
1H-Pyrazolo[3,4-b]pyridines: Synthesis and Biomedical Applications

 , ,
, ,  and
and
Abstract
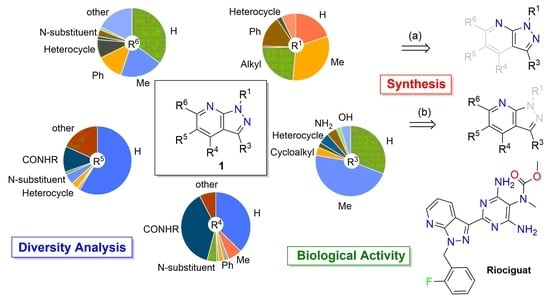
:
1. Introduction
- (a)
- The review will cover only those compounds that present a fully unsaturated pyridine ring, not taking into account other degrees of unsaturation. Nevertheless, the presence of hydroxy groups at C4 or C6 will be covered, and consequently, the corresponding pyridone tautomers will be included due to the greater stability of the 4-oxo or 6-oxo derivatives.
- (b)
- In the case of pyrazolo[3,4-b]pyridines not substituted at the nitrogen atoms of the pyrazole ring, two tautomeric forms are possible: the 1H- (1, R1 = H) and the 2H-pyrazolo[3,4-b]pyridine (2, R2 = H) (Figure 2). Although such tautomerism could complicate the diversity analysis of such compounds, the AM1 calculations of Alkorta and Elguero clearly showed the greater stability of the 1H-tautomer by a difference of 37.03 kJ/mol (almost 9 kcal/mol) [9].
- (c)
- To the best of our knowledge, there are only five specific reviews on pyrazolopyridines prior to this review. Three of them are devoted to the synthesis of such compounds [6,7,8], the most recent being from 2012 [6]. The other two cover biological aspects, either from a general perspective (a review from 1985, [16]) or a very specific point of view (kinase inhibitors, 2013 [17]). Furthermore, 5591 references cover the 300,000 1H-pyrazolo[3,4-b]pyridines included in SciFinder, but 3005 of them (almost 54%) are from 2012 or later (1413 being patents).
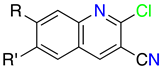
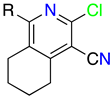
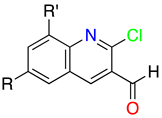
2. Structural Features of 1H-Pyrazolo[3,4-b]pyridines: Substitution Patterns
2.1. Substitution Pattern at N1
2.2. Substitution Pattern at C3

2.3. Substitution Pattern at C4, C5, and C6
| R4 | Structures 1 (%) | Number of References | Selected References |
|---|---|---|---|
| H | 37.33 | 3003 | [37,38] |
| Me | 6.59 | 389 | [18,27] |
| Ph | 2.29 | 660 | [10,39] |
| Heterocycle | 2.42 | 355 | [27,40] |
| OH | 0.89 | 312 | [41,42] |
| N-substituent | 4.54 | 980 | [43,44] |
| CONHR | 38.30 | 148 | [29,45] |
| Other | 7.63 | - | - |
| R5 | Structures 1 (%) | Number of References | Selected References |
|---|---|---|---|
| H | 58.61 | 2753 | [26,34] |
| Me | 0.84 | 87 | [46,47] |
| Ph | 0.66 | 211 | [19,48] |
| Heterocycle | 3.18 | 349 | [49,50] |
| N-substituent | 4.90 | 231 | [51,52] |
| Halogen | 1.34 | 583 | [18,19] |
| CONHR | 12.13 | 430 | [53,54] |
| Other | 18.34 | - | - |
| R6 | Structures 1 (%) | Number of References | Selected References |
|---|---|---|---|
| H | 35.10 | 3270 | [19,55] |
| Me | 20.15 | 679 | [18,27] |
| Ph | 12.44 | 509 | [33,56] |
| Heterocycle | 8.65 | 300 | [57,58] |
| OH | 1.52 | 334 | [59,60] |
| N-substituent | 3.22 | 565 | [61,62] |
| Carbonyl group | 1.27 | 92 | [63,64] |
| Other | 17.64 | - | - |
