1. Introduction
The lipophilicity of compounds allows for the prediction of a compound’s fate in living organisms and indicates the types of transport and accumulation of the drug in the body. Lipophilicity is useful as an essential property of drugs at the time of their design so as to obtain the optimal properties required to achieve a molecular target [
1,
2]. The knowledge of this parameter is extremely important in metabolic transformations with the participation of bioactive molecules and their affinity for the protein target. Lipophilicity is believed to regulate the transport of a biologically active substance in its environment. Therefore, optimization of lipophilicity allows us to find the optimal drug structure in terms of quantification, structure-activity relationship studies (QSAR) [
3,
4,
5].
The definition of IUPAC shows lipophilicity as the affinity a molecule or moiety has for a lipophilic or non-polar environment [
6]. Additionally, lipophilicity is one of the fundamental properties of compounds required to assess absorption, distribution, metabolism, and elimination (ADME parameters) in biological systems, in addition to their solubility, stability, and acid-base nature (
Figure 1). Before the molecule reaches its pharmacological target, the lipophilicity of a compound indicates that the structure is similar to its lipophilic environment, allowing it to be transported across protein–lipid membranes into the biological system, forming complexes between the compound and the receptor binding site [
7,
8].
Lipophilicity also belongs to one of the factors determining the bioavailability of the drug in Lipinski’s, Ghose’s, and Veber’s rules [
9,
10,
11,
12].
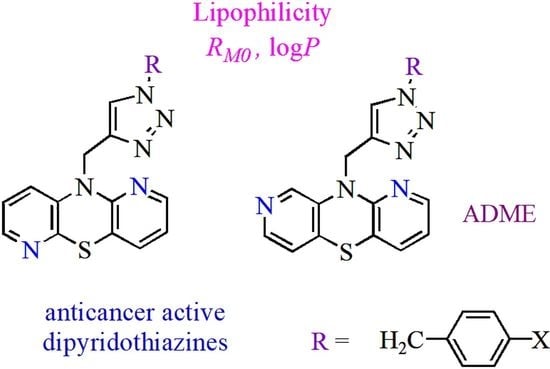
Dipyridothiazines are modified phenothiazine structures into which two pyridine rings have been introduced instead of two benzene rings [
13]. In recent years, significant and highly promising anticancer activities of these heterocyclic systems have been proven [
14,
15,
16,
17]. Additionally, selected derivatives of this group showed immunomodulatory and antioxidant potential [
18,
19]. The biological activity of selected dipyridothiazines has been shown to depend on lipophilicity and in some way correlates with ADME parameters [
20,
21,
22].
Recently, the synthesis of dipyridothiazine derivatives with 1,2,3-triazole substituents (these being 1,2,3-triazole-dipyridothiazine hybrids) and their promising anticancer activities have been published [
23]. These compounds showed in vitro anticancer activity against cancer cell lines: glioblastoma SNB-19, colorectal carcinoma Caco-2, lung cancer A549, and breast cancer MDA-MB231. In our research, dipyridothiazine hybrids were divided into two batches: the first containing 2,7- and 3,6-diazaphenothiazines in their structure, and the second containing 1,6- and 1,8-diazaphenothiazines in their structure. Thorough tests of lipophilicity and ADME parameters were performed for both groups. The results of the first part of the study show the influence of the above parameters on activity [
24].
The results presented in this paper are a continuation of previous research [
24] focused on 1,6- and 1,8-diazaphenothiazine derivatives. We investigated the lipophilicity of two series of 1,2,3-triazole-1,6-diazaphenothiazine (
1–
5) and 1,2,3-triazole-1,8-diazaphenothiazine (
6–
10) hybrids by RP TLC methodology, calculated programs, and studying the established relationships between their lipophilicity and ADME properties. The structures of the investigated compounds are presented in
Figure 2. The lipophilicity was studied with the intention that it would provide a better insight into the differences in biological activity and also to deeper trace the influence of lipophilicity in reaching a molecular target.
2. Results
In the first stage of the study, eleven popular computer programs (VCCLAB and SwissADME [
25,
26,
27,
28]) based on different algorithms were used. The log
Pcalcd values for the substituted dipirydothiazine-1,2,3-triazole hybrids
1–
10 were different depending on the substituents in 1,2,3-triazole rings, places of nitrogen atoms in the dipyridothiazine system, and on the program used. The log
Pcalcd values varied significantly from 2.08 to 5.09 (
Table 1). The highest lipophilicity in the group of 1,6-diaphenothiazine derivatives was demonstrated according to the ALOGP module for compound
3 (log
Pcalcd = 5.09) with a
p-chlorophenyl substituent in its structure. On the other hand, the lowest lipophilicity in this group was calculated for derivative
4 with a
p-cyanobenzyl (log
Pcalcd = 2.08) according to the MLOGP program. Both of these programs predicted similar results in the 1,8-diazphenothiazine group, where the highest lipophilicity characterized compound
8 with a
p-chlorobenzyl (log
Pcalcd = 4.55), and the lowest derivative
9 with a
p-cyanobenzyl (log
Pcalcd = 2.08).
