3.2. Synthesis
General Procedure for Castagnoli–Cushman reaction (2a–2f). A round-bottom flask under an argon atmosphere was charged with the corresponding imine
1 (0.100 mmol, 1.0 eq.) in TFE (3 mL) and then charged with homophthalic anhydride
IVa (24.3 mg, 0.150 mmol, 1.5 eq.) at −40 °C. The reaction mixture was stirred at −40 °C until the starting material was consumed, as indicated by TLC analysis. The mixture was concentrated in vacuo, and the residue was purified by preparative TLC to afford the pure product. Purification: CH
2Cl
2/MeOH (95:5). Copies of
1H and
13C spectra for all prepared are available in the
supplementary materials.
4-(benzyloxy)benzaldehyde (0c): The title compound was prepared following the literature procedure [
31] using
p-anisaldehyde (2.30 g, 18.846 mmol, 1.0 eq.) and benzyl bromide (3.36 mL, 28.269 mmol, 1.5 eq.). Product
0c was isolated in 93% overall yield (3.74 g) in one step as a white powder. Mp: 68–70 °C; IR (ν): 1738, 1604, 1507, 1451, 1266, 1061, 708, 617 cm
−1;
1H NMR (300 MHz, CDCl
3): d (ppm) 9.91 (s, 1H), 7.87 (d,
J = 8.9 Hz, 2H), 7.48–7.37 (m, 5H), 7.10 (d,
J = 8.9 Hz, 2H), 5.18 (s, 2H);
13C NMR (125 MHz, CDCl
3): d (ppm) 190.8, 163.7, 136.0, 132.0, 130.1, 128.7, 128.3, 127.5, 115.1, 70.2; HRMS (E.S.I.
+, m/z) calcd for C
14H
13O
2+ (M + H)
+: 213.0910, found: 213.0887.
N-(benzo[d][1,3]dioxol-5-yl)-1-(4-methoxy phenyl)methanimine (1a): To a stirred solution of benzo[d][1,3]dioxol-5-amine (1.28 g, 9.326 mmol, 1.0 eq.) in dry MeOH (40 mL) with molecular sieve 4Å were added p-anisaldehyde (1.13 mL, 9.326 mmol, 1.0 eq.) and glacial acetic acid (53.4 µL, 0.933 mmol, 0.1 eq.). The reaction mixture was stirred overnight at reflux. After cooling, the mixture was filtered on celite and concentrated under reduced pressure. The residue was purified by recrystallisation (heptane/AcOEt, 95:5) to afford the pure product 1a. Brown crystals (1.71 g, 72%). Mp: 107–108 °C; IR (ν): 2889, 1601, 1574, 1499, 1479, 1241, 1170, 1028, 924, 816 cm−1; 1H NMR (500 MHz, CDCl3): d (ppm) 8.30 (s, 1H), 7.75 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.7 Hz, 2H), 6.74 (d, J = 8.3 Hz, 1H), 6.73 (d, J = 2.1 Hz, 1H), 6.65 (dd, J = 8.3 Hz, J = 2.1 Hz, 1H), 5.90 (s, 2H), 3.79 (s, 3H); 13C NMR (125 MHz, CDCl3): d (ppm) 162.1, 158.1, 148.2, 146.9, 145.8, 130.3 (2C), 129.3, 114.6, 114.2 (2C), 108.3, 101.9, 101.3, 55.4; HRMS (E.S.I.+, m/z) calcd for C15H14NO3+ (M + H)+: 256.0968, found: 256.0955.
4-((benzo[d][1,3]dioxol-5-ylimino)methyl) phenol (1b): To a stirred solution of benzo[d][1,3]dioxol-5-amine (13.7 mg, 0.100 mmol, 1.0 eq.) in dry MeOH (2 mL) with molecular sieve 4Å were added 4-hydroxybenzaldehyde (10.0 µL, 0.100 mmol, 1.0 eq.) and one drop of glacial acetic acid (0.010 mmol, 0.1 eq.). The reaction mixture was stirred overnight at reflux. After cooling, the mixture was concentrated under reduced pressure to afford the brut product 1b without further purification. The imine was used in Castagnoli–Cushman reactions directly after preparation
N-(benzo[d][1,3]dioxol-5-yl)-1-(4-(benzyloxy) phenyl)methanimine (1c): To a stirred solution of benzo[d][1,3]dioxol-5-amine (193.0 mg, 1.407 mmol, 1.0 eq.) in dry MeOH (7 mL) with molecular sieve 4Å were added aldehyde 0c (298.6 mg, 1.407 mmol, 1.0 eq.) and glacial acetic acid (8.1 µL, 0.141 mmol, 0.1 eq.). The reaction mixture was stirred for 1 h at reflux. After cooling, the mixture was filtered on celite and concentrated under reduced pressure to afford the pure product 1c. Brown solid (462.8 mg, quant.). Mp: 102–104 °C; IR (ν): 2962, 2922, 1605, 1571, 1506, 1479, 1257, 1085, 1012, 791 cm−1; 1H NMR (500 MHz, DMSO-d6): d (ppm) 8.53 (s, 1H), 7.85 (d, J = 8.7 Hz, 2H), 7.49–7.32 (m, 5H), 7.13 (d, J = 8.7 Hz, 2H), 6.96 (d, J = 2.1 Hz, 1H), 6.92 (d, J = 8.1 Hz, 1H), 6.76 (dd, J = 8.1 Hz, J = 2.1 Hz, 1H), 6.04 (s, 2H), 5.19 (s, 2H); 13C NMR (125 MHz, DMSO-d6): d (ppm) 160.7, 158.1, 147.9, 146.0, 145.4, 136.7, 130.1 (2C), 129.2, 128.4 (2C), 127.9, 127.7 (2C), 115.2, 115.0 (2C), 108.4, 101.4, 101.2, 69.4; HRMS (E.S.I.+, m/z) calcd for C21H18NO3+ (M + H)+: 332.1281, found: 322.0998.
N-(4-methoxybenzyl)-1-(4-methoxyphenyl) methanimine (1d): The title compound was prepared following the literature procedure [
32] using (4-methoxyphenyl)methanamine (620.0 µL, 4.746 mmol, 1.0 eq.) and
p-anisaldehyde (577.5 µL, 4.746 mmol, 1.0 eq.). Product
1d was isolated in quant. overall yield (1.20 g) in one step as a brown solid. Mp: 36–37 °C; IR (ν): 2830, 2910, 1510, 1245, 1035, 810 cm
−1;
1H NMR (300 MHz, CDCl
3): d (ppm) 8.32 (s, 1H), 7.73 (d,
J = 8.8 Hz, 2H), 7.27 (d,
J = 8.8 Hz, 2H), 6.95 (d,
J = 8.7 Hz, 2H), 6.91 (d,
J = 8.7 Hz, 2H), 4.75 (s, 2H), 3.85 (s, 3H), 3.81 (s, 3H);
13C NMR (75 MHz, CDCl
3): d (ppm) 161.7, 160.9, 158.7, 131.7, 129.8 (2C), 129.2 (2C), 114.0 (2C), 113.9 (2C), 64.4, 55.3, 55.2; HRMS (E.S.I.
+, m/z) calcd for C
16H
18NO
2+ (M + H)
+: 256.1332, found: 256.1342.
N-tert-butyl-1-(4-methoxyphenyl)methanimine (1e): To a stirred solution of tert-butylamine (10.7 µL, 0.100 mmol, 1.0 eq.) in dry MTBE (2 mL) with molecular sieve 4Å was added p-anisaldehyde (12.2 µL, 0.100 mmol, 1.0 eq.). The reaction mixture was stirred for 24 h at room temperature. The mixture was concentrated under reduced pressure to afford the brut product 1e without further purification. The imine was used in Castagnoli–Cushman reactions directly after preparation.
2-methyl-N-phenylpropan-1-imine (1f): To a stirred solution of aniline (9.1 µL, 0.100 mmol, 1.0 eq.) in dry CH2Cl2 (1 mL) with MgSO4 (18.1 mg, 0.150 mmol, 1.5 eq) was added isobutyraldehyde (13.7 µL, 0.150 mmol, 1.5 eq.). The reaction mixture was stirred for 1 h at reflux. After cooling, the mixture was filtered and concentrated under reduced pressure to afford the brut product 1f without further purification. The imine was used in Castagnoli–Cushman reactions directly after preparation.
N-cyclopropyl-1-(thiophen-2-yl)methanimine (1g): The title compound was prepared following the literature procedure [
33] using cyclopropanamine (6.9 µL, 0.100 mmol, 1.0 eq.) and thiophene-2-carbaldehyde (9.4 µL, 0.100 mmol, 1.0 eq.). The brut product
1g was used in Castagnoli–Cushman reactions without further purification directly after preparation. The imine was used in Castagnoli–Cushman reactions directly after preparation.
N-butyl-1-(4-nitrophenyl)methanimine (1h): To a stirred solution of butan-1-amine (9.9 µL, 0.100 mmol, 1.0 eq.) in dry toluene (1 mL) with MgSO4 (18.1 mg, 0.150 mmol, 1.5 eq) was added 4-nitrobenzaldehyde (15.1 mg, 0.100 mmol, 1.0 eq.). The reaction mixture was stirred overnight at 80 °C. After cooling, the mixture was filtered and concentrated under reduced pressure to afford the brut product 1h without further purification. The imine was used in Castagnoli–Cushman reactions directly after preparation.
1-(4-methoxyphenyl)-N-(4-(trifluoromethyl) phenyl)methanimine (1i): To a stirred solution of 4-(trifluoromethyl)aniline (12.6 µL, 0.100 mmol, 1.0 eq.) in dry MeOH (1 mL) with molecular sieve 4Å were added 4-methoxybenzaldehyde (12.2 µL, 0.100 mmol, 1.0 eq.) and one drop of glacial acetic acid (0.010 mmol, 0.1 eq.). The reaction mixture was stirred overnight at reflux. After cooling, the mixture was filtered and concentrated under reduced pressure to afford the brut product 1i without further purification. The imine was used in Castagnoli–Cushman reactions directly after preparation.
Ethyl 2-((4-methoxyphenyl)imino)acetate (1j): The title compound was prepared following the literature procedure [
34] using 4-methoxyaniline (12.3 mg, 0.100 mmol, 1.0 eq.) and ethyl 2-oxoacetate (20.4 µL, 0.100 mmol, 1.0 eq., 50% in toluene). The brut product
1j was used without further purification. The imine was used in Castagnoli–Cushman reactions directly after preparation.
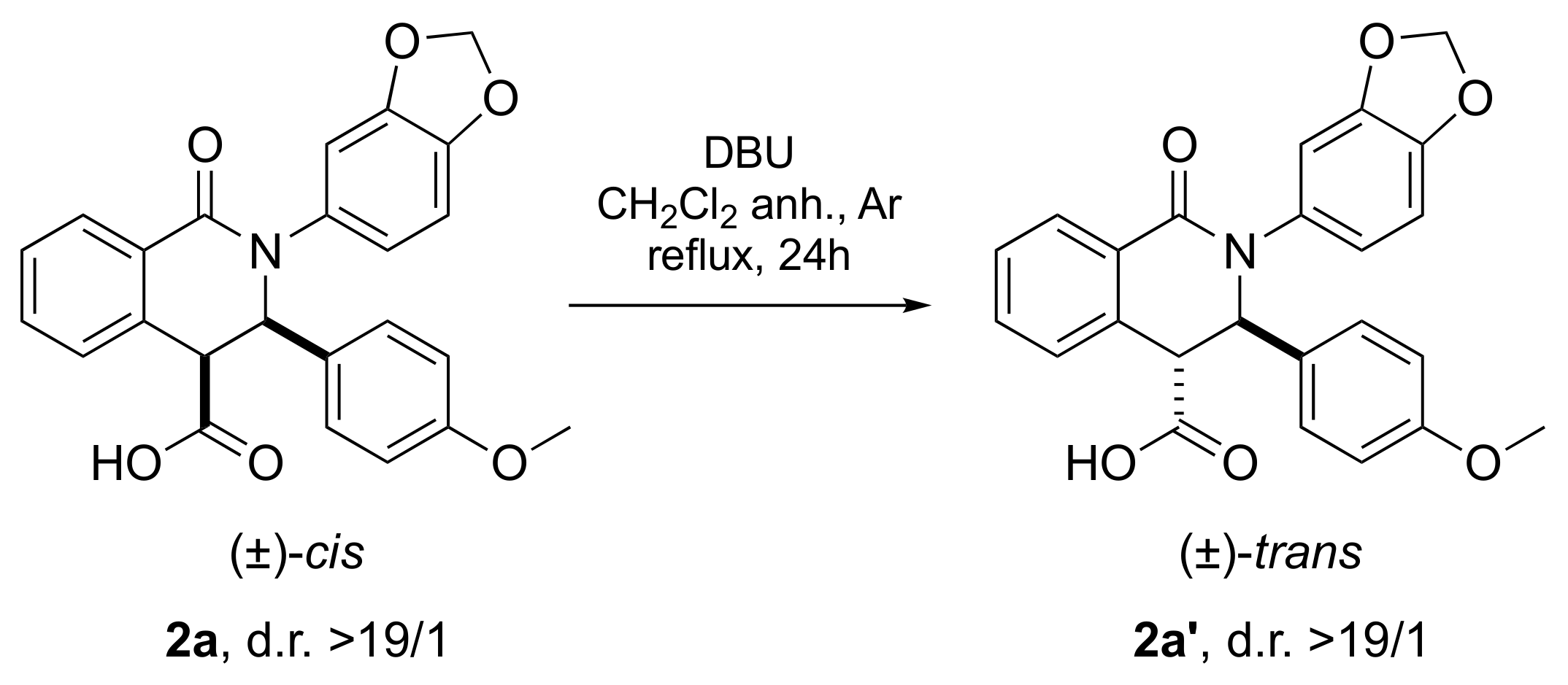
Cis-2-(benzo[d][1,3]dioxol-5-yl)-3-(4-metho xyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2a): The title compound was prepared following the general procedure described above using 1a (25.5 mg, 0.100 mmol, 1.0 eq.) and IVa (24.3 mg, 0.150 mmol, 1.5 eq.) in 15 min of reaction time. The product 2a was isolated in 81% yield (33.8 mg, d.r. (cis/trans): >19/1) as a brown powder. When the reaction was performed at room temperature in 2 min of reaction time: 30.1 mg, 72%, d.r. (cis/trans): >19/1. Mp: 158–159 °C; IR (ν): 3321, 2902, 1717, 1609, 1485, 1246, 1175, 1032, 926, 796, 731 cm−1; 1H NMR (300 MHz, CDCl3): d (ppm) 8.30 (dd, J = 7.0 Hz, J = 1.2 Hz, 1H), 7.60–7.48 (m, 3H), 6.97 (dd, J = 6.7 Hz, J = 2.0 Hz, 2H), 6.75 (d, J = 8.0 Hz, 1H), 6.70 (dd, J = 6.8 Hz, J = 2.0 Hz, 2H), 6.63 (d, J = 2.0 Hz, 1H), 6.59 (dd, J = 8.0 Hz, J = 2.0 Hz, 1H), 5.96 (s, 2H), 5.28 (d, J = 6.0 Hz, 1H), 4.92 (d, J = 6.0 Hz, 1H), 3.74 (s, 3H); 13C NMR (125 MHz, CDCl3): d (ppm) 172.0, 163.9, 159.7, 147.8, 146.7, 135.4, 132.6, 132.5, 129.2, 129.1 (2C), 128.8, 128.3, 128.1, 127.7, 120.6, 113.9 (2C), 108.8, 108.2, 101.5, 65.3, 55.1, 49.5; HRMS (E.S.I.+, m/z) calcd for C24H20NO6+ (M + H)+: 418.1285, found: 418.1352.
Trans-2-(benzo[d][1,3]dioxol-5-yl)-3-(4-methoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2a’): A round-bottom flask under an argon atmosphere was charged with 2a (41.7 mg, 0.100 mmol, 1 eq) in dry CH2Cl2 (2 mL), and DBU (5.6 µL, 0.050 mmol, 0.5 eq) was added. The reaction mixture was stirred for 24 h at reflux. The resultant solution was concentrated under reduced pressure, and the residue was purified by preparative TLC to afford the pure product 2a’. Purification: CH2Cl2/MeOH (95/5). White amorphous solid (35.5 mg, 85%), d.r. (trans/cis) >19/1. IR (ν): 3321, 2902, 1717, 1609, 1485, 1246, 1175, 1032, 926, 796, 731 cm−1; 1H NMR (300 MHz, CDCl3): δ (ppm) 8.18 (dd, J = 6.5 Hz, J = 2.7 Hz, 1H), 7.42 (dd, J = 6.5 Hz, J = 5.4 Hz, 2H), 7.22 (dd, J = 5.4 Hz, J = 2.7 Hz, 1H), 7.00 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 1.7 Hz, 1H), 6.76 (dd, J = 8.3 Hz, J = 1.7 Hz, 1H), 6.72 (d, J = 8.6 Hz, 2H), 6.69 (d, J = 8.3 Hz, 1H), 5.90 (d, J = 1.4 Hz, 2H), 5.49 (s, 1H), 3.94 (s, 1H), 3.72 (s, 3H); 13C NMR (75 MHz, CDCl3): δ (ppm) 173.2, 163.6, 159.3, 147.7, 146.5, 136.1, 132.5, 132.4, 130.8, 129.6, 129.3, 128.5, 128.4, 127.6 (2C), 120.3, 114.1 (2C), 108.6, 108.2, 101.4, 64.7, 55.2, 53.4; (E.S.I.+, m/z) calcd for C24H20NO6+ (M + H)+: 418.1285, found: 418.1352.
Cis-2-(benzo[d][1,3]dioxol-5-yl)-3-(4-hydroxy phenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2b): The title compound was prepared following the general procedure described above using the brut 1b (0.100 mmol, 1.0 eq.) and IVa (24.3 mg, 0.150 mmol, 1.5 eq.) in 20 min of reaction time. The product 2b was isolated in 71% yield (28.6 mg, d.r. (cis/trans): 15.5/1) as a beige powder. When the reaction was performed at room temperature in 2 min of reaction time: 29.9 mg, 74%, d.r. (cis/trans): 15.5/1. Mp: 205–206 °C; IR (ν): 3389, 2904, 1717, 1591, 1557, 1513, 1483, 1433, 1249, 1197, 1175, 1034, 927, 786, 703 cm−1; 1H NMR (300 MHz, CD3OD): d (ppm) 8.14 (dd, J = 7.6 Hz, J = 1.1 Hz, 1H), 7.68 (d, J = 7.6 Hz, 1H), 7.58 (td, J = 7.5 Hz, J = 1.1 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.9 Hz, 1H), 6.62 (d, J = 8.9 Hz, 2H), 6.57 (d, J = 8.5 Hz, 2H), 5.94 (d, J = 2.8 Hz, 2H), 5.31 (d, J = 5.9 Hz, 1H), 4.82 (d, J = 5.9 Hz, 1H); 13C NMR (75 MHz, CD3OD): d (ppm) 172.7, 166.3, 158.7, 149.2, 148.2, 136.6, 136.2, 133.8, 130.7 (2C), 130.3, 129.3, 129.2, 128.8, 122.2, 116.4, 116.0 (2C), 109.9, 108.9, 102.9, 67.0, 51.0; HRMS (E.S.I.+, m/z) calcd for C23H18NO6+ (M + H)+: 404.1129, found: 404.1090.
Cis-2-(benzo[d][1,3]dioxol-5-yl)-3-(4-(benzyloxy)phenyl)-1-oxo-1,2,3,4tetrahydroisoquinoline -4-carboxylic acid (2c): The title compound was prepared following the general procedure described above using 1c (33.1 mg, 0.100 mmol, 1.0 eq.) and IVa (24.3 mg, 0.150 mmol, 1.5 eq.) in 45 min of reaction time. The product 2c was isolated in 72% yield (35.5 mg, d.r. (cis/trans): 13/1) as a brown powder. When the reaction was performed at room temperature in 8 min of reaction time: 40.0 mg, 81%, d.r. (cis/trans): 13/1. Mp: 177–178 °C; IR (ν): 3289, 3059, 1715, 1644, 1600, 1504, 1487, 1384, 1246, 1036, 1007, 792, 733, 699 cm−1; 1H NMR (300 MHz, CD3OD): d (ppm) 8.08 (dd, J = 7.7 Hz, J = 1.3 Hz, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.52 (td, J = 7.5 Hz, J = 1.4 Hz, 1H), 7.42–7.26 (m, 6H), 7.01 (d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.8 Hz, 2H), 6.72 (d, J = 7.7 Hz, 1H), 6.66 (s, 1H), 6.63 (d, J = 2.2 Hz, 1H), 5.93 (d, J = 4.9 Hz, 2H), 5.33 (d, J = 5.9 Hz, 1H), 4.97 (s, 2H), 4.61 (d, J = 5.9 Hz, 1H); 13C NMR (75 MHz, CD3OD): d (ppm) 174.2, 165.3, 158.4, 147.6, 146.5, 137.6, 137.3, 135.6, 135.5, 134.6, 132.0, 129.9, 129.4 (2C), 128.8, 128.6, 128.0, 127.3, 127.2, 127.1, 126.4, 120.5, 113.9 (2C), 108.5, 107.4, 101.4, 69.5, 66.5, 52.8; HRMS (E.S.I.+, m/z) calcd for C30H24NO6+ (M + H)+: 494.1598, found: 494.1574.
2-(4-methoxybenzyl)-3-(4-methoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2d): The title compound was prepared following the general procedure described above using 1d (25.5 mg, 0.100 mmol, 1.0 eq.) and IVa (24.3 mg, 0.150 mmol, 1.5 eq.) in 20 min of reaction time. The product 2d was isolated in 63% yield (26.3 mg, d.r. (cis/trans): >19/1) as a light amorphous solid. When the reaction was performed at room temperature in 2 min of reaction time: 40.5 mg, 97%, d.r. (cis/trans): 2.2/1 with separation. Cis product (2d): light amorphous solid (27.8 mg, 67%). Trans product (2d’): light amorphous solid (12.5 mg, 30%). Cis product 2d: IR (ν): 2928, 1726, 1613, 1572, 1511, 1470, 1249, 1176, 1031, 832, 735 cm−1; 1H NMR (300 MHz, CD3OD): d (ppm) 8.00 (dd, J = 7.8 Hz, J = 1.5 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.41–7.20 (m, 3H), 7.11 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 6.77 (d, J = 8.7 Hz, 2H), 6.59 (d, J = 8.7 Hz, 2H), 5.31 (d, J = 14.5 Hz, 1H), 4.88 (d, J = 6.4 Hz, 1H) 4.21 (d, J = 6.4 Hz, 1H), 3.67 (s, 3H), 3.60 (s, 3H), 3.56 (d, J = 14.5 Hz, 1H); 13C NMR (75 MHz, CD3OD): d (ppm) 166.6, 160.9, 160.7, 138.2, 136.1, 133.3, 131.8, 130.6 (2C), 130.5 (2C), 130.2, 129.9, 128.8, 128.4, 127.8, 115.1 (2C), 114.6 (2C), 62.5, 55.7, 55.6, 53.5, 48.9; HRMS (E.S.I.+, m/z) calcd for C25H24NO5+ (M + H)+: 418.1649, found: 418.1667. Trans product 2d’: IR (ν): 2960, 2926, 1728, 1611, 1574, 1510, 1466, 1246, 1174, 1029, 799, 733 cm−1; 1H NMR (300 MHz, CDCl3): d (ppm) 8.16 (dd, J = 6.9 Hz, J = 2.2 Hz, 1H), 7.41–7.31 (m, 3H), 7.10 (d, J = 8.6 Hz, 2H), 7.01 (dd, J = 6.5 Hz, J = 1.8 Hz, 1H), 6.87 (d, J = 8.6 Hz, 2H), 6.67 (d, J = 8.7 Hz, 2H), 6.65 (d, J = 8.7 Hz, 2H), 5.50 (d, J = 14.5 Hz, 1H), 4.98 (s, 1H), 3.75 (s, 1H), 3.66 (s, 3H), 3.63 (s, 3H), 3.56 (d, J = 14.5 Hz, 1H); 13C NMR (75 MHz, CDCl3): d (ppm) 175.0, 163.7, 159.3, 159.1, 132.1, 131.5, 130.2 (2C), 129.4, 129.1, 128.8, 128.6, 128.3, 127.5 (2C), 114.2 (2C), 113.7 (2C), 59.5, 55.2, 55.1, 51.1, 48.2; HRMS (E.S.I.+, m/z) calcd for C25H24NO5+ (M + H)+: 418.1649, found: 418.1609.
Cis-2-(tert-butyl)-3-(4-methoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2e): The title compound was prepared following the general procedure described above using the brut 1e (0.100 mmol, 1.0 eq.) and IVa (24.3 mg, 0.150 mmol, 1.5 eq.) in 40 min of reaction time. The product 2e was isolated in 69% yield (24.4 mg, d.r. (cis/trans): >19/1) as a white powder. When the reaction was performed at room temperature in 2 min of reaction time: 27.9 mg, 79%, d.r. (cis/trans): >19/1. Mp: 60–62 °C; IR (ν): 2923, 2510, 1716, 1653, 1603, 1511, 1390, 1254, 1161, 1025, 788, 756 cm−1; 1H NMR (500 MHz, CD3OD): d (ppm) 8.03 (d, J = 7.7 Hz, 1H), 7.72 (d, J = 7.7 Hz, 1H), 7.41 (dd, J = 7.7 Hz, J = 7.7 Hz, 1H), 7.34 (dd, J = 7.7 Hz, J = 7.7 Hz, 1H), 6.99 (d, J = 8.6 Hz, 2H), 6.67 (d, J = 8.6 Hz, 2H), 5.58 (d, J = 5.6 Hz, 1H), 4.43 (d, J = 5.6 Hz, 1H), 3.70 (s, 3H), 1.52 (s, 9H); 13C NMR (125 MHz, CDCl3): d (ppm) 170.8, 162.3, 144.4, 132.7, 132.0, 130.6 (2C), 130.1, 129.7, 128.0, 127.7, 114.7, 114.3 (2C), 60.7, 60.3, 55.6, 54.6, 29.2 (3C); HRMS (E.S.I.+, m/z) calcd for C21H24NO4+ (M + H)+: 354.1700, found: 354.1745.
Cis-3-isopropyl-1-oxo-2-phenyl-1,2,3,4-tetra hydroisoquinoline-4-carboxylic acid (2f): The title compound was prepared following the general procedure described above using the brut 1f (0.100 mmol, 1.0 eq.) and IVa (24.3 mg, 0.150 mmol, 1.5 eq.) in 40 min of reaction time. The product 2f was isolated in 41% yield (12.8 mg, d.r. (cis/trans): >19/1) as a light amorphous solid. When the reaction was performed at room temperature in 5 min of reaction time: 12.4 mg, 40%, d.r. (cis/trans): >19/1. IR (ν): 3398, 2963, 1710, 1599, 1580, 1552, 1497, 1381, 1261, 1027, 799, 755 cm−1; 1H NMR (300 MHz, CD3OD): d (ppm) 7.96 (dd, J = 7.5 Hz, J = 1.5 Hz, 2H), 7.58 (dd, J = 8.1 Hz, J = 1.5 Hz, 2H), 7.52 (dd, J = 7.5 Hz, J = 1.3 Hz, 1H), 7.5 (t, J = 7.5 Hz, 2H), 7.37 (dd, J = 8.1 Hz, J = 1.3 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 4.55 (dd, J = 5.6 Hz, J = 3.3 Hz, 1H), 4.50 (d, J = 5.6 Hz, 1H), 3.66 (b-s, 1H, OH), 2.27 (ddd, J = 7.1 Hz, J = 7.1 Hz, J = 3.3 Hz, 1H), 0.73 (d, J = 7.1 Hz, 3H), 0.68 (d, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CD3OD): d (ppm) 176.6, 166.6, 144.8, 140.0, 133.2, 130.9, 129.7 (2C), 129.2, 128.8 (2C), 128.5, 127.7, 127.6, 68.6, 52.5, 32.5, 23.0, 19.8; HRMS (E.S.I.+, m/z) calcd for C19H20NO3+ (M + H)+: 310.1438, found: 310.1452.
Cis-2-cyclopropyl-1-oxo-3-(thiophen-2-yl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2g): A round-bottom flask under an argon atmosphere was charged with the brut 1g (0.100 mmol, 1.0 eq.) in TFE (3 mL) and then charged with homophthalic anhydride IVa (24.3 mg, 0.150 mmol, 1.5 eq.). The reaction mixture was stirred for 8 min at room temperature. The mixture was concentrated in vacuo, and the residue was purified by preparative TLC to afford the pure products 2g and 2g’. Purification: CH2Cl2/MeOH (93:7). The product 2g (cis product) was isolated in 58% yield (18.2 mg, white powder). The product 2g’ (trans product) was isolated in 32% yield (10.1 mg, white powder). Cis product 2g: Mp: 228–229 °C; IR (ν): 3428, 3075, 3011, 1714, 1642, 1598, 1462, 1423, 1359, 1301, 1244, 1226, 1027, 907, 827, 697 cm−1; 1H NMR (500 MHz, CD3OD): d (ppm) 8.09 (d, J = 7.6 Hz, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.55 (t, J = 7.6 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 4.7 Hz, 1H), 6.89 (d, J = 2.7 Hz, 1H), 6.83 (dd, J = 4.7 Hz, J = 2.7 Hz, 1H), 5.50 (d, J = 5.7 Hz, 1H), 4.55 (d, J = 5.7 Hz, 1H), 2.68–2.63 (m, 1H), 1.06–1.01 (m, 1H), 0.91–0.85 (m, 2H), 0.85–0.80 (m, 1H); 13C NMR (125 MHz, CD3OD): d (ppm) 173.6, 168.2, 141.6, 136.7, 133.6, 130.2, 130.0, 128.7, 128.4, 128.3, 126.9, 126.4, 61.6, 51.7, 30.8, 9.7, 6.4; HRMS (E.S.I.+, m/z) calcd for C17H16NO3S+ (M + H)+: 314.0845, found: 314.0853. Trans product 2g’: Mp: 238–239 °C; IR (ν): 3395, 3076, 3011, 1709, 1638, 1598, 1580, 1465, 1432, 1359, 1245, 1156, 1028, 966, 826, 728, 698 cm−1; 1H NMR (500 MHz, CD3OD): d (ppm) 8.00 (d, J = 7.6 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 4.8 Hz, 1H), 6.87 (d, J = 2.8 Hz, 1H), 6.85 (dd, J = 4.8 Hz, J = 2.8 Hz, 1H), 5.68 (s, 1H), 3.93 (s, 1H), 2.83–2.78 (m, 1H), 1.05–0.97 (m, 1H), 0.88–0.82 (m, 2H), 0.82–0.76 (m, 1H); 13C NMR (125 MHz, CD3OD): d (ppm) 176.0, 167.9, 145.8, 138.2, 133.4, 131.1, 130.0, 128.3 (2C), 127.4, 126.1, 125.5, 62.2, 55.5, 31.0, 9.6, 6.5; HRMS (E.S.I.+, m/z) calcd for C17H16NO3S+ (M + H)+: 314.0845, found: 314.0839.
Trans-2-butyl-3-(4-nitrophenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2h): A round-bottom flask under an argon atmosphere was charged with the brut 1h (0.100 mmol, 1.0 eq.) in TFE (3 mL) and then charged with homophthalic anhydride IVa (24.3 mg, 0.150 mmol, 1.5 eq.). The reaction mixture was stirred for 2 min at room temperature. The mixture was concentrated in vacuo, and residue was purified by preparative TLC to afford the pure products 2h and 2h’. Purification: CH2Cl2/MeOH (93:7). The product 2h (trans product) was isolated in 48% yield (17.5 mg, white powder). The product 2h’ (cis product) was isolated in 29% yield (10.8 mg, white powder). Trans product 2h: Mp: 223–224 °C; IR (ν): 3415, 2959, 2932, 2871, 1708, 1636, 1599, 1518, 1472, 1344, 1263, 1164, 1109, 908, 853, 712 cm−1; 1H NMR (300 MHz, CD3OD): d (ppm) 8.09 (d, J = 8.8 Hz, 2H), 8.00 (dd, J = 7.1 Hz, J = 2.1 Hz, 1H), 7.42–7.32 (m, 2H), 7.38 (d, J = 8.8 Hz, 2H), 7.11 (dd, J = 6.5 Hz, J = 2.1 Hz, 1H), 5.59 (d, J = 1.0 Hz, 1H), 4.04 (dt, J = 13.3 Hz, J = 8.0 Hz, 1H), 3.86 (d, J = 1.0 Hz, 1H), 2.96 (ddd, J = 13.3 Hz, J = 8.0 Hz, J = 6.5 Hz, 1H), 1.72–1.61 (m, 2H), 1.41–1.33 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CD3OD): d (ppm) 174.4, 165.0, 148.0, 147.2, 135.5, 131.7, 129.1, 128.6, 127.0 (3C), 126.6, 123.1 (2C), 62.5, 53.8, 46.6, 29.4, 19.8, 12.6; HRMS (E.S.I.+, m/z) calcd for C20H21N2O5+ (M + H)+: 369.1445, found: 369.1440. Cis product 2h’: Mp: 232–233 °C; IR (ν): 3386, 2958, 2931, 2872, 1727, 1633, 1598, 1519, 1471, 1377, 1345, 1313, 1255, 1163, 1110, 1014, 909, 855, 799, 729, 715 cm−1; 1H NMR (500 MHz, CD3OD): d (ppm) 8.08 (d, J = 7.4 Hz, 1H), 8.03 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 7.4 Hz, 1H), 7.49 (dd, J = 7.4 Hz, J = 7.4 Hz, 1H), 7.41 (dd, J = 7.4 Hz, J = 7.4 Hz, 1H), 7.32 (d, J = 8.3 Hz, 2H), 5.29 (d, J = 6.2 Hz, 1H), 4.55 (d, J = 6.2 Hz, 1H), 4.02 (ddd, J = 14.4 Hz, J = 8.0 Hz, J = 8.0 Hz, 1H), 2.91 (ddd, J = 14.4 Hz, J = 8.0 Hz, J = 5.5 Hz, 1H), 1.71–1.59 (m, 2H), 1.44–1.35 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CD3OD): d (ppm) 175.5, 166.4, 149.0, 146.9, 137.3, 133.4, 130.5 (2C), 130.1, 130.0, 128.4, 128.1, 124.0 (2C), 63.6, 53.6, 47.6, 31.2, 21.2, 14.1; HRMS (E.S.I.+, m/z) C20H21N2O5+ (M + H)+: 369.1445, found: 369.1447.
Cis-3-(4-methoxyphenyl)-1-oxo-2-(4-(trifluoromethyl)phenyl)-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2i): A round-bottom flask under an argon atmosphere was charged with the brut 1i (0.100 mmol, 1.0 eq.) in TFE (3 mL) and then charged with homophthalic anhydride IVa (24.3 mg, 0.150 mmol, 1.5 eq.). The reaction mixture was stirred for 8 min at room temperature. The mixture was concentrated in vacuo, and the residue was purified by preparative TLC to afford the pure product 2i. Purification: CH2Cl2/MeOH (93:7). The product 2i was isolated in 80% yield (28.3 mg, d.r. (cis/trans): 5/1) as a white powder. Mp: 238–239 °C; IR (ν): 3427, 3075, 2935, 2839, 1719, 1648, 1603, 1513, 1460, 1391, 1322, 1249, 1162, 1115, 1060, 1018, 930, 734, 693 cm−1; 1H NMR (300 MHz, CD3OD): d (ppm) 8.13 (dd, J = 7.7 Hz, J = 1.1 Hz, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.60 (d, J = 8.3 Hz, 2H), 7.57 (ddd, J = 7.7 Hz, J = 7.7 Hz, J = 1.1 Hz, 1H), 7.45 (dd, J = 7.7 Hz, J = 7.7 Hz, 1H), 7.42 (d, J = 8.3 Hz, 2H), 7.05 (d, J = 8.8 Hz, 2H), 6.69 (d, J = 8.8 Hz, 2H), 5.52 (d, J = 5.6 Hz, 2H), 4.64 (d, J = 5.6 Hz, 1H), 3.69 (s, 3H); 13C NMR (75 MHz, CD3OD): d (ppm) 174.6, 160.9, 146.6, 138.3, 136.2, 133.8, 132.4, 131.5, 130.8 (2C), 130.4, 130.0, 129.6 (2C), 129.0, 128.3, 126.8, 126.7, 114.6 (2C), 66.8, 55.6, 53.4; 19F NMR (280 MHz, CD3OD): d (ppm) -63.96 (s, CF3); HRMS (E.S.I.+, m/z) calcd for C24H19F3NO4+ (M + H)+: 442.1261, found: 442.1253.
Trans-3-(ethoxycarbonyl)-2-(4-methoxyphenyl)-1-oxo-1,2,3,4-tetrahydroisoquinoline-4-carboxylic acid (2j): A round-bottom flask under an argon atmosphere was charged with the brut 1j (0.100 mmol, 1.0 eq.) in TFE (3 mL) and then charged with homophthalic anhydride IVa (24.3 mg, 0.150 mmol, 1.5 eq.). The reaction mixture was stirred 8 min at room temperature. The mixture was concentrated in vacuo, and residue was purified by preparative TLC to afford the pure product 2j. Purification: CH2Cl2/MeOH (93:7). The product 2j was isolated in 68% yield (25.0 mg, d.r. (trans/cis): >19/1) as a brown powder. Mp: 224–225 °C; IR (ν): 3386, 2960, 2936, 1738, 1634, 1599, 1509, 1462, 1429, 1364, 1299, 1241, 1178, 1026, 912, 834, 711 cm−1; 1H NMR (300 MHz, CD3OD): d (ppm) 8.00 (dd, J = 7.5 Hz, J = 1.2 Hz, 1H), 7.75 (ddd, J = 7.5 Hz, J = 7.5 Hz, J = 1.4 Hz, 1H), 7.44 (d, J = 8.9 Hz, 2H), 7.46–7.40 (m, 2H), 6.98 (d, J = 8.9 Hz, 2H), 5.20 (d, J = 1.7 Hz, 1H), 4.24 (d, J = 1.7 Hz, 1H), 4.08 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 1.11 (t, J = 7.1 Hz, 3H); 13C NMR (175 MHz, CD3OD): d (ppm) 175.8, 172.7, 166.6, 160.2, 138.6, 136.6, 133.2, 130.5, 129.9, 129.7 (2C), 128.6, 128.2, 115.1 (2C), 67.8, 62.7, 55.9, 51.7, 14.3; HRMS (E.S.I.+, m/z) calcd for C20H20NO6+ (M + H)+: 370.1285, found: 370.1283.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