3. Synthetic Approaches to 1H-Pyrazolo[3,4-b]pyridines
3.1. Pyridine Formation onto a Preexisting Pyrazole Ring
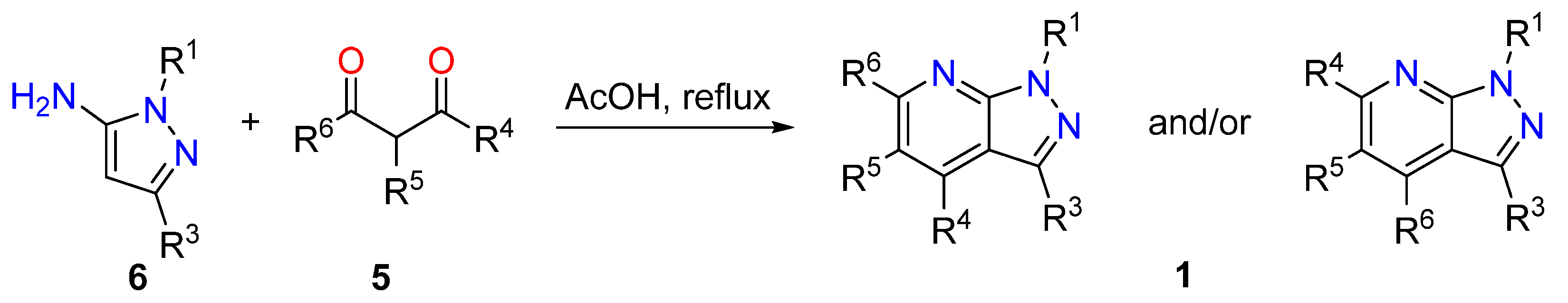
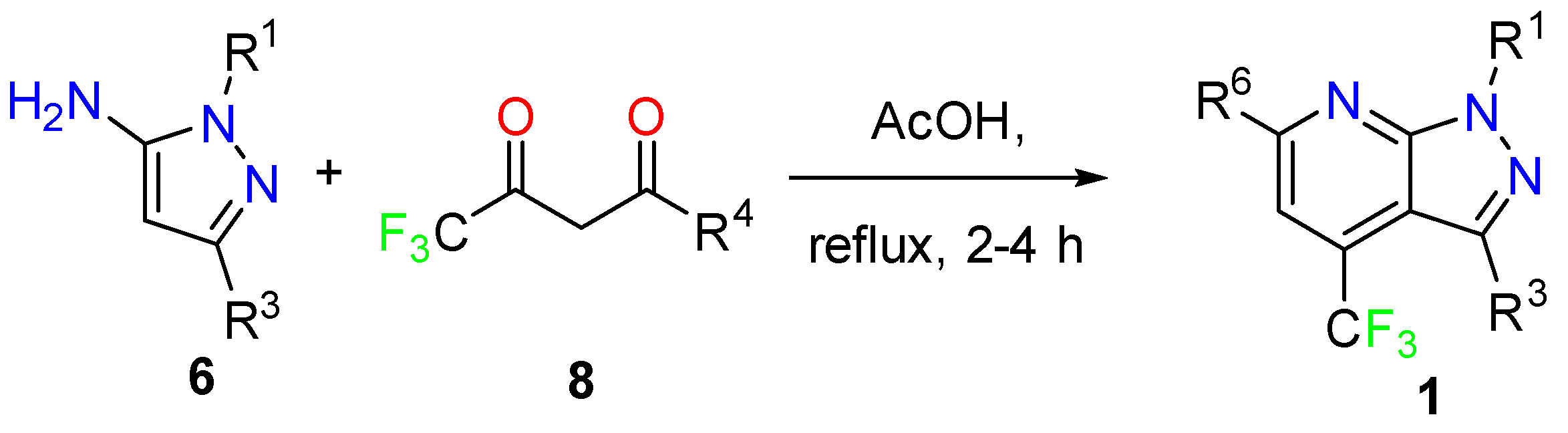
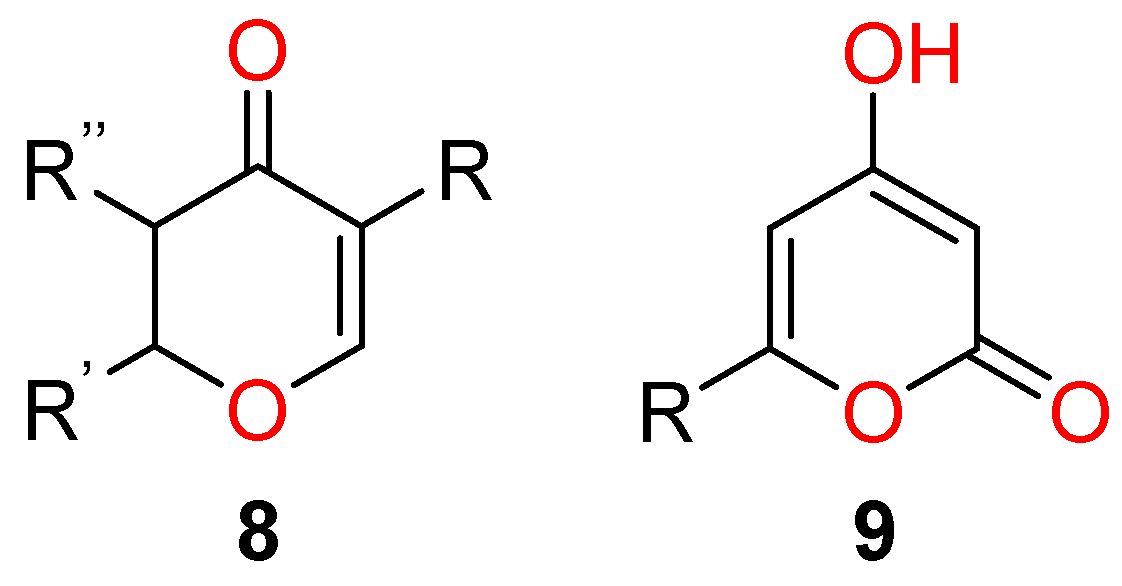
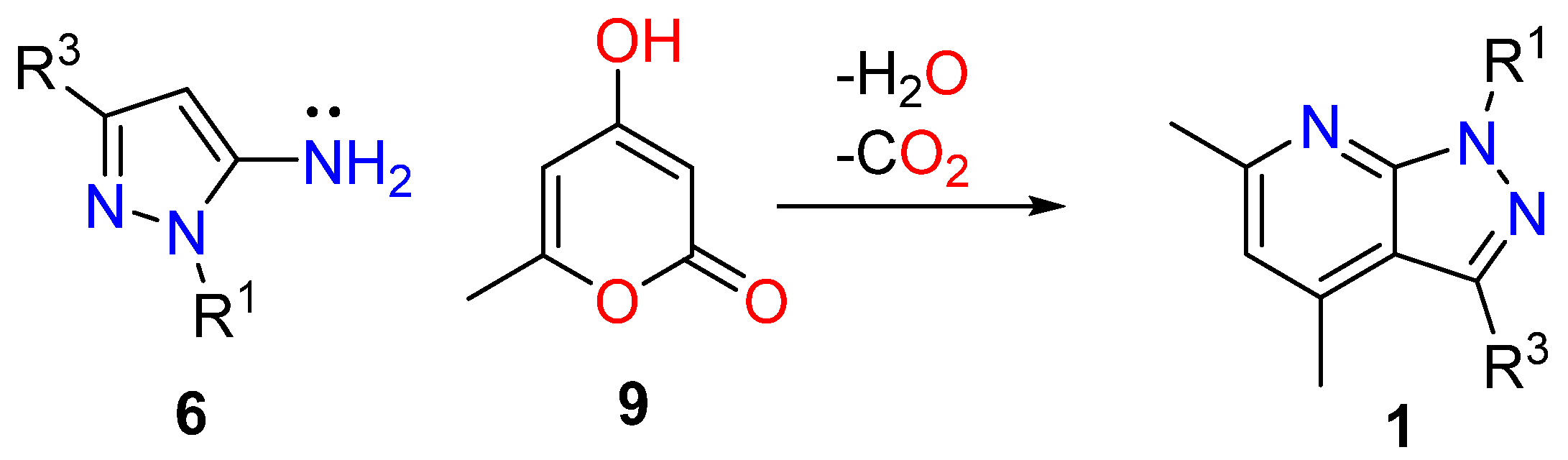
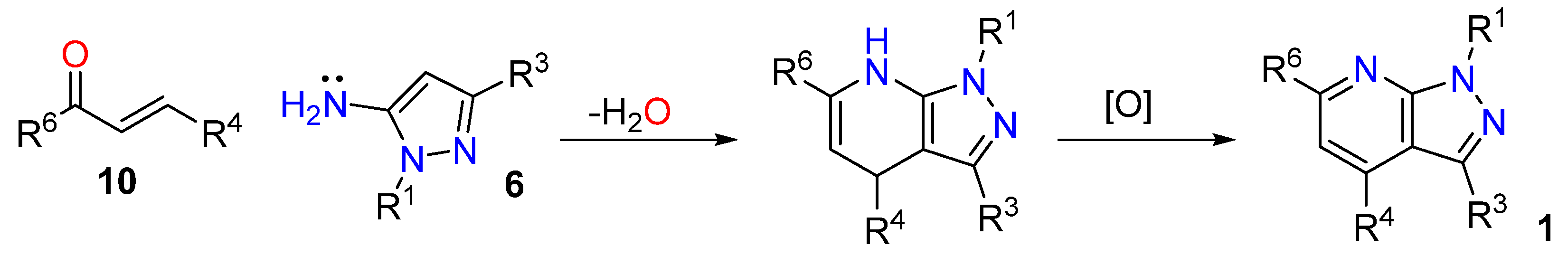
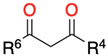
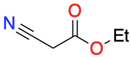
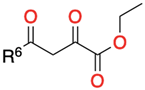
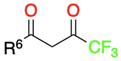
3.1.1. 1,3-Dicarbonyl Compounds and Derivatives as 1,3-CCC-Biselectrophiles
3.1.2. Michael Acceptors Used as 1,3-CCC-Biselectrophiles
3.1.3. Diethyl 2-(Ethoxymethylene)malonate as 1,3-CCC-Biselectrophile (Gould–Jacobs Reaction)
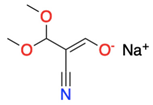
3.1.4. In Situ Formation of the 1,3-CCC-Biselectrophiles or the 1,3-NCC-Dinucleophiles
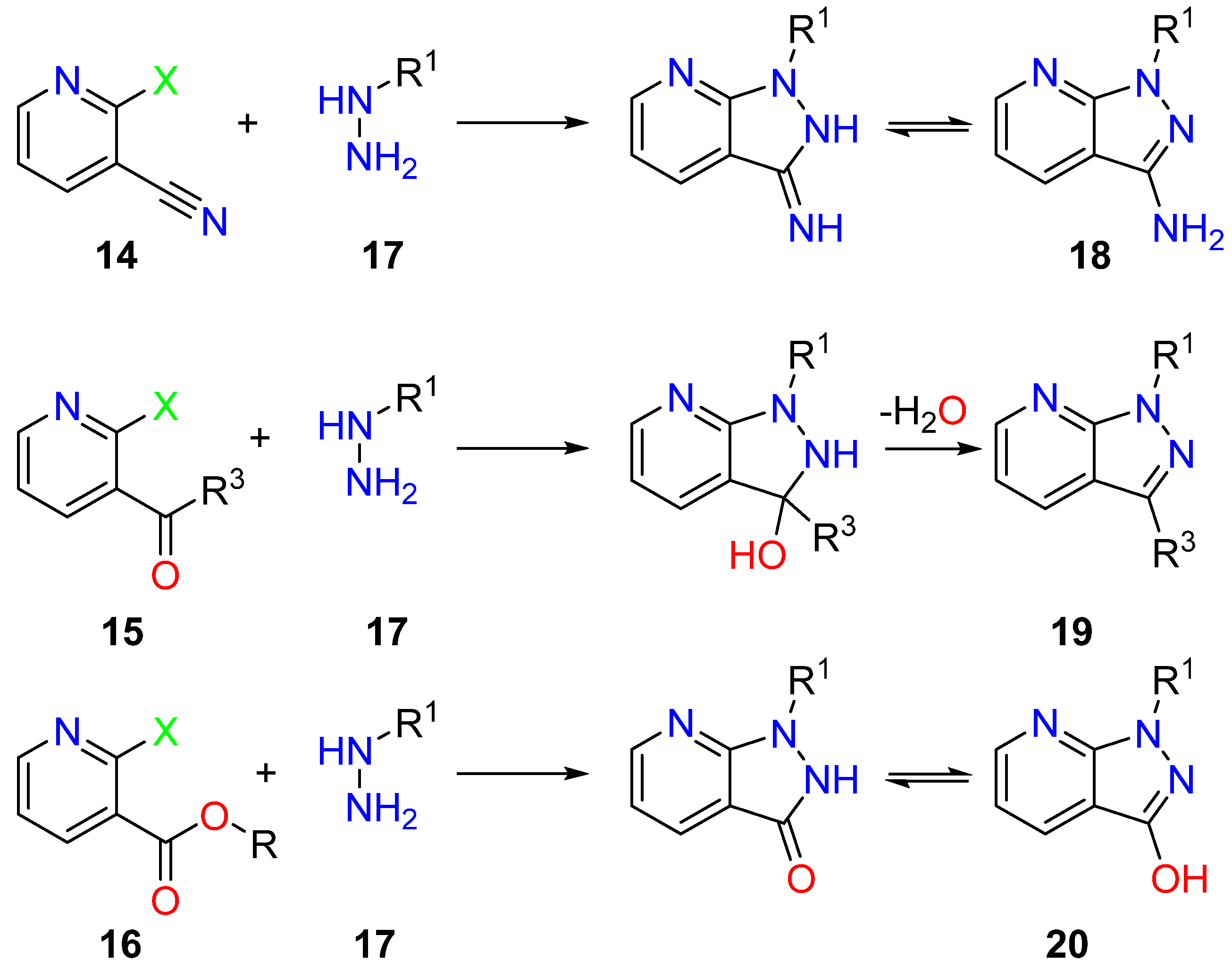
3.2. Pyrazole Formation onto a Preexisting Pyridine Ring
3.3. Other Reactions
4. Biomedical Applications of 1H-Pyrazolo[3,4-b]pyridines
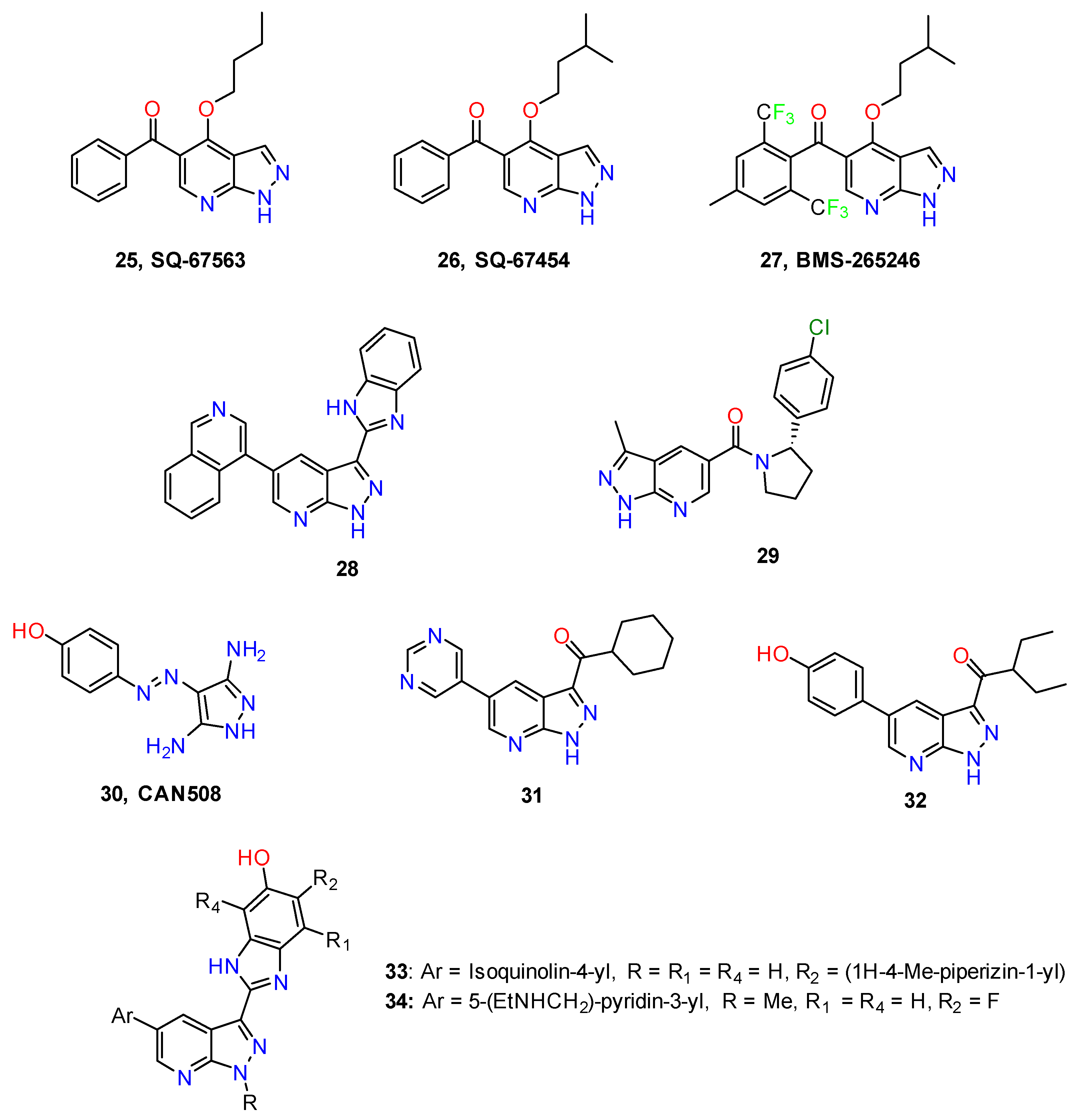
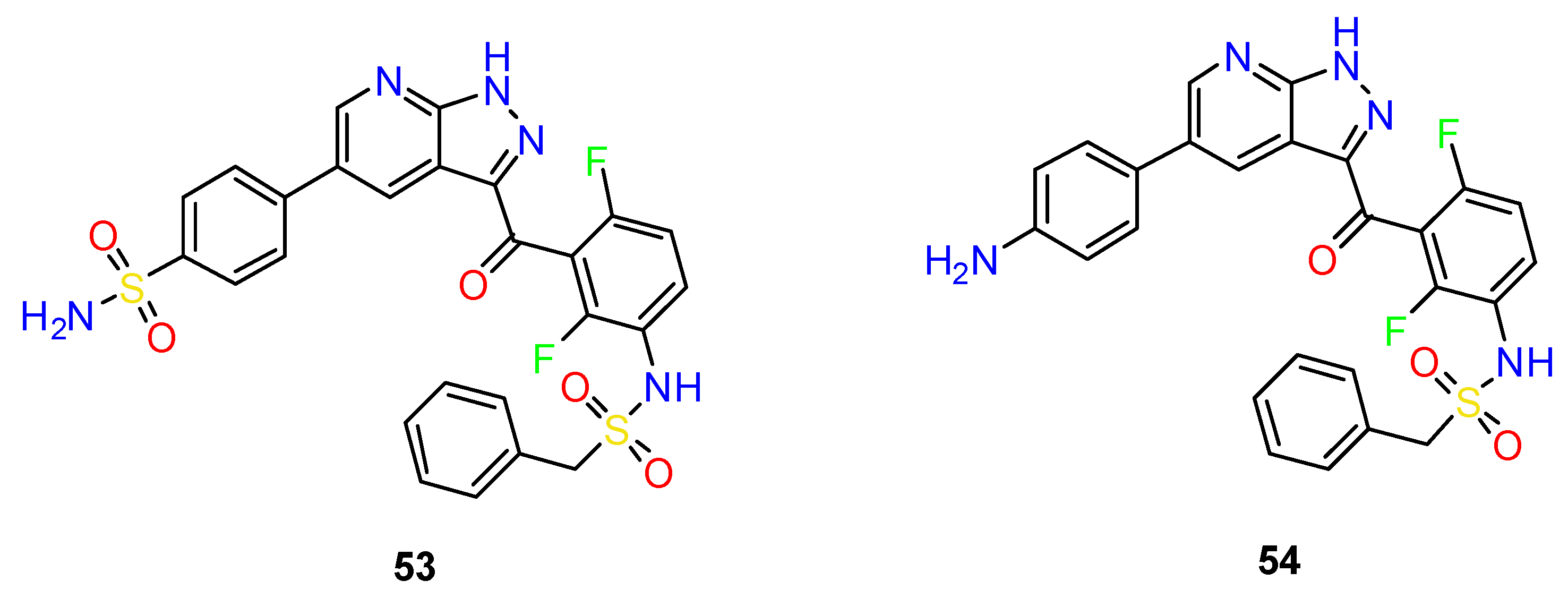
4.1. Antitumor Agents
4.2. Anti-Inflammatory Agents
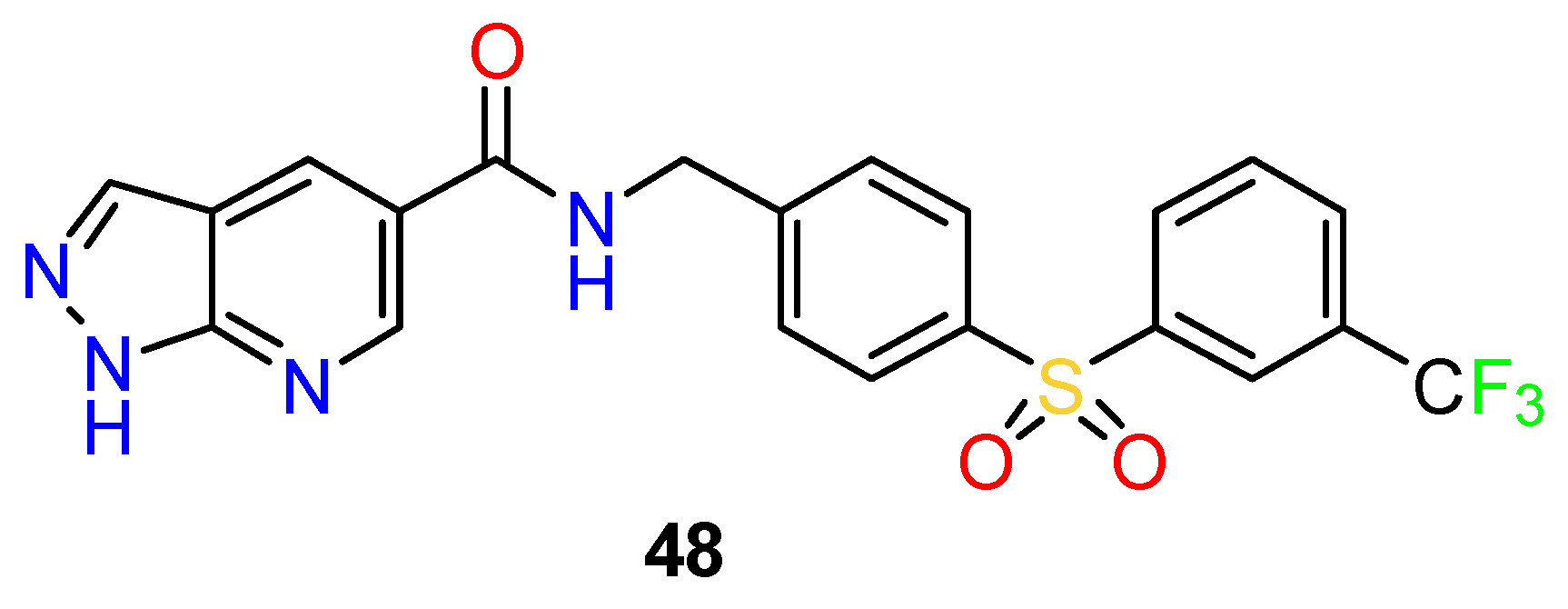
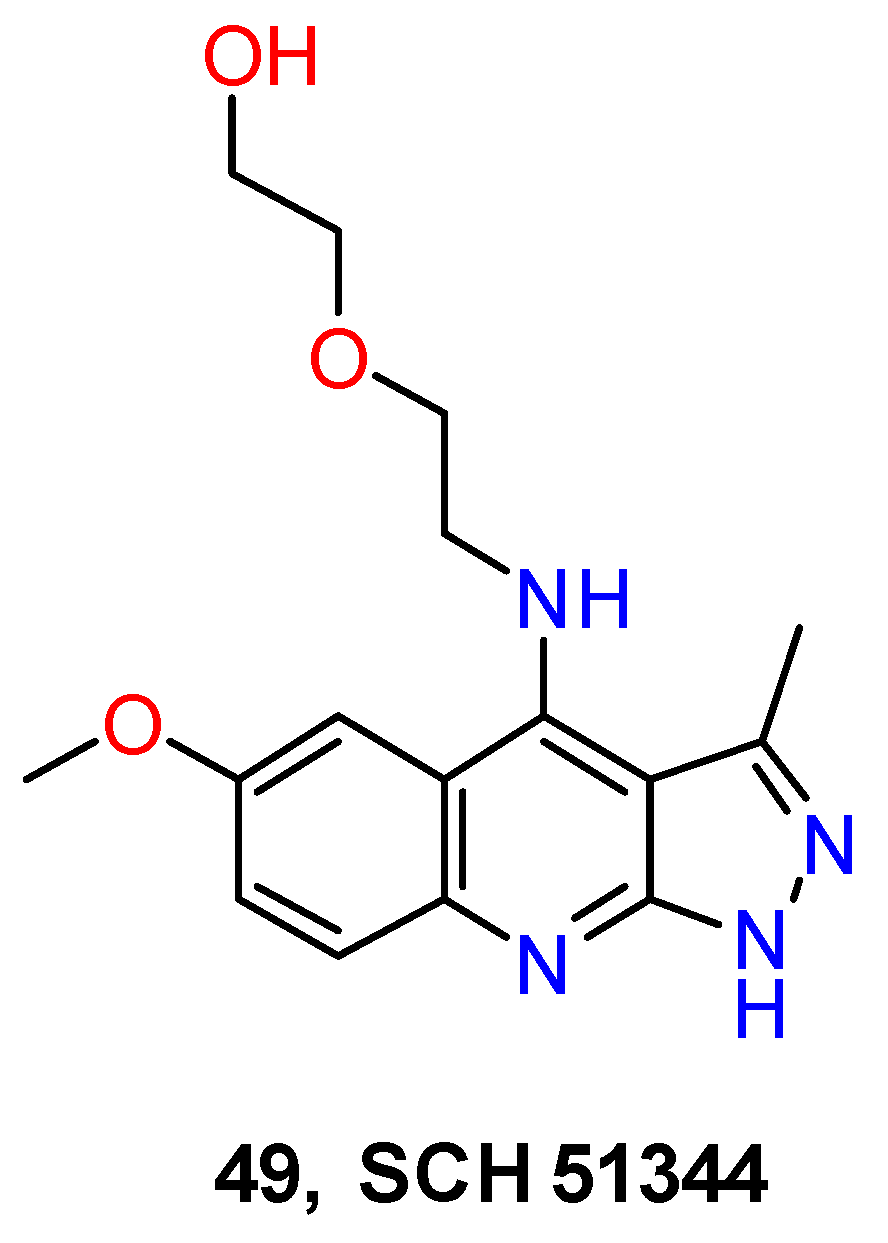
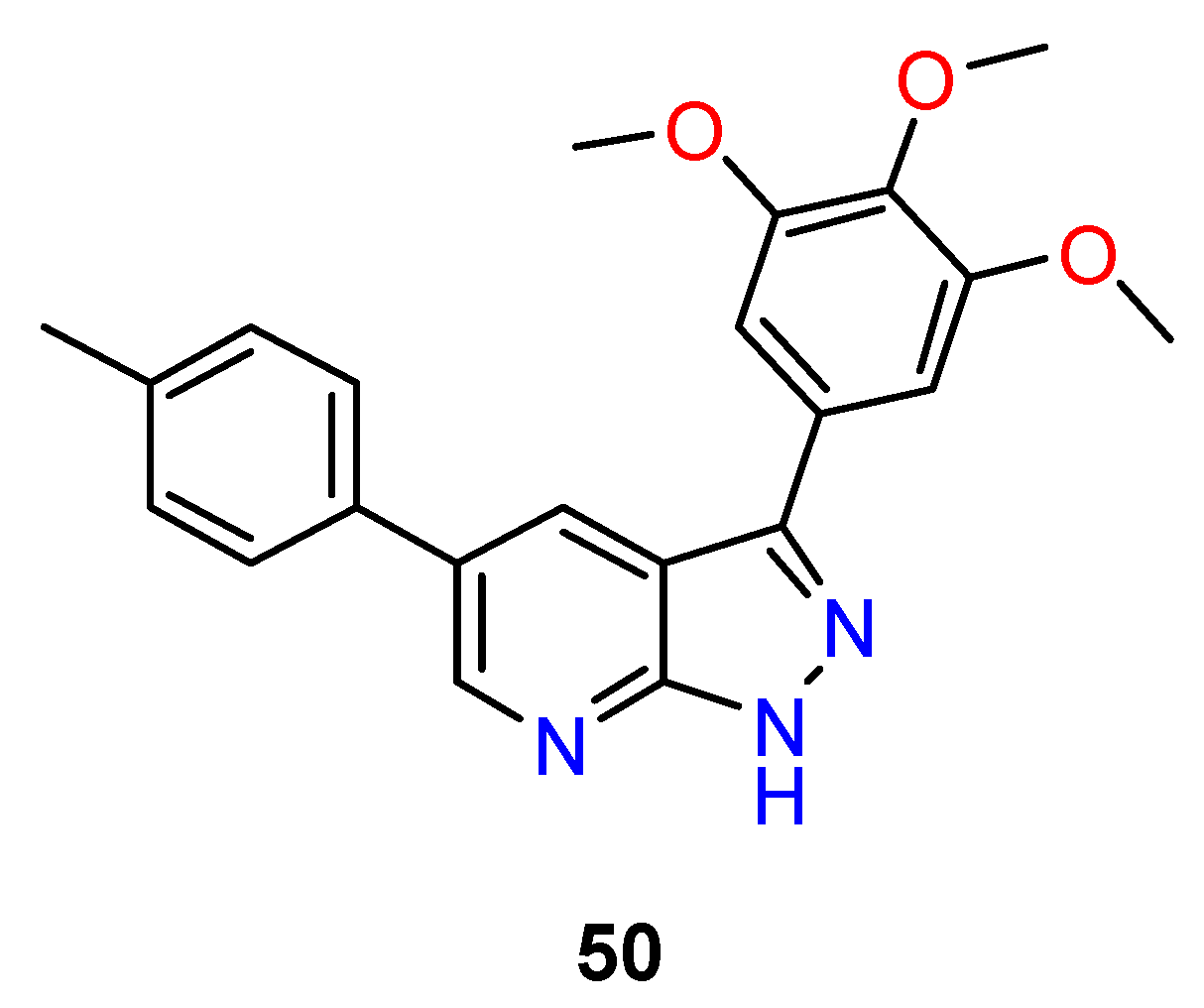
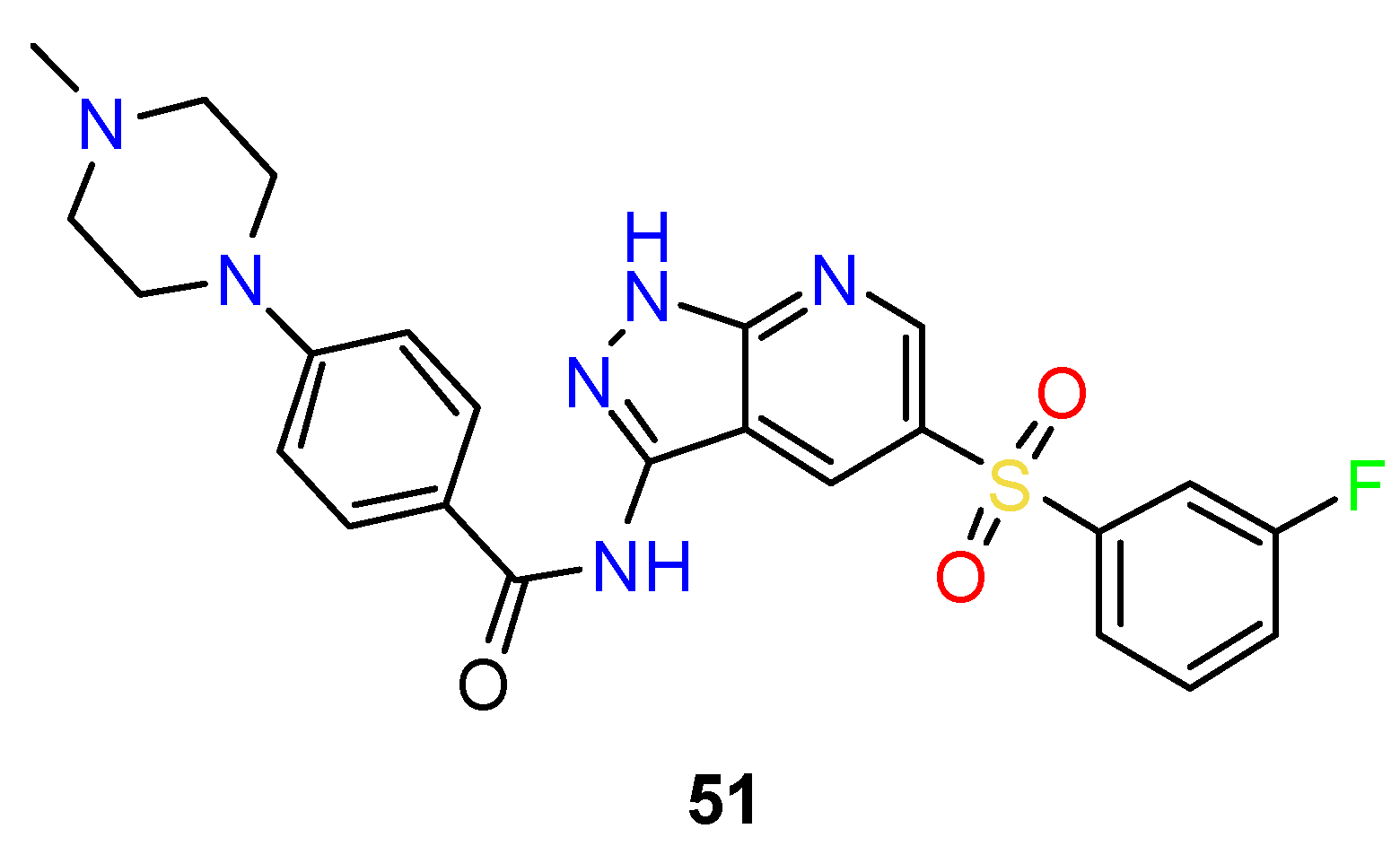
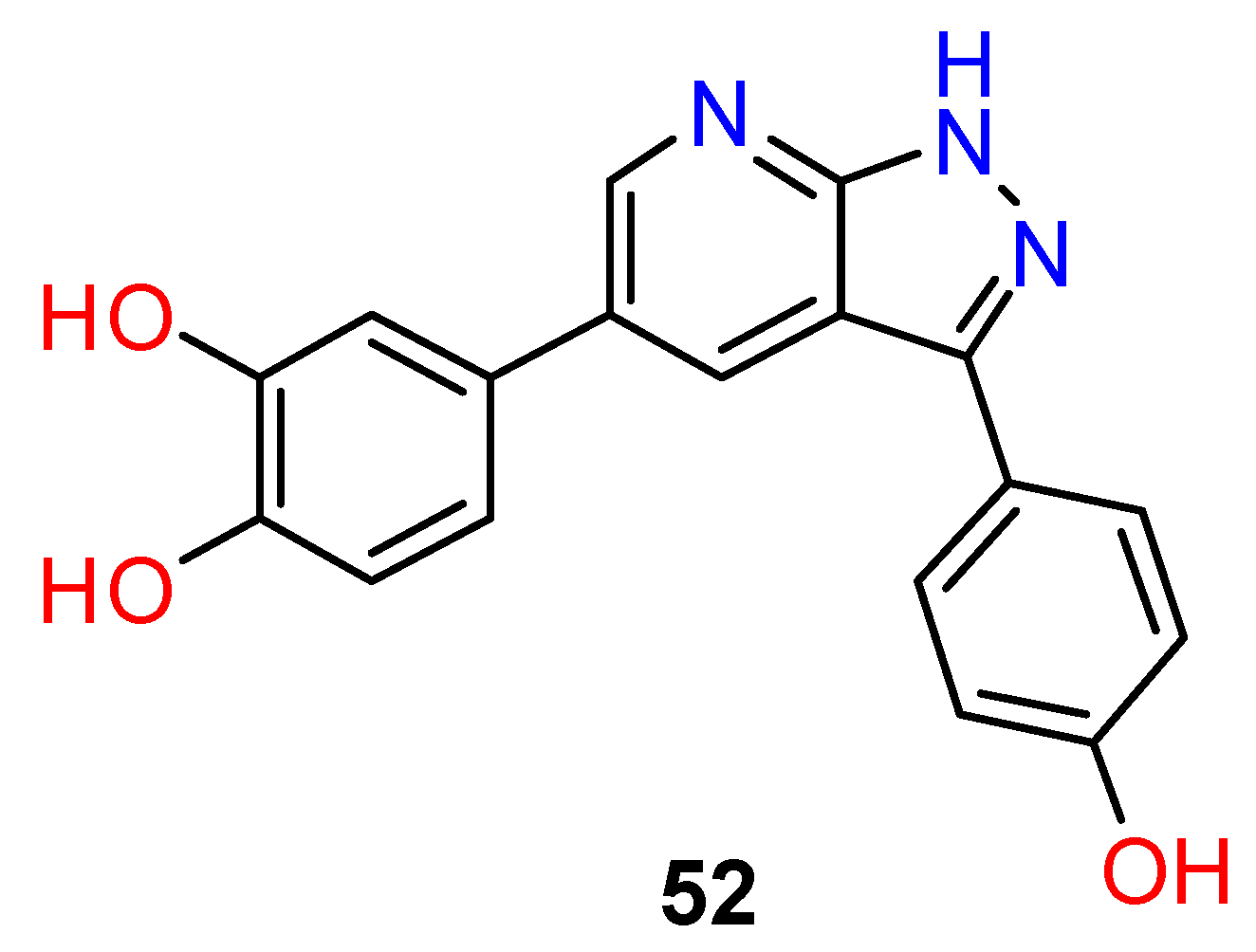
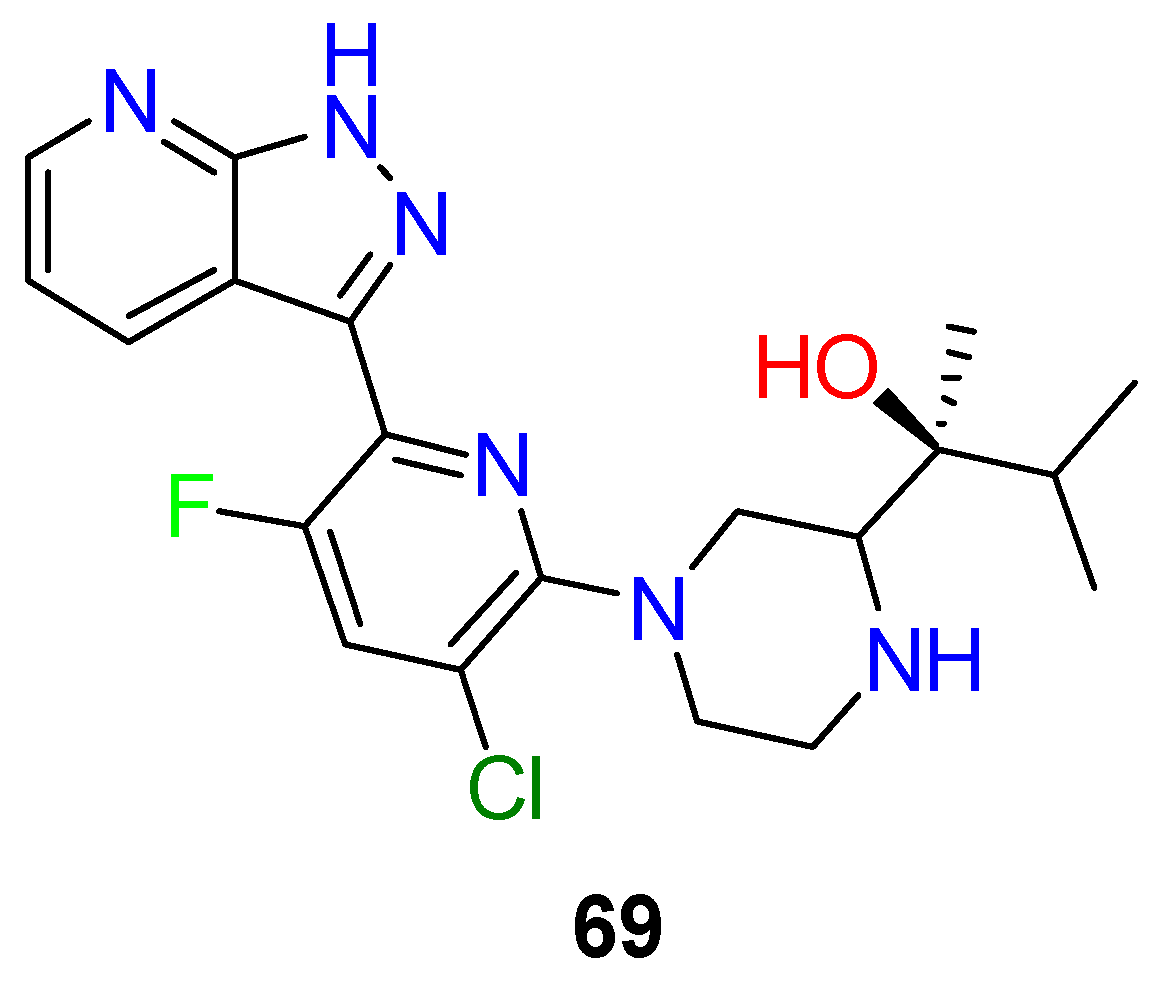
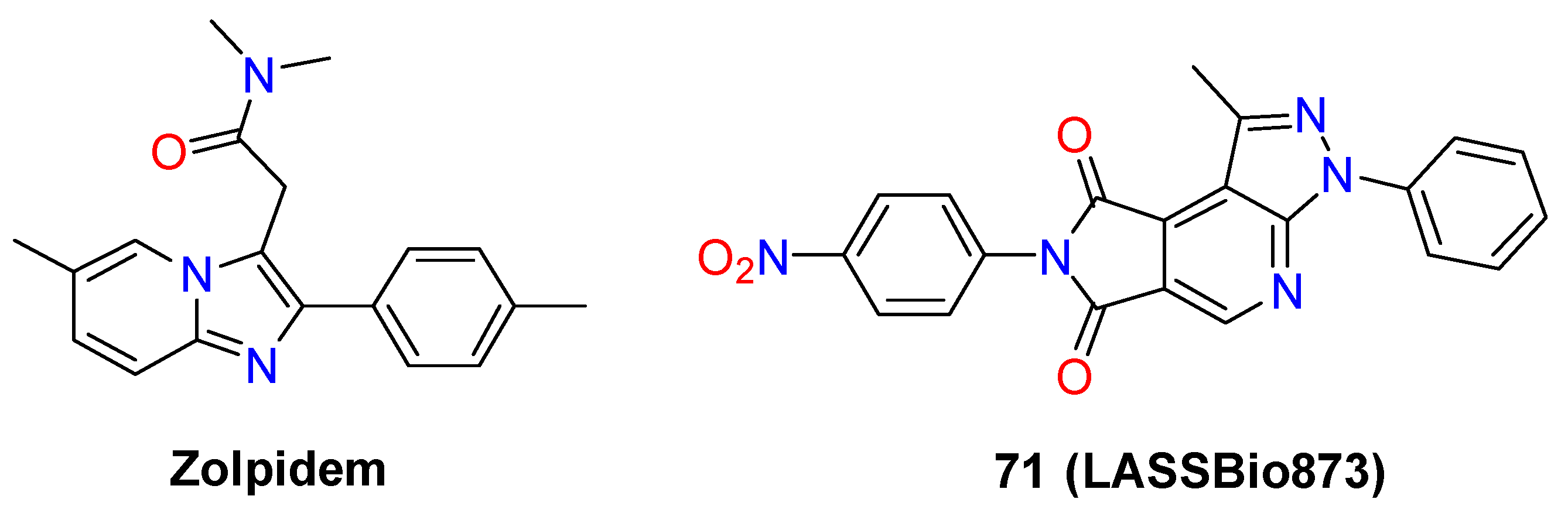
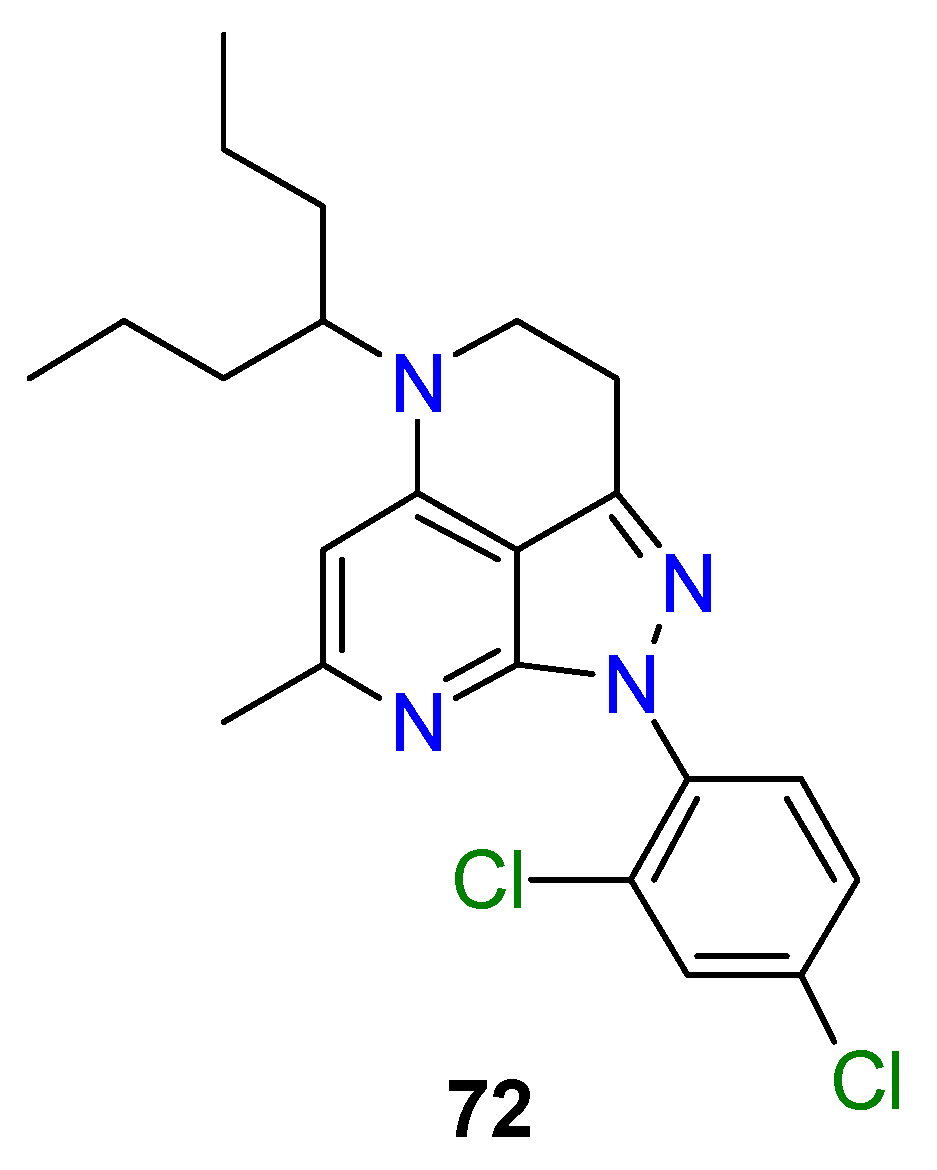
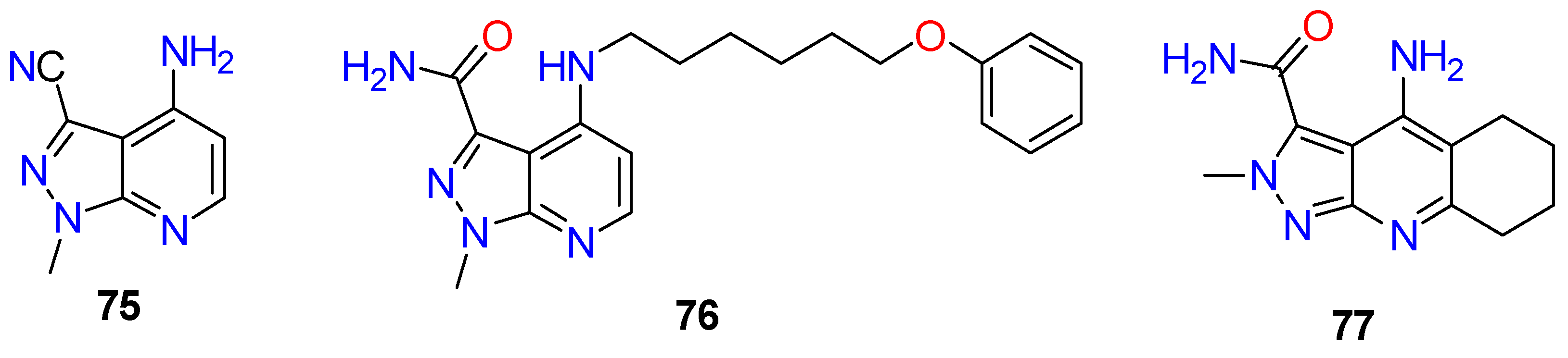
4.3. Nervous System Agents
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Buckley, B.R. Bicyclic 5-6 Systems: Three Heteroatoms 2:1. In Comprehensive Heterocyclic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2008; Volume 10, pp. 431–491. ISBN 9780080449920. [Google Scholar]
- Ortoleva, G. New Compound Obtained by the Action of Iodine on Benzalphenylhydrazone in Pyridine Solution. Gazz. Chim. Ital. 1908, 37, 71–82. [Google Scholar]
- Bulow, C. Synthesis of Derivatives of 1,2,7-Pyrazopyridine. A New Variety of Homo-(C. C)-Condensed, Bisheterocyclic Compounds. Ber. Dtsch. Chem. Ges. 1911, 43, 3401. [Google Scholar]
- Chemical Abstracts Service. Scifinder, Version 2019; Chemical Abstracts Service: Columbus, OH, USA, 2019. [Google Scholar]
- Sanghvi, Y.S.; Larson, S.B.; Robins, R.K.; Revankar, G.R. Synthesis of Certain C-4 Substituted Pyrazolo[3,4-b]Pyridine Nucleosides Structurally Related to Adenosine and Inosine by the Sodium Salt Glycosylation Procedure. Nucleosides Nucleotides 1989, 8, 887–890. [Google Scholar] [CrossRef]
- Dodiya, D.K.; Trivedi, A.R.; Kataria, V.B.; Shah, V.H. Advances in the Synthesis of Pyrazolo[3,4-b]Pyridines. Curr. Org. Chem. 2012, 16, 400–417. [Google Scholar] [CrossRef]
- Rao, R.P. Synthesis of New Pyrazolopyridines and Pyrazolopyrimidines. Rec. Chem. Prog. 1968, 29, 103. [Google Scholar]