In further research, in order to obtain reliable values, the relative lipophilicities of derivatives 1–10 expressed by the chromatographic values of RM0 were measured by the experimental RP-TLC method.
The experimental RP TLC method provided the retention parameter
RM (calculated from the R
F values) using the following equation:
The values of
RM decreased linearly, with an increasing concentration of acetone in the mobile phase (
r = 0.9885–0.9981). The extrapolation to 0% concentration of acetone gave the relative lipophilicity parameter (
RM0) values, which showed the partitioning between the non-polar stationary and polar mobile phases, using the equation:
where C is the concentration of acetone. The
RM0 values were found to be within the range of 1.975–2.701 (
Table 2).
The presented 1,2,3-triazole and dipyridothiazine hybrid derivatives
1–
10 belong to another group of isomeric dipyridothiazines of structure 1,6- and 1,8-diazaphenothiazines. Therefore, they are isomers of the hybrids described above [
24]. Structurally, they differ only in the location of nitrogen atoms in the azaphenothiazine core. These compounds do not show substantial differences in molecular descriptors, nevertheless the ADME parameters are substantially different (
Table 3 and
Table 4). All tested derivatives meet the requirements of Lipinski’s rule of five as well as the rules of Ghose and Veber [
27] (
Table 3).
In order to determine the pharmacokinetic properties of the tested group of compounds, the PreADMET server was used to calculate the following parameters: BBB, Caco-2, HIA, MDCK, PPB and SP (
Table 4) [
29]. Caco-2 and MDCK (Madin-Darby dog kidney) cell models have been calculated and are recommended as highly reliable in vitro models for predicting oral drug absorption. Another in silico human intestinal absorption (HIA) and skin permeability (SP) model predicts and identifies potential drugs for oral and transdermal administration. The parameter BBB (blood–brain barrier penetration) informs about the possibility of the compound acting in the central nervous system, and the PPB model (binding plasma proteins) indicates the binding efficiency [
30,
31]. These studies also used prothipendyl, a weak centrally acting neuroleptic, as the reference compound. The values of the
RM0 parameter were correlated with molecular descriptors and ADME activities (
Table 5)
Then a calibration curve was created using analogous measuring conditions. The set of reference substances
A–
E with literature values of log
Plit were used in the range of 1.21–3.54 (
Table 6). This curve made it possible to convert the values of the relative lipophilicity parameter
RM0 of the tested hybrids into the value of the absolute lipophilicity parameter log
PTLC.
The log
PTLC values for all new anticancer hybrids (
1–
10) are collected in
Table 7.
3. Discussion
This work focuses on the assessment of the lipophilicity of new, anticancer active dipyridothiazines linked to the 1,2,3-triazole ring (
1–
10), which are recognized in chemical literature as hybrids of both heterocycles. Two series of dipyridothiazines (1,6- and 1,8-diazaphenothiazines) contain a 1,2,3-triazole ring in which various benzyl substituents and a phenylthiomethyl substituent have been introduced (
Figure 2).
These compounds showed promising anticancer activity in vitro against the tumor cell lines SNB-19 glioblastoma, Caco-2 colorectal carcinoma, A549 lung carcinoma and MDA-MB231 breast cancer, and low cytotoxicity against NHDF normal human fibroblasts. This group included derivatives
3 and
8 with
p-chlorobenzyl substituents that showed highly promising activities against Caco-2, MDA-MB231 and A549 (IC
50 in the range of 0.25–0.51 μM) [
23]. The most active derivative,
3, was analyzed for the expression of genes influencing the neoplastic process (
H3,
TP53,
CDKN1A,
BCL-2 and
BAX). These studies have shown the activation of the mitochondrial apoptosis pathway and disruptions in the proper formation of DNA histones [
23].
We started our research with in silico lipophilicity calculations using the available VCCLAB and SwissADME internet servers. The calculated lipophilicity within these modules varies greatly, which is most likely related to the different mathematical models used to calculate it.
The most lipophilic compound was derivative
3 (log
Pcalcd = 5.09), but the isomeric compound
10 (log
Pcalcd = 4.55) was slightly less lipophilic, both with a
p-chlorobenzyl substituent at the triazole ring. The least lipophilic compounds were compound
4 and
9 (log
Pcalcd = 2.08), which are isomers and contain a
p-cyanobenzyl substituent in their structure. The results of these measurements are summarized in
Table 1, and the graphical visualization of the calculated log
P values of each compound is shown in
Figure 3 and
Figure 4. In the studies, large differences of over two units were observed for each compound. The most inflated results for the studied group of derivatives were indicated by the ALOGP program. Such large discrepancies in results were observed in our previous studies related to 2,7-diaza- and 3,6-diazaphenothiazines derivatives [
22,
23,
34]. It is also an indication of the need to perform experimental measurements in order to correctly and accurately determine the lipophilicity parameter.
In the next stage of the research, we started to determine the relative lipophilicity parameter of RM0 according to the procedure described in chapters two and four. The highest relative value of lipophilicity RM0 was characteristic for compound 3 (with a p-chlorobenzyl substituent in the 1,2,3-triazole ring and in 1,6-diazaphenothiazine) (RM0 =2.872). Interestingly, isomer 8 (1,8-diazaphenothiazin) showed lower lipophilicity (RM0 =2.464). It should be noted that in the 1,8-diazaphenothiazines group this compound was the most lipophilic among all derivatives. Compound 9 (with a p-cyanobenzyl substituent) from the 1,8-diazaphenothiazine series was characterized by the least lipophilic character.
It can be seen that all the isomeric 1,8-derivatives
6–
10 exhibit substantially lower relative lipophilicity parameters (
Table 2).
The interdependence between the relative lipophilicity parameter
RM0 and the specific hydrophobic surface
b for all compounds
1–
10 is given by the equation:
This relationship indicated the existence of structurally expected congeneric subgroups:
the 1,6-diazaphenothiazine derivatives 1–5 RM0 = −58.614b + 0.4567 r = 0.9741.
the 1,8-diazaphenothiazine derivatives 6–10 RM0 = −98.997b − 1.0412 r = 0.9781.
These relationships are closely related to the location of nitrogen atoms in the dipyridothiazine system. Similar situations were previously observed for hybrids of isomeric 2,7- and 3,6-diazaphenothiazines [
24].
A calibration curve was performed to determine the absolute lipophilicity parameter log
P. The standard substances were compounds with the known log
P parameter: acetanilide, acetophenone, 4-bromoacetophenone, benzophenone, and antracene for which in the literature, log
Plit values are in the range 1.21–5.53 (
Table 6) [
32,
33].
The relative lipophilicity parameter RM0 for the reference substance was determined under the same conditions as for hybrids 1–10.
The standard curve equation is as follows:
logPTLC = 0.9862RM0 + 0.1957 (r = 0.9949, s = 0.2246, F = 359.97, p = 0.0002)
On the basis of the calibration curve, the absolute
logPTLC parameter of all tested compounds
1–
10 was determined. They fall within the scope of: 2.159–3.027 (
Table 7).
Compound 3 was characterized by the highest lipophilicity, and the lowest for hybrid 6. In the 1,6-diazaphenothiazines group, derivative 3 was the most lipophilic, whereas compound 4 was the least lipophilic. In the 1,8-diazaphenothiazines group, derivative 8 showed the highest lipophilicity and compound 9 the lowest. On this basis, it is noted that the p-chlorobenzyl substituent in both isomers increases the lipophilicity and the p-cyanobenzyl substituent lowers the lipophilicity.
Comparing the lipophilicity of the described 1,6- and 1,8-diazaphenothiazine derivatives
1–
10 with the previously described group of 2,7- and 3,6-diazphenothiazine hybrids is illustrated in
Figure 5. It can be noticed that the 2,7-diazaphenothiazine derivatives were the least lipophilic group of all isomers. Their lipophilicity was in the range of 1.408–2.569 [
24]. It can be observed that the isomeric 1,6-diazaphenothiazines were characterized by the highest lipophilicity. It should be noted that in the group of tested compounds, the highest anticancer activity was demonstrated by the 1,6-diazaphenothiazine hybrid with a triazo ring and
p-chlorobenzyl substituent
3 [
23]. When these facts are compared with those of other isomeric hybrids, it can be assumed that this type of activity was not determined by lipophilicity.
Analysis of ADME parameters of compounds
1–
10 compared with the reference compound
11 showed interesting information (
Table 4). The tested compounds have BBB indices in the range of 0.352–2.156 which are substantially lower than those of reference compound
11 (3.103), which may indicate poor migration across the blood–brain barrier and low neurotoxicity. The permeability of Caco-2 cells was different among the tested derivatives. Compounds
1,
2,
4,
6 and
7 have a comparable affinity to reference compound
11. However, derivatives
3,
5,
8,
9 and
10 were characterized by substantially higher indexes, which may indicate their stronger cellular affinity. All tested compounds exhibited a high HIA index, which was in the range of 97–99. The permeability of MDCK cells was variable and ranged from 1.78–48.87. Derivatives
1–
5 exhibited lower parameters than derivatives
6–
10. The PPB parameter for the tested group of compounds is substantially higher than for the reference compound, which may indicate an increased ability to bind to plasma proteins. All the tested derivatives showed a poor SP index, which was comparable to the reference compound. The calculated ADME parameters showed the similarity of the tested derivatives to the drug substance.
In our research, we made attempts to correlate the relative lipophilicity parameter R
M0 with molecular descriptors and ADME parameters (
Table 5). These correlations showed moderate r values in the range of 0.3265–0.6892. These results may suggest that lipophilicity is one of the many factors directly influencing biological activity. Additionally, they may indicate that lipophilicity depends on the conformation of molecules, their ionic interactions or van der Walls interactions.
Moreover, all tested derivatives meet the requirements of Lipinski’s rule of five as well as the rules of Ghose and Veber, which point out that derivatives can become a drug with the ability for orally active use. The presented results are promising and encourage further continuation.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}