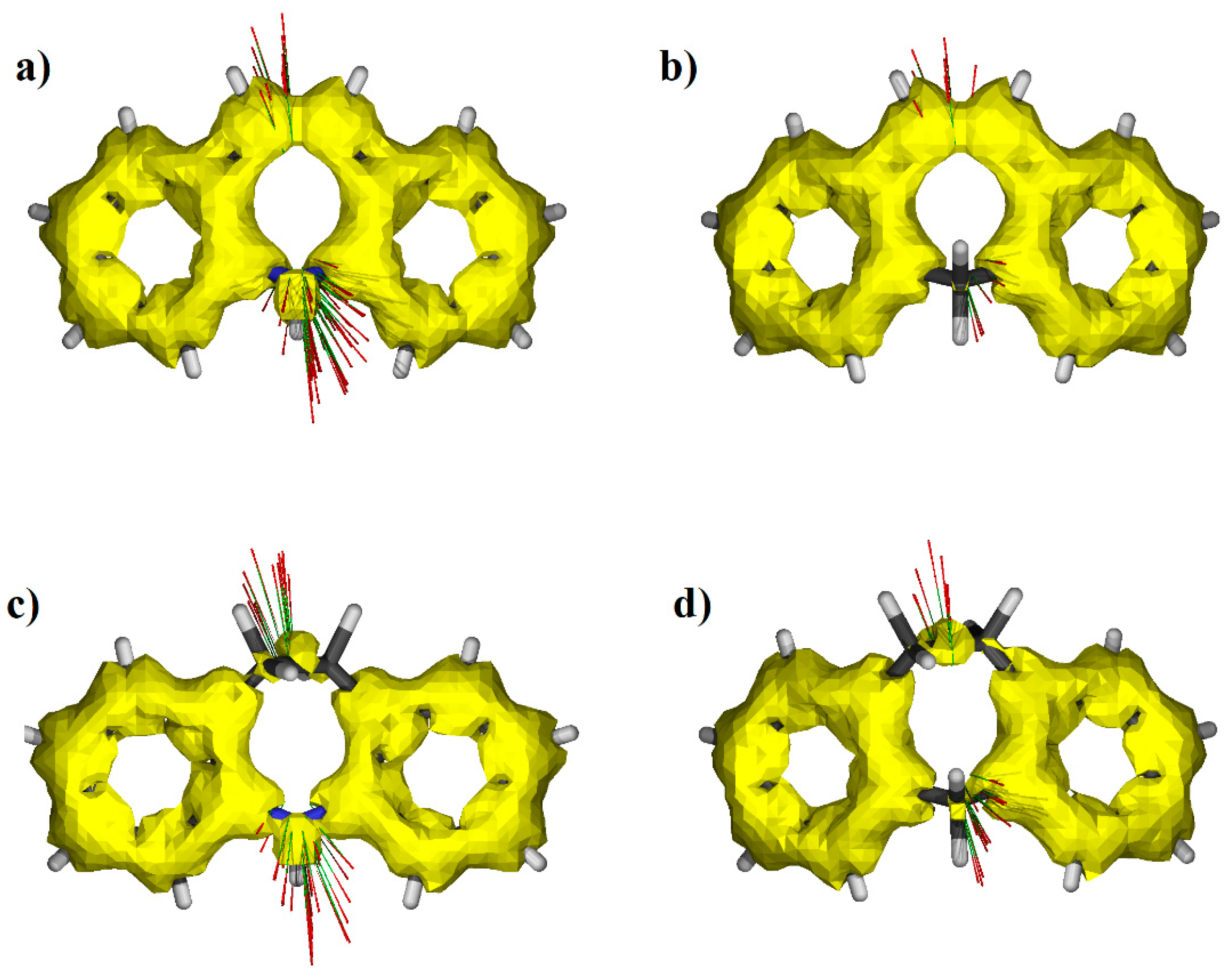
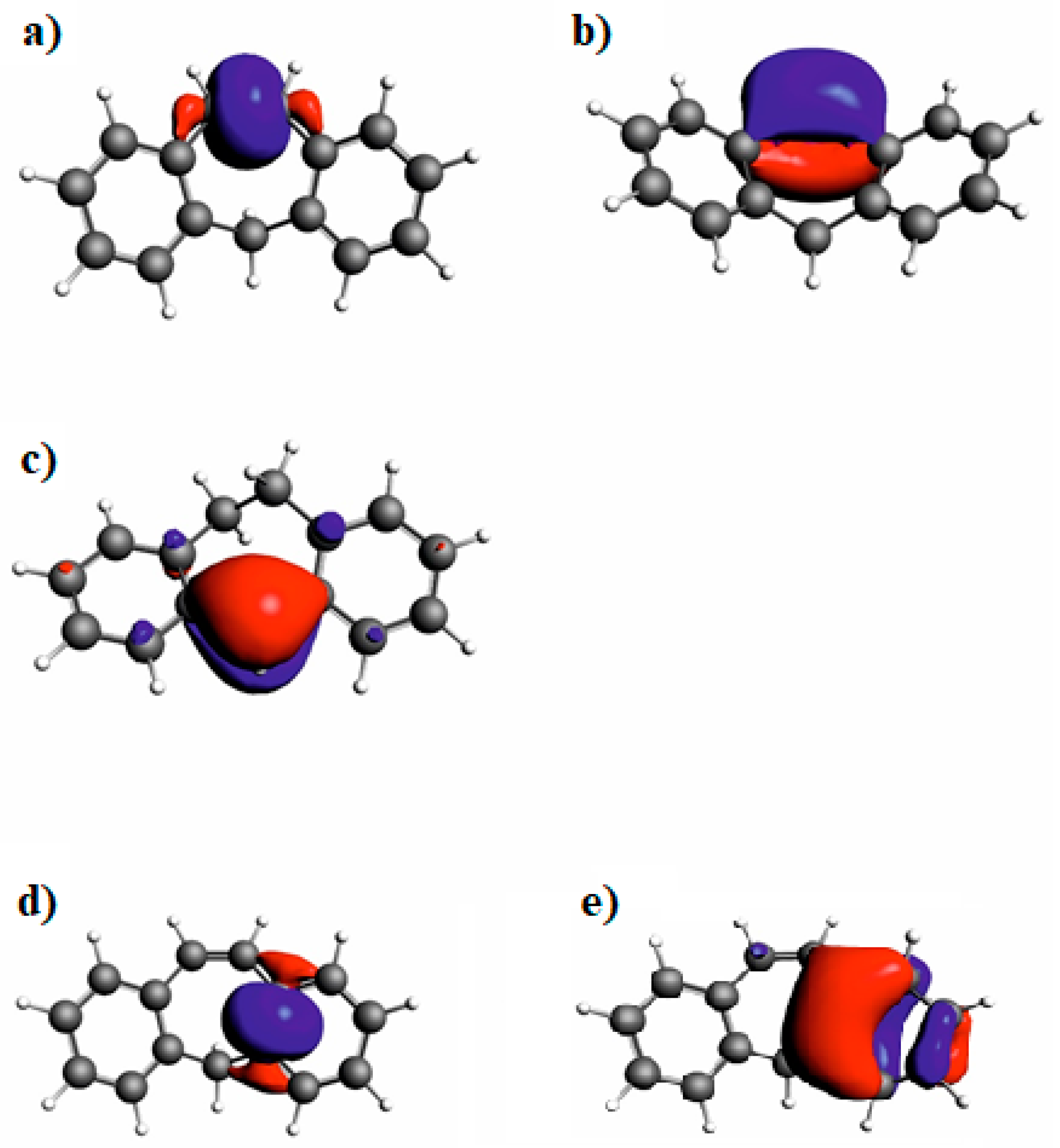
3.4. NBO Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Szymańska, M.; Majerz, I. Geometry and electron density of phenothazines. J. Mol. Struct. 2020, 1200, 127095. [Google Scholar] [CrossRef]
- Prisinzano, T.E. Medicinal Chemistry: A Molecular and Biochemical Approach. J. Med. Chem. 2006, 49, 3428. [Google Scholar] [CrossRef]
- Post, M.L.; Kennard, O.; Horn, A.S. The tricyclic antidepressants: Imipramine hydrochloride. The crystal and molecular structure of 5-(3-dimethylaminopropyl)-10,1l-dihydro-5H-dibenz[b,f]azepine hydrochloride. Acta Cryst. Sect. B Struct. Sci. 1975, 31, 1008–1013. [Google Scholar] [CrossRef] [Green Version]
- Kaur, N.; Fang, Y.-C.; Lee, H.-Y.; Singh, A.; Nepali, K.; Lin, M.-H.; Yeh, T.-K.; Lai, M.-J.; Chan, L.; Tu, Y.-K.; et al. Protective effects of 10,11-dihydro-5H-dibenzo[b,f]azepine hydroxamates on vascular cognitive impairment. Eur. J. Med. Chem. 2020, 187, 111915. [Google Scholar] [CrossRef]
- Vaghi, L.; Gaudino, E.; Cravotto, G.; Palmisano, G.; Penoni, A. A Structurally Diverse Heterocyclic Library by Decoration of Oxcarbazepine Scaffold. Molecules 2013, 181, 13705–13722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, K.; Kurebayashi, H.; Miyachi, H.; Tobe, M.; Onishi, M.; Isobe, Y. Synthesis and structure–activity relationship of tricyclic carboxylic acids as novel anti-histamines. Bioorg. Med. Chem. 2011, 19, 3005–3021. [Google Scholar] [CrossRef]
- Walden, J.; Grunze, H.; Bingmann, D.; Liu, Z.; Düsing, R. Calcium antagonistic effects of carbamazepine as a mechanism of action in neuropsychiatric disorders: Studies in calcium dependent model epilepsies. Eur. Neuropsychopharmacol. 1992, 2, 455–462. [Google Scholar] [CrossRef]
- Okada, M.; Hirano, T.; Mizuno, K.; Kawata, Y.; Wada, K.; Murakami, T.; Tasaki, H.; Kaneko, S. Effects of carbamazepine on hippocampal serotonergic system. Epilepsy Res. 1998, 31, 187–198. [Google Scholar] [CrossRef]
- Blom, S. Trigeminal neuralgia: Its treatment with a new anticonvulsant drug (G-32883). Lancet 1962, 279, 839–840. [Google Scholar] [CrossRef]
- Pierre, T.; Maron, M.; Rascle, C.; Cottencin, O.; Vaiva, G.; Goudemand, M. Carbamazepine in the treatment of neuroleptic malignant syndrome. Biol. Psychiatry 1998, 43, 303–305. [Google Scholar] [CrossRef]
- Adam, R.W.; Al-Labban, H.M.Y.; Aljanaby, A.A.J.; Abbas, N.A. Synthesis, Characterization and Antibacterial Activity of Some New of Novel 1,2,3-Triazole-Chalcone Derivatives from N-Acetyl-5H-Dibenzo [b,f] Azepine-5-Carboxamide. Nano Biomed. Eng. 2019, 11, 99–110. [Google Scholar] [CrossRef]
- Kumar, H.V.; Gnanendra, C.R.; Naik, N.; Gowda, C.D. In Vitro Antioxidant Activity of Dibenz[b,f]azepine and its Analogues. E-J. Chem. 2008, 5, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.V.; Ambati, R.R.; Sadineni, V.; Naik, N. Evaluation of In Vitro Antioxidant Activity of 5H-dibenz[b,f]azepine and Its Analogues. J. Phys. Sci. 2010, 21, 79–92. [Google Scholar]
- Perri, R.D.; Maill, F.; Bramanti, P. The effects of amineptine on the mood and nocturnal sleep of depressed patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 1987, 11, 65–70. [Google Scholar] [CrossRef]
- Platzek, J. & Snatzke, G. Synthesis of potically active 10,11-dihydro-5Hdibenzo[a,d]cycloheptenes. Tetrahedron 1987, 43, 4947–4968. [Google Scholar] [CrossRef]
- Ting, P.C.; Lee, J.F.; Solomon, D.M.; Smith, S.R.; Terminelli, C.A.; Jakway, J.P.; Zambas, D.N.; Lee, J.F.; Solomon, D.M.; Smith, S.R.; et al. Synthesis of dibenzo[a,d]cycloheptanes as cytokine biosynthesis inhibitors. Bioorg. Med. Chem. Lett. 1995, 5, 2749–2754. [Google Scholar] [CrossRef]
- Căproiu, M.T.; Dumitrascu, F.; Shova, S.; Chirită, I.C.; Missir, A.V.; Cioroianu, D.-M. Synthesis of new 10,11-dihydrodibenzo[a,d]cycloheptene S-thiocarbamate derivatives via a benzylic Newman–Kwart rearrangement. Tetrahedron Lett. 2014, 55, 4011–4013. [Google Scholar] [CrossRef]
- Villani, F.J.; Daniels, P.J.; Ellis, C.A.; Mann, T.A.; Wang, K.-C.; Wefer, E.A. Derivatives of 10,11-dihydro-5H-dibenzo(a,d)cycloheptene and related compounds. 6. Aminoalkyl derivatives of the aza isosteres. J. Med. Chem. 1972, 15, 750–754. [Google Scholar] [CrossRef]
- Socea, L.I.; Barbuceanu, S.F.; Iscrulescu, L.; Socea, B.; Hrubaru, M.; Pahontu, E.M.; Diaconu, C.C.; Bratu, O.G.; Olaru, O.T. New N-acylhydrazones with Potential Cytotoxic Activity. Rev. Chim. 2018, 69, 3341–3344. [Google Scholar] [CrossRef]
- Socea, L.I.; Visan, D.C.; Barbuceanu, S.F.; Apostol, T.V.; Bratu, O.G.; Socea, B. The Antioxidant Activity of Some Acylhydrazones with Dibenzo[a,d][7]annulene Moiety. Rev. Chim. 2018, 69, 795–797. [Google Scholar] [CrossRef]
- Socea, L.-I.; Saramef, G.; Mihalcea, F.; Apostol, T.V.; Andreescu, C.; Draghici, C.; Socea, B. New 1,2,4-triazoles and 1,3,4-oxadiazoles Derivatives with a 5H-dibenzo[a,d] [7] Annulene Moieties with Potential Antimicrobial Activity. Rev. Chim. 2014, 65, 156–159. [Google Scholar]
- Kopanski, C.; Turck, M.; Schultz, J. Effects of long-term treatment of rats with antidepressants on adrenergic-receptor sensitivity in cerebral cortex: Structure activity study. Neurochem. Int. 1983, 5, 649–659. [Google Scholar] [CrossRef]
- van Rossum, J.M. The Relation Between Chemical Structure and Biological Activity. J. Pharm. Pharmacol. 1963, 15, 285–316. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H. The Cambridge Structural Database: A Quarter of a Million Crystal Structures and Rising. Acta Cryst. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian Inc 16; Revision, A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 15, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Boudhar, K.; Debieche, M.; Serhane, A.; Zeghdaoui, A. Crystal structure, Raman spectroscopy study and quantum chemical DFT calculations of N-phenyl-3-para nitro phenyl isoxazolidine-5-carbonitrile. J. Mol. Struct. 2021, 1246, 1–9. [Google Scholar] [CrossRef]
- Abkari, A.; Chaabane, I.; Guidara, K. DFT (B3LYP/LanL2DZ and B3LYP/6311G+(d,p)) comparative vibrational spectroscopic analysis of organic–inorganic compound bis(4-acetylanilinium) tetrachlorocuprate(II). Phys. E Low-Dimens. Syst. Nanostructures 2016, 81, 136–144. [Google Scholar] [CrossRef]
- Herges, R.; Geuenich, D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T.J. Chemistry with ADF. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; teVelde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- ADF2019.302, SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands.
- Raczyńska, E.D.; Hallman, M.; Kolczyńska, K.; Stępniewski, T.M. On the Harmonic Oscillator Model of Electron Delocalization (HOMED) Index and its Application to Heteroatomic π-Electron Systems. Symmetry 2010, 2, 1485–1509. [Google Scholar] [CrossRef] [Green Version]
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric Equilibria in Relation to Pi-Electron Delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef] [PubMed]
- Reboul, J.P.; Cristau, B.; Soyfer, J.C.; Astier, J.P. 5H-Dibenz[b,f]azépinecarboxamide-5 (Carbamazépine). Acta Crystallogr. Sect. B 1981, 37, 1844–1848. [Google Scholar] [CrossRef]
- Lisgarten, J.N.; Palmer, R.A.; Saldanha, J.W. Crystal and molecular structure of 5-carbamyl-5H-dibenzo[b,f] azepine. J. Crystallogr. Spectrosc. Res. 1989, 19, 641–649. [Google Scholar] [CrossRef]
- Lowes, M.M.J.; Caira, M.R.; Lötter, A.P.; Van Der Watt, J.G. Physicochemical Properties and X-ray Structural Studies of the Trigonal Polymorph of Carbamazepine. J. Pharm. Sci. 1987, 76, 744–752. [Google Scholar] [CrossRef]
- Himes, V.L.; Mighell, A.D.; De Camp, W.H. Structure of carbamazepine: 5H-dibenz[b,f]azepine-5-carboxamide. Acta Crystallogr. Sect. 1981, 37, 2242–2245. [Google Scholar] [CrossRef] [Green Version]
- Grzesiak, A.L.; Lang, M.; Kim, K.; Matzger, A.J. Comparison of the Four Anhydrous Polymorphs of Carbamazepine and the Crystal Structure of Form I. J. Pharm. Sci. 2003, 92, 2260–2271. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.; Kampf, J.W.; Matzger, A.J. Form IV of Carbamazepine. J. Pharm. Sci. 2002, 91, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, P.; Shankland, K.; Florence, A.J.; Shankland, N.; Johnston, A. Solving Molecular Crystal Structures from X-ray Powder Diffraction Data: The Challenges Posed by γ-Carbamazepine and Chlorothiazide N,N,-Dimethylformamide (1/2) Solvate. J. Pharm. Sci. 2007, 96, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Eccles, K.S.; Stokes, S.P.; Daly, C.A.; Barry, N.M.; McSweeney, S.P.; O’Neill, D.J.; Kelly, D.M.; Jennings, W.B.; Ní Dhubhghaill, O.M.; Moynihan, H.A.; et al. Evaluation of the Bruker SMART X2S: Crystallography for the nonspecialist? J. Appl. Crystallogr. 2011, 44, 213–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlin, J.-B.; Price, L.S.; Price, S.L.; Florence, A.J. A strategy for producing predicted polymorphs: Catemeric carbamazepine form V. Chem. Commun. 2011, 47, 7074. [Google Scholar] [CrossRef] [Green Version]
- El Hassan, N.; Ikni, A.; Gillet, J.-M.; Spasojevic-de Biré, A.; Ghermani, N.E. Electron Properties of Carbamazepine Drug in Form III. Cryst. Growth Des. 2013, 13, 2887–2896. [Google Scholar] [CrossRef]
- Horstman, E.M.; Goyal, S.; Pawate, A.; Lee, G.; Zhang, G.G.Z.; Gong, Y.; Kenis, P.J.A. Crystallization Optimization of Pharmaceutical Solid Forms with X-ray Compatible Microfluidic Platforms. Cryst. Growth Des. 2015, 15, 1201–1209. [Google Scholar] [CrossRef]
- Sovago, I.; Gutmann, M.J.; Senn, H.M.; Thomas, L.H.; Wilson, C.C.; Farrugia, L.J. Electron density, disorder and polymorphism: High-resolution diffraction studies of the highly polymorphic neuralgic drug carbamazepine. Acta Crystallogr. Sect. B 2016, 72, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Nievergelt, P.P.; Spingler, B. Growing single crystals of small molecules by thermal recrystallization, a viable option even for minute amounts of material? CrystEngComm 2017, 19, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Van Genderen, E.; Clabbers, M.T.B.; Das, P.P.; Stewart, A.; Nederlof, I.; Barentsen, K.C.; Portillo, Q.; Pannu, N.S.; Nicolopoulos, S.; Gruene, T.; et al. Ab initio structure determination of nanocrystals of organic pharmaceutical compounds by electron diffraction at room temperature using a Timepix quantum area direct electron detector. Acta Crystallogr. Sect. A 2016, 72, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.G.; Martynowycz, M.W.; Hattne, J.; Tyler, J.; Fulton, T.J.; Stoltz, B.M.; Rodriguez, J.A.; Nelson, H.M.; Gonen, T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent. Sci. 2018, 4, 1587–1592. [Google Scholar] [CrossRef]
- Kolb, U.; Krysiak, Y.; Plana-Ruiz, S. Automated electron diffraction tomography—Development and applications. Acta Crystallogr. Sect. B 2019, 75, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, J.E. Functionalized Acenes and Heteroacenes for Organic Electronics. Chem. Rev. 2006, 106, 5028–5048. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Ahrens, L.; Brosius, V.; Freudenberg, J.; Bunz, U.H.F. Unusual Stabilization of Larger Acenes and Heteroacenes. J. Mater. Chem. 2019, 7, 14011–14034. [Google Scholar] [CrossRef]
- Schleyer, P.; von Rague Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.; von Rague Schleyer, P.; Manoharan, M.; Wang, Z.-X.; Kiran, B.; Jiao, H.; Puchta, R.; van Eikema Hommes, N.J.R. Dissected Nucleus-Independent Chemical Shift Analysis of π-Aromaticity and Antiaromaticity. Org. Lett. 2001, 3, 2465–2468. [Google Scholar] [CrossRef] [PubMed]
- Cyrański, M.K. Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef]
- Matito, E.; Duran, M.; Solà, M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization. J. Chem. Phys. 2005, 122, 14109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poater, J.; Fradera, X.; Duran, M.; Solà, M. The Delocalization Index as an Electronic Aromaticity Criterion: Application to a Series of Planar Polycyclic Aromatic Hydrocarbons. Chem. –A Eur. J. 2003, 9, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Dominikowska, J.; Palusiak, M. EL: The new aromaticity measure based on one-electron density function. Struct. Chem. 2012, 23, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Krygowski, T.M. Crystallographic Studies of Inter- and Intramolecular Interactions Reflected in Aromatic Character of π-Electron Systems. J. Chem. Inf. Comput. Sci. 1993, 24. [Google Scholar] [CrossRef]
- Reetz, M.T.; Hütte, S.; Goddard, R.; Minet, U. Tetrabutylammonium salts of carbazole and dibenzoazepine: Synthesis, crystal structures and use in anionic polymerization. J. Chem. Soc. Chem. Commun. 1995, 2, 275–277. [Google Scholar] [CrossRef]
- Florence, A.J.; Leech, C.K.; Shankland, N.; Shankland, K.; Johnston, A. Control and prediction of packing motifs: A rare occurrence of carbamazepine in a catemeric configuration. CrystEngComm 2006, 8, 746–747. [Google Scholar] [CrossRef]
- Aljohani, M.; Pallipurath, A.R.; McArdle, P.; Erxleben, A. A Comprehensive Cocrystal Screening Study of Chlorothiazide. Cryst. Growth Des. 2017, 17, 5223–5232. [Google Scholar] [CrossRef]
- Briceño, A.; Dorta, R. cis-Dichloridobis{[(S)-N-(3,5-dioxa-4-phosphacyclohepta [2,1-a;3,4-a′]dinaphthalen-4-yl]dibenz[b,f]azepin-κP}palladium(II) deuterochloroform disolvate. Acta Crystallogr. Sect. E 2007, 63, m1718–m1719. [Google Scholar] [CrossRef]
- Leitner, T.D.; Gmeinder, Y.; Röhricht, F.; Herges, R.; Mena-Osteritz, E.; Bäuerle, P. Twisted Thienylene–Phenylene Structures: Through-Space Orbital Coupling in Toroidal and Catenated Topologies. Eur. J. Org. Chem. 2020, 3, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Reck, G.; Thiel, W. Crystal structures of the adducts carbamazepine-ammonium chloride and carbamazepine-ammonium bromide and their transformation to carbamazepine dihydrate. Pharmazie 1991, 46, 509–512. [Google Scholar]
- Nicolaï, B.; Fournier, B.; Dahaoui, S.; Gillet, J.-M.; Ghermani, N.-E. Crystal and Electron Properties of Carbamazepine-Aspirin Co-crystal. Cryst. Growth Des. 2019, 19, 1308–1321. [Google Scholar] [CrossRef]
- Faudone, S.N.; Paschoal, A.R.; Carvalho, P.S.; Ellena, J.; Martins, F.T.; Cuffini, S.L.; Ayala, A.P.; Sperandeo, N.R. X-ray diffraction, vibrational and thermal study of dibenzazepinodione, a pharmacopeial impurity of oxcarbazepine. J. Mol. Struct. 2019, 1182, 204–212. [Google Scholar] [CrossRef]
- Wang, W.; Yang, M.; Han, D.; He, Q.; Fan, R. Tandem Palladium Catalysis for Rapid Construction of 3,4-Fused Tricyclic Indoles. Adv. Synth. Catal. 2020, 362, 1281–1285. [Google Scholar] [CrossRef]
- Loya, J.D.; Li, S.J.; Unruh, D.K.; Hutchins, K.M. Mechanochemistry as a Tool for Crystallizing Inaccessible Solids from Viscous Liquid Components. Cryst. Growth Des. 2022, 22, 285–292. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Fan, R. 1,2- and 1,4-Additions of 2-Alkynylcyclohexadienimines with Aromatic Amines To Access 4-Amino-N-arylindoles and -azepinoindoles. Org. Lett. 2012, 14, 6076–6079. [Google Scholar] [CrossRef] [PubMed]
- Cordoneanu, A.; Drewitt, M.J.; Bavarian, N.; Baird, M.C. Synthesis and characterization of weakly coordinating anion salts of a new, stable carbocationic reagent, the dibenzosuberenyl (dibenzotropylium) ion. New J. Chem 2008, 32, 1890–1898. [Google Scholar] [CrossRef]
- Fu, X.; Han, H.; Zhang, D.; Yu, H.; He, Q.; Zhao, D. Polycyclic aromatic hydrocarbon diradical with pH-responsive magnetic properties. Chem. Sci. 2020, 11, 5565–5571. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Löwdin, P.-O. Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Phys. Rev. 1955, 97, 1474–1489. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tautomer | ΔE [kcal۰mol−1] | ||
|---|---|---|---|
| 5H-dibenzo[b,f]azepine | 1 |  | 0 |
| 2 | 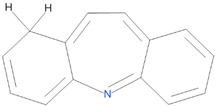 | 47.71 | |
| 3 | 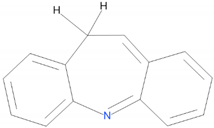 | 21.11 | |
| 5H-dibenzo[a,d][7]annulene | 1 |  | 0 |
| 2 |  | 47.29 | |
| 3 | 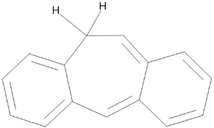 | 22.18 |
| Name | Minimum | Maximum | Mean | Variance | Std. Dev | Mean. Dev | Median |
|---|---|---|---|---|---|---|---|
| α angle | |||||||
| 5H-dibenzo[b,f]azepine | 6.478 | 76.212 | 55.102 | 47.040 | 6.859 | 3.706 | 54.891 |
| 5H-dibenzo[a,d][7]annulene | 1.196 | 72.078 | 54.257 | 192.096 | 13.860 | 9.081 | 58.611 |
| 10,11-dihydro-5H-dibenzo[b,f]azepine | 7.035 | 82.424 | 53.577 | 180.502 | 13.435 | 9.313 | 57.400 |
| 10,11-dihydro-5H-dibenzo[a,d][7]annulene | 5.028 | 83.931 | 59.767 | 206.198 | 14.36 | 11.258 | 60.169 |
| β1,β2 angle | |||||||
| 5H-dibenzo[b,f]azepine | 0.183 | 8.312 | 3.155 | 1.975 | 1.405 | 1.053 | 3.135 |
| 5H-dibenzo[b,f]azepine | 0.273 | 9.489 | 3.117 | 2.428 | 1.558 | 1.178 | 3.107 |
| 5H-dibenzo[a,d][7]annulene | 0.041 | 15.744 | 3.399 | 3.374 | 1.837 | 1.428 | 3.202 |
| 5H-dibenzo[a,d][7]annulene | 0.044 | 11.373 | 3.449 | 3.161 | 1.778 | 1.41 | 3.251 |
| 10,11-dihydro-5H-dibenzo[b,f]azepine | 0.416 | 19.911 | 3.094 | 6.373 | 2.525 | 1.627 | 2.42 |
| 10,11-dihydro-5H-dibenzo[b,f]azepine | 0.441 | 15.258 | 2.967 | 5.078 | 2.253 | 1.508 | 2.266 |
| 10,11-dihydro-5H-dibenzo[a,d][7]annulene | 0.136 | 12.738 | 3.433 | 4.439 | 2.107 | 1.608 | 3.062 |
| 10,11-dihydro-5H-dibenzo[a,d][7]annulene | 0.072 | 12.512 | 3.21 | 4.658 | 2.158 | 1.636 | 2.736 |
| Distance to the C12,C13,C14,C15 plane | |||||||
| 5H-dibenzo[b,f]azepine C10 | 0.004 | 0.884 | 0.537 | 0.008 | 0.087 | 0.056 | 0.527 |
| 5H-dibenzo[b,f]azepine C11 | 0.028 | 0.864 | 0.536 | 0.007 | 0.087 | 0.054 | 0.534 |
| 5H-dibenzo[b,f]azepine N | 0.105 | 0.833 | 0.626 | 0.006 | 0.077 | 0.037 | 0.629 |
| 5H-dibenzo[a,d][7]annulene C10 | 0.002 | 1.003 | 0.525 | 0.025 | 0.158 | 0.114 | 0.556 |
| 5H-dibenzo[a,d][7]annulene C11 | 0.003 | 0.891 | 0.523 | 0.024 | 0.156 | 0.112 | 0.550 |
| 5H-dibenzo[a,d][7]annulene C5 | 0.005 | 0.818 | 0.632 | 0.025 | 0.158 | 0.101 | 0.677 |
| 10,11-dihydro-5H-dibenzo[b,f]azepine C10 | 0.000 | 0.873 | 0.532 | 0.034 | 0.184 | 0.125 | 0.587 |
| 10,11-dihydro-5H-dibenzo[b,f]azepine C11 | 0.015 | 0.980 | 0.554 | 0.103 | 0.32 | 0.295 | 0.514 |
| 10,11-dihydro-5H-dibenzo[b,f]azepine N | 0.015 | 1.008 | 0.611 | 0.100 | 0.316 | 0.294 | 0.698 |
| 10,11-dihydro-5H-dibenzo[a,d][7]annulene C10 | 0.003 | 1.247 | 0.642 | 0.111 | 0.333 | 0.296 | 0.727 |
| 10,11-dihydro-5H-dibenzo[a,d][7]annulene C11 | 0.001 | 1.235 | 0.693 | 0.098 | 0.314 | 0.268 | 0.806 |
| 10,11-dihydro-5H-dibenzo[a,d][7]annulene C5 | 0.011 | 0.930 | 0.627 | 0.035 | 0.186 | 0.137 | 0.644 |
| C10C11 bond length | |||||||
| 5H-dibenzo[b,f]azepine C10=C11 | 1.240 | 1.473 | 1.35 | 0.001 | 0.034 | 0.023 | 1.341 |
| 5H-dibenzo[a,d][7]annulene C10=C11 | 1.252 | 1.499 | 1.371 | 0.002 | 0.044 | 0.040 | 1.348 |
| 10,11-dihydro-5H-dibenzo[b,f]azepine C10-C11 | 1.332 | 1.606 | 1.519 | 0.001 | 0.038 | 0.022 | 1.524 |
| 10,11-dihydro-5H-dibenzo[a,d][7]annulene C10-C11 | 1.299 | 1.711 | 1.524 | 0.002 | 0.041 | 0.026 | 1.526 |
| a substituent in the 5-position | 5H-dibenzo[b,f]azepine | 5H-dibenzo[a,d][7]annulene | ||||||||
| α | C10C11 bond length | distances of the atoms from the A plane | α | C10C11 bond length | distances of the atoms from the A plane | |||||
| C10 | C11 | N5 | C10 | C11 | C5 | |||||
| unsubstituted structure | 45.37 | 1.345 | 0.442 | 0.442 | 0.511 | 56.79 | 1.351 | 0.527 | 0.527 | 0.688 |
| CH3 | 50.49 | 1.349 | 0.449 | 0.449 | 0.592 | 51.23 | 1.349 | 0.439 | 0.439 | 0.636 |
| CH2CH3 | 51.46 | 1.349 | 0.452 | 0.456 | 0.605 | 51.41 | 1.349 | 0.440 | 0.436 | 0.640 |
| C(CH3)3 | 52.89 | 1.349 | 0.498 | 0.498 | 0.606 | 46.15 | 1.346 | 0.380 | 0.380 | 0.576 |
| CHO | 50.80 | 1.348 | 0.480 | 0.483 | 0.587 | 54.38 | 1.350 | 0.493 | 0.500 | 0.662 |
| COOH | 52.73 | 1.350 | 0.489 | 0.504 | 0.612 | 55.35 | 1.350 | 0.512 | 0.514 | 0.666 |
| NO2 | 50.42 | 1.351 | 0.476 | 0.476 | 0.582 | 52.08 | 1.350 | 0.494 | 0.494 | 0.614 |
| NH2 | 52.45 | 1.350 | 0.450 | 0.447 | 0.653 | |||||
| OH | 53.35 | 1.352 | 0.466 | 0.466 | 0.659 | |||||
| Cl | 47.70 | 1.348 | 0.440 | 0.440 | 0.564 | |||||
| a substituent in the 5-position | 10,11-dihydro-5H-dibenzo[b,f]azepine | 10,11-dihydro-5H-dibenzo[a,d][7]annulene | ||||||||
| α | C10C11 bond length | distances of the atoms from the A plane | α | C10C11 bond length | distances of the atoms from the A plane | |||||
| C10 | C11 | N5 | C10 | C11 | C5 | |||||
| unsubstituted structure | 22.02 | 1.544 | 0.464 | 0.470 | 0.003 | 55.90 | 1.538 | 0.891 | 0.213 | 0.614 |
| CH3 | 51.82 | 1.534 | 0.885 | 0.143 | 0.523 | 43.34 | 1.534 | 0.759 | 0.046 | 0.480 |
| CH2CH3 | 51.79 | 1.536 | 0.856 | 0.151 | 0.534 | 51.59 | 1.536 | 0.838 | 0.125 | 0.571 |
| C(CH3)3 | 56.55 | 1.536 | 0.232 | 0.911 | 0.582 | 41.90 | 1.529 | 0.698 | 0.053 | 0.481 |
| CHO | 54.37 | 1.537 | 0.211 | 0.901 | 0.558 | 49.30 | 1.535 | 0.082 | 0.828 | 0.542 |
| COOH | 57.06 | 1.539 | 0.911 | 0.271 | 0.597 | 53.88 | 1.536 | 0.180 | 0.876 | 0.585 |
| NO2 | 53.94 | 1.539 | 0.897 | 0.227 | 0.551 | 50.59 | 1.535 | 0.858 | 0.155 | 0.530 |
| NH2 | 44.95 | 1.535 | 0.774 | 0.022 | 0.498 | |||||
| OH | 51.59 | 1.536 | 0.838 | 0.125 | 0.571 | |||||
| Cl | 45.09 | 1.534 | 0.812 | 0.047 | 0.463 | |||||
| Refcode. | Bond Length C10-C11 | Distance C10 to the A Plane | Distance C11 to the A Plane | R-factor | T | Space Group |
|---|---|---|---|---|---|---|
| CBMZPN01 [37] | 1.330 | 0.543 | 0.514 | 3.5 | rt | P21/c |
| CBMZPN02 [38] | 1.325 | 0.515 | 0.556 | 8.4 | rt | P21/n |
| CBMZPN03 [39] | 1.3456 | 0.549 | 0.540 | 6.9 | rt | R-3 |
| CBMZPN10 [40] | 1.331 | 0.520 | 0.546 | 3.9 | rt | P21/n |
| CBMZPN11 [41] | 1.337 | 0.528 | 0.536 | 5.06 | 158 | P-1 |
| CBMZPN12 [42] | 1.340 | 0.492 | 0.469 | 3.57 | 158 | C2/c |
| CBMZPN13 [43] | 1.376 | 0.547 | 0.531 | 17.96 | 160 | P-1 |
| CBMZPN14 [44] | 1.336 | 0.543 | 0.518 | 4.04 | rt | P21/n |
| CBMZPN16 [45] | 1.347 | 0.505 | 0.571 | 4.5 | 123 | Pbca |
| CBMZPN17 [46] | 1.350 | 0.542 | 0.509 | 4 | rt | P21/n |
| CBMZPN18 [46] | 1.352 | 0.543 | 0.510 | 1.08 | 100 | P21/n |
| CBMZPN19 [46] | 1.352 | 0.543 | 0.510 | 0 | P21/n | |
| CBMZPN20 [47] | 1.333 | 0.539 | 0.527 | 3.95 | rt | P21 |
| CBMZPN21 [48] | 1.353 | 0.545 | 0.509 | 6.79 | 100 | P21/n |
| CBMZPN22 [48] | 1.351 | 0.543 | 0.510 | 4.07 | 100 | P21/n |
| CBMZPN23 [48] | 1.352 | 0.545 | 0.512 | 2.4 | 100 | P21/n |
| CBMZPN27 [49] | 1.344 | 0.511 | 0.543 | 4.26 | 183 | P21/n |
| CBMZPN28 [50] | 1.243 | 0.650 | 0.514 | 25.45 | rt | P21/n |
| CBMZPN29 [50] | 1.344 | 0.430 | 0.490 | 40.54 | rt | P21/n |
| CBMZPN30 [50] | 1.260 | 0.451 | 0.452 | 36.9 | rt | P21/n |
| CBMZPN31 [51] | 1.396 | 0.483 | 0.545 | 19.21 | rt | P21/n |
| CBMZPN32 [52] | 1.338 | 0.347 | 0.395 | 43.85 | rt | P21/n |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymańska, M.; Majerz, I. Theoretical Study of the Geometry of Dibenzoazepine Analogues. Molecules 2022, 27, 790. https://doi.org/10.3390/molecules27030790
Szymańska M, Majerz I. Theoretical Study of the Geometry of Dibenzoazepine Analogues. Molecules. 2022; 27(3):790. https://doi.org/10.3390/molecules27030790
Chicago/Turabian StyleSzymańska, Małgorzata, and Irena Majerz. 2022. "Theoretical Study of the Geometry of Dibenzoazepine Analogues" Molecules 27, no. 3: 790. https://doi.org/10.3390/molecules27030790