- Komarova, E.S.; Makarov, V.A.; Granik, V.G.; Parkanyi, C. Synthesis of Pyrazolo[3,4-b]Pyridin-6-Ones. J. Heterocycl. Chem. 2012, 49, 969–998. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Theoretical Estimation of the Annular Tautomerism of Indazoles. J. Phys. Org. Chem. 2005, 18, 719–724. [Google Scholar] [CrossRef]
- Aggarwal, R.; Singh, G.; Kumar, S. Molecular Iodine Mediated Transition-Metal-Free Oxidative Dehydrogenation of 4,7-Dihydropyrazolo[3,4-b]Pyridines. Synth. Commun. 2021, 51, 3601–3609. [Google Scholar] [CrossRef]
- Lemmers, J.G.H.; Deretey, E.; Klomp, J.P.G.; Cals, J.M.G.; Oubrie, A. Preparation of 4-(4-Phenoxyphenyl)-4,5,6,7-Tetrahydro-2H-Pyrazolo[3,4-b]Pyridin-6-One Derivatives and Related Compounds as Estrogen-Related Receptor Alpha (ERRα) Modulators. WO Patent 2021074365A1, 22 April 2021. [Google Scholar]
- Bou-Petit, E.; Plans, A.; Rodríguez-Picazo, N.; Torres-Coll, A.; Puigjaner, C.; Font-Bardia, M.; Teixidó, J.; Ramon, Y.; Cajal, S.; Estrada-Tejedor, R.; et al. C4-C5 Fused Pyrazol-3-Amines: When the Degree of Unsaturation and Electronic Characteristics of the Fused Ring Controls Regioselectivity in Ullmann and Acylation Reactions. Org. Biomol. Chem. 2020, 18, 5145–5156. [Google Scholar] [CrossRef] [PubMed]
- Almansa, C.; Virgili, M.; Carceller, E.; Grima-Poveda, P. Versatile Three Component Coupling for the Synthesis of Pyrazolopyridines and Other Pyrido Fused Systems. Heterocycles 2008, 75, 1695–1709. [Google Scholar] [CrossRef]
- Grandberg, I.I. Pyrazoles. XIX. Aminopyrazoles as the Amino Components in the Skraup Reaction. Synthesis of Pyrazolopyridines. Zhurnal Obs. Khimii 1961, 31, 2307–2310. [Google Scholar]
- Bhatt, A.; Singh, R.K.; Kant, R.; Chauhan, P.K. Synthesis, Characterization and Biological Evaluation of Some Novel Pyrazolo[3,4-b]Pyridin-3-Amine Derivatives. Chem. Biol. Interface 2016, 6, 357–365. [Google Scholar]
- Patel, J.B.; Malick, J.B.; Salama, A.I.; Goldberg, M.E. Pharmacology of Pyrazolopyridines. Pharmacol. Biochem. Behav. 1985, 23, 675–680. [Google Scholar] [CrossRef]
- Wenglowsky, S. Pyrazolo[3,4-b]Pyridine Kinase Inhibitors: A Patent Review (2008–Present). Expert Opin. Ther. Pat. 2013, 23, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, E.; Mehdhar, F.S.; Abdel-Ghani, G.E. Synthesis of New Polyheterocyclic Ring Systems Derived from 3-Amino-5-Bromo-4,6-Dimethyl-1H-Pyrazolo[3,4-b]Pyridine. Polycycl. Aromat. Compd. 2021, 41, 333–346. [Google Scholar] [CrossRef]
- Park, A.; Hwang, J.; Lee, J.Y.; Heo, E.J.; Na, Y.J.; Kang, S.; Jeong, K.S.; Kim, K.Y.; Shin, S.J.; Lee, H. Synthesis of Novel 1H-Pyrazolo[3,4-b]Pyridine Derivatives as DYRK 1A/1B Inhibitors. Bioorganic Med. Chem. Lett. 2021, 47, 128226. [Google Scholar] [CrossRef]
- Li, Z.; Qin, J.; Sun, X.; Jin, Y.; Su, W. Fast, Solvent-Free, and Highly Efficient Synthesis of Pyrazolo[3,4-b]Pyridines Using Microwave Irradiation and Khso4 as A Reusable Green Catalyst. Heterocycles 2019, 98, 1408–1422. [Google Scholar] [CrossRef]
- Urvashi; Tandon, V.; Das, P.; Kukreti, S. Synthesis of 3,6-Diaryl-1H-Pyrazolo[3,4-b]Pyridines via One-Pot Sequential Suzuki-Miyaura Coupling. RSC Adv. 2018, 8, 34883–34894. [Google Scholar] [CrossRef]
- Xing, Y.; Zuo, J.; Krogstad, P.; Jung, M.E. Synthesis and Structure-Activity Relationship (SAR) Studies of Novel Pyrazolopyridine Derivatives as Inhibitors of Enterovirus Replication. J. Med. Chem. 2018, 61, 1688–1703. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Shekarrao, K.; Saikia, P.; Gogoi, S.; Boruah, R.C. Palladium Mediated Regioselective Intramolecular Heck Reaction: Synthesis of 1,3,4-Trisubstituted Pyrazolo[3,4-b]Pyridines, 3H-Pyrazolo[3,4-c]Isoquinolines and 3H-Pyrazolo[4,3-f][1,7]Naphthyridines. RSC Adv. 2015, 5, 21099–21102. [Google Scholar] [CrossRef]
- Hao, S.Y.; Qi, Z.Y.; Wang, S.; Wang, X.R.; Chen, S.W. Synthesis and Bioevaluation of N-(3,4,5-Trimethoxyphenyl)-1H-Pyrazolo[3,4-b]Pyridin-3-Amines as Tubulin Polymerization Inhibitors with Anti-Angiogenic Effects. Bioorganic Med. Chem. 2021, 31, 115985. [Google Scholar] [CrossRef] [PubMed]
- Merchant, R.R.; Lang, S.B.; Yu, T.; Zhao, S.; Qi, Z.; Suzuki, T.; Bao, J. A General One-Pot Protocol for Hindered N-Alkyl Azaheterocycles from Tertiary Carboxylic Acids. Org. Lett. 2020, 22, 4180–4184. [Google Scholar] [CrossRef] [PubMed]
- Mekky, A.E.M.; Sanad, S.M.H. Synthesis, Characterization, and Antimicrobial Evaluation of Novel Thiohydrazonates and Pyrazolo[3,4-b]Pyridines. Polycycl. Aromat. Compd. 2021, 41, 936–949. [Google Scholar] [CrossRef]
- Aggarwal, R.; Kumar, S.; Sadana, R.; Guzman, A.; Kumar, V. Multicomponent Synthesis, in Vitro Cytotoxic Evaluation and Molecular Modelling Studies of Polyfunctionalized Pyrazolo[3,4-b]Pyridine Derivatives against Three Human Cancer Cell Lines. Synth. Commun. 2021, 51, 3308–3324. [Google Scholar] [CrossRef]
- Pfaffenrot, B.; Klövekorn, P.; Juchum, M.; Selig, R.; Albrecht, W.; Zender, L.; Laufer, S.A. Design and Synthesis of 1H-Pyrazolo[3,4-b]Pyridines Targeting Mitogen-Activated Protein Kinase Kinase 4 (MKK4)—A Promising Target for Liver Regeneration. Eur. J. Med. Chem. 2021, 218, 113371. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.L.S.; Soares, J.C.A.V.; Portapilla, G.B.; Providello, M.V.; Lima, C.H.S.; Muri, E.M.F.; de Albuquerque, S.; Dias, L.R.S. Trypanocidal Activity of New 1,6-Diphenyl-1H-Pyrazolo[3,4-b]Pyridine Derivatives: Synthesis, in Vitro and in Vivo Studies. Bioorganic Med. Chem. 2021, 29, 115855. [Google Scholar] [CrossRef]
- Luo, D.; Guo, Z.; Zhao, X.; Wu, L.; Liu, X.; Zhang, Y.; Zhang, Y.; Deng, Z.; Qu, X.; Cui, S.; et al. Novel 5-Fluorouracil Sensitizers for Colorectal Cancer Therapy: Design and Synthesis of S1P Receptor 2 (S1PR2) Antagonists. Eur. J. Med. Chem. 2022, 227, 113923. [Google Scholar] [CrossRef]
- Miyachi, H.; Yuzuriha, T.; Tabata, R.; Fukuda, S.; Nunomura, K.; Lin, B.; Kobayashi, T.; Ishimoto, K.; Doi, T.; Tachibana, K. Structural Development of 1H-Pyrazolo-[3,4-b]Pyridine-4-Carboxylic Acid Derivatives as Human Peroxisome Proliferator-Activated Receptor Alpha (PPARα)-Selective Agonists. Bioorganic Med. Chem. Lett. 2019, 29, 2124–2128. [Google Scholar] [CrossRef]
- Doi, T.; Tachibana, K.; Kobayashi, T.; Yuzuriha, T.; Ishimoto, K.; Miyachi, H. PPARα Transcriptional Activator Containing Pyrazolopyridine Derivative, and Use Thereof. WO Patent 2017082377A1, 18 May 2017. [Google Scholar]
- Pal, A.; Banik, B.K. Microwave-Induced Suzuki-Coupling toward Pyrazoles. Heterocycl. Lett. 2021, 11, 19–23. [Google Scholar]
- Bhukya, B.; Guguloth, H. Synthesis and Anticancer Activity of Novel Oxadiazole Functionalized Pyrazolo[3,4-b]Pyridine Derivatives. Asian J. Chem. 2021, 33, 1331–1335. [Google Scholar] [CrossRef]
- Shen, L.-Q.; Tang, Y.; Wu, A.-Q.; Pan, D.; Diao, K.-S. Synthesis, X-ray Crystal Structure, and Computational Study of Novel Pyrazolo[3,4-b]Pyridin-3-Ol Derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2014, 189, 674–686. [Google Scholar] [CrossRef]
- Chang, Y.; Lu, X.; Shibu, M.A.; Dai, Y.B.; Luo, J.; Zhang, Y.; Li, Y.; Zhao, P.; Zhang, Z.; Xu, Y.; et al. Structure Based Design of N-(3-((1H-Pyrazolo[3,4-b]Pyridin-5-Yl)Ethynyl)Benzenesulfonamides as Selective Leucine-Zipper and Sterile-α Motif Kinase (ZAK) Inhibitors. J. Med. Chem. 2017, 60, 5927–5932. [Google Scholar] [CrossRef] [PubMed]
- Sumesh, R.V.; Kumar, R.R.; Almansour, A.I.; Kumar, R.S.; Ashraf, M.K.M. Pyrano[2,3-f]Pyrazolo[3,4-b]Quinoline-3-Carbonitriles: A Three-Component Synthesis and AChE Inhibitory Studies. Synth. Commun. 2021, 51, 1058–1065. [Google Scholar] [CrossRef]
- Teixeira, F.C.; Lucas, C.; Curto, M.J.M.; André, V.; Duarte, M.T.; Teixeira, A.P.S. Synthesis of Novel Pyrazolo[3,4-b]Quinolinebisphosphonic Acids and an Unexpected Intramolecular Cyclization and Phosphonylation Reaction. Org. Biomol. Chem. 2021, 19, 2533–2545. [Google Scholar] [CrossRef] [PubMed]
- Hamza, E.K.; Hamdy, N.A.; Zarie, E.S.; Fakhr, I.M.I.; Elwahy, A.H.M.; Awad, H.M. Synthesis and in Vitro Evaluation of Novel Tetralin-Pyrazolo[3,4-b]Pyridine Hybrids as Potential Anticancer Agents. J. Heterocycl. Chem. 2020, 57, 182–196. [Google Scholar] [CrossRef]
- Jadhav, C.; Nipate, A.; Chate, A.; Gill, C. Triethylammonium Hydrogen Sulfate [Et3NH][HSO4]-Catalyzed Rapid and Efficient Multicomponent Synthesis of Pyrido[2,3-d]Pyrimidine and Pyrazolo[3,4-b]Pyridine Hybrids. ACS Omega 2021, 6, 18215–18225. [Google Scholar] [CrossRef]
- Patnaik, S.; Basu, D.; Southall, N.; Dehdashti, S.; Wan, K.K.; Zheng, W.; Ferrer, M.; Taylor, M.; Engel, D.A.; Marugan, J.J. Identification, Design and Synthesis of Novel Pyrazolopyridine Influenza Virus Nonstructural Protein 1 Antagonists. Bioorganic Med. Chem. Lett. 2019, 29, 1113–1119. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Rizk, H.F.; El-Borai, M.A.; Sadek, M.E. Green Routes for the Synthesis of New Pyrazole Bearing Biologically Active Imidiazolyl, Pyridine and Quinoxaline Derivatives as Promising Antimicrobial and Antioxidant Agents. J. Iran. Chem. Soc. 2021, 18, 1391–1404. [Google Scholar] [CrossRef]
- Teleb, M.A.M.; Hassaneen, H.M.; Abdelhamid, I.A.; Saleh, F.M. 5-Aminopyrazole-4-Carbonitriles as Precursors to Novel 4-Aminotetrahydropyrazolo[3,4-b]Quinolin-5-Ones and N-(4-Cyanopyrazol-5-Yl)Pyridine-3-Carbonitrile. Synth. Commun. 2021, 51, 2357–2364. [Google Scholar] [CrossRef]
- Mohamed, S.S.; Shabaan, S.N.; Abdelghaffar, N.F.; Dauoud, N.T.; Sayed, G.H.; Anwer, K.E. Synthesis and Exploring Novel Annulated 1,3-Diphenylpyrazole Derivatives as Antimicrobial and Anticancer Agents. J. Basic Environ. Sci. 2021, 8, 124–139. [Google Scholar]
- Ramzan, A.; Siddiqui, S.; Irfan, A.; Al-Sehemi, A.G.; Ahmad, A.; Verpoort, F.; Chughtai, A.H.; Khan, M.A.; Munawar, M.A.; Basra, M.A.R. Antiplatelet Activity, Molecular Docking and QSAR Study of Novel N’-Arylmethylidene-3-Methyl-1-Phenyl-6-p-Chlorophenyl-1H-Pyrazolo[3,4-b]Pyridine-4-Carbohydrazides. Med. Chem. Res. 2018, 27, 388–405. [Google Scholar] [CrossRef]
- Orlikova, B.; Chaouni, W.; Schumacher, M.; Aadil, M.; Diederich, M.; Kirsch, G. Synthesis and Bioactivity of Novel Amino-Pyrazolopyridines. Eur. J. Med. Chem. 2014, 85, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Arokiaraj, S.R.; Tajuddin, N.B.; Muthusamy, K.; Jayaraj, J.M.; Alagumuthu, M. TRAF2 and NCK-Interacting Kinase Inhibitors for Colorectal Cancer: In Vitro and Theoretical Validations. ACS Comb. Sci. 2020, 22, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Mostinski, Y.; Heynen, G.J.J.E.; Lopez-Alberca, M.P.; Paul, J.; Miksche, S.; Radetzki, S.; Schaller, D.; Shanina, E.; Seyffarth, C.; Kolomeets, Y.; et al. From Pyrazolones to Azaindoles: Evolution of Active-Site SHP2 Inhibitors Based on Scaffold Hopping and Bioisosteric Replacement. J. Med. Chem. 2020, 63, 14780–14804. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, S.A.; El-Emary, T.I. New Pyrazolo[3,4-b]Pyridines: Synthesis and Antimicrobial Activity. Pharma Chem. 2018, 10, 44–51. [Google Scholar]
- Hu, L.; Li, L.; Chang, Q.; Fu, S.; Qin, J.; Chen, Z.; Li, X.; Liu, Q.; Hu, G.; Li, Q. Discovery of Novel Pyrazolo[3,4-b]Pyridine Derivatives with Dual Activities of Vascular Remodeling Inhibition and Vasodilation for the Treatment of Pulmonary Arterial Hypertension. J. Med. Chem. 2020, 63, 11215–11234. [Google Scholar] [CrossRef]
- Adib, M.; Peytam, F. An Efficient Synthesis of Fully Substituted Pyrazolo[3,4-b]Pyridin-5-Amines from α-Azidochalcones. Tetrahedron 2018, 74, 2414–2420. [Google Scholar] [CrossRef]
- Rao, H.S.P.; Adigopula, L.N.; Ramadas, K. One-Pot Synthesis of Densely Substituted Pyrazolo[3,4-b]-4,7-Dihydropyridines. ACS Comb. Sci. 2017, 19, 279–285. [Google Scholar] [CrossRef]
- Czodrowski, P.; Mallinger, A.; Wienke, D.; Esdar, C.; Poeschke, O.; Busch, M.; Rohdich, F.; Eccles, S.A.; Ortiz-Ruiz, M.-J.; Schneider, R.; et al. Structure-Based Optimization of Potent, Selective, and Orally Bioavailable CDK8 Inhibitors Discovered by High-Throughput Screening. J. Med. Chem. 2016, 59, 9337–9349. [Google Scholar] [CrossRef]
- Aly, A.A.; El-Emary, T.I.; Mourad, A.-F.E.; Khallaf Alyan, Z.; Brase, S.; Nieger, M. 5-Carbohydrazide and 5-Carbonylazide of Pyrazolo[3,4-b]Pyridines as Reactive Intermediates in the Synthesis of Various Heterocyclic Derivatives. J. Chem. Res. 2019, 43, 219–229. [Google Scholar] [CrossRef]
- Alam, R.M.; Keating, J.J. An Expedient Approach to Pyrazolo[3,4-b]Pyridine-3-Carboxamides via Palladium-Catalyzed Aminocarbonylation. Synthesis 2021, 53, 4709–4722. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Zhang, L.; Chen, D.; Dong, Z.; Zhao, C.; Liu, Z.; Xia, Q.; Wu, J.; Chen, Y.; et al. Design, Synthesis, and Biological Evaluation of Indazole Derivatives as Selective and Potent FGFR4 Inhibitors for the Treatment of FGF19-Driven Hepatocellular Cancer. Eur. J. Med. Chem. 2021, 214, 113219. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, T.; Takahashi, F.; Kato, K.; Sugano, Y.; Yamaki, S.; Moritomo, A.; Kubo, S.; Nakamura, K.; Yamagami, K.; Hamakawa, N.; et al. Optimization and in Vivo Evaluation of Pyrazolopyridines as a Potent and Selective PI3Kδ Inhibitor. Bioorganic Med. Chem. 2018, 26, 3917–3924. [Google Scholar] [CrossRef] [PubMed]
- Ravula, S.; Bobbala, R.R.; Kolli, B. Synthesis of Novel Isoxazole Functionalized Pyrazolo[3,4-b]Pyridine Derivatives; Their Anticancer Activity. J. Heterocycl. Chem. 2020, 57, 2535–2538. [Google Scholar] [CrossRef]
- Hansen, B.B.; Jepsen, T.H.; Larsen, M.; Sindet, R.; Vifian, T.; Burhardt, M.N.; Larsen, J.; Seitzberg, J.G.; Carnerup, M.A.; Jerre, A.; et al. Fragment-Based Discovery of Pyrazolopyridones as JAK1 Inhibitors with Excellent Subtype Selectivity. J. Med. Chem. 2020, 63, 7008–7032. [Google Scholar] [CrossRef]
- Labhade, K.R.; Jachak, M.N.; Labhade, S.R.; Kale, A.S.; Gaikwad, V.B. Synthesis and in Vitro Antimicrobial Evaluation of Novel Trifluoromethyl Substituted Pyrazolo[3,4-b]Pyridine 6-One Derivatives. Indo Am. J. Pharm. Res. 2016, 6, 6442–6447. [Google Scholar]
- Sanad, S.M.H.; Abdel-Fattah, A.M.; Attaby, F.A.; Elneairy, M.A.A. Pyridine-2(1H)-Thiones: Versatile Precursors for Novel Pyrazolo[3,4-b]Pyridine, Thieno[2,3-b]Pyridines, and Their Fused Azines. J. Heterocycl. Chem. 2019, 56, 651–662. [Google Scholar] [CrossRef]
- Mohamed, L.W.; Shaaban, M.A.; Zaher, A.F.; Alhamaky, S.M.; Elsahar, A.M. Synthesis of New Pyrazoles and Pyrozolo[3,4-b] Pyridines as Anti-Inflammatory Agents by Inhibition of COX-2 Enzyme. Bioorganic Chem. 2019, 83, 47–54. [Google Scholar] [CrossRef]
- Ghaedi, A.; Bardajee, G.R.; Mirshokrayi, A.; Mahdavi, M.; Shafiee, A.; Akbarzadeh, T. Facile, Novel and Efficient Synthesis of New Pyrazolo[3,4-b]Pyridine Products from Condensation of Pyrazole-5-Amine Derivatives and Activated Carbonyl Groups. RSC Adv. 2015, 5, 89652–89658. [Google Scholar] [CrossRef]
- El-borai, M.A.; Rizk, H.F.; Abd-Aal, M.F.; El-Deeb, I.Y. Synthesis of Pyrazolo[3,4-b]Pyridines under Microwave Irradiation in Multi-Component Reactions and Their Antitumor and Antimicrobial Activities. Part 1. Eur. J. Med. Chem. 2012, 48, 92–96. [Google Scholar] [CrossRef]
- Volochnyuk, D.M.; Ryabukhin, S.V.; Plaskon, A.S.; Dmytriv, Y.V.; Grygorenko, O.O.; Mykhailiuk, P.K.; Krotko, D.G.; Pushechnikov, A.; Tolmachev, A.A. Approach to the Library of Fused Pyridine-4-Carboxylic Acids by Combes-Type Reaction of Acyl Pyruvates and Electron-Rich Amino Heterocycles. J. Comb. Chem. 2010, 12, 510–517. [Google Scholar] [CrossRef]
- Emelina, E.E.; Petrov, A.A.; Selivanov, S.I.; Filyukov, D.V. α-Aminoazoles in Synthesis of Heterocycles: III. 4-Trifluoromethylpyrazolo[3,4-b]Pyridines: Synthesis and Structure. Russ. J. Org. Chem. 2008, 44, 251–256. [Google Scholar] [CrossRef]
- Wenglowsky, S.; Ren, L.; Ahrendt, K.A.; Laird, E.R.; Aliagas, I.; Alicke, B.; Buckmelter, A.J.; Choo, E.F.; Dinkel, V.; Feng, B.; et al. Pyrazolopyridine Inhibitors of B-RafV600E. Part 1: The Development of Selective, Orally Bioavailable, and Efficacious Inhibitors. ACS Med. Chem. Lett. 2011, 2, 342–347. [Google Scholar] [CrossRef]
- Mityuk, A.P.; Hrebonkin, A.; Lebed, P.S.; Grabchuk, G.P.; Volochnyuk, D.M.; Ryabukhin, S.V. Efficient Route for the Synthesis of Diverse Heteroannelated 5-Cyanopyridines. Synthesis 2021, 53, 2133–2141. [Google Scholar] [CrossRef]
- Jouha, J.; Loubidi, M.; Bouali, J.; Hamri, S.; Hafid, A.; Suzenet, F.; Guillaumet, G.; Dagcı, T.; Khouili, M.; Aydın, F.; et al. Synthesis of New Heterocyclic Compounds Based on Pyrazolopyridine Scaffold and Evaluation of Their Neuroprotective Potential in MPP+-Induced Neurodegeneration. Eur. J. Med. Chem. 2017, 129, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Iaroshenko, V.O.; Mkrtchyan, S.; Gevorgyan, A.; Vilches-Herrera, M.; Sevenard, D.V.; Villinger, A.; Ghochikyan, T.V.; Saghiyan, A.; Sosnovskikh, V.Y.; Langer, P. Synthesis of Heteroannulated 3-Nitro- and 3-Aminopyridines by Cyclocondensation of Electron-Rich Aminoheterocycles with 3-Nitrochromone. Tetrahedron 2012, 68, 2532–2543. [Google Scholar] [CrossRef]
- Iaroshenko, V.O.; Gevorgyan, A.; Mkrtchyan, S.; Grigoryan, T.; Movsisyan, E.; Villinger, A.; Langer, P. Regioselective Direct Arylation of Fused 3-Nitropyridines and Other Nitro-Substituted Heteroarenes: The Multipurpose Nature of the Nitro Group as a Directing Group. ChemCatChem 2015, 7, 316–324. [Google Scholar] [CrossRef]
- Iaroshenko, V.O.; Mkrtchyan, S.; Gevorgyan, A.; Miliutina, M.; Villinger, A.; Volochnyuk, D.; Sosnovskikh, V.Y.; Langer, P. 2,3-Unsubstituted Chromones and Their Enaminone Precursors as Versatile Reagents for the Synthesis of Fused Pyridines. Org. Biomol. Chem. 2012, 10, 890–894. [Google Scholar] [CrossRef]
- Stepaniuk, O.O.; Stepaniuk, O.O.; Rudenko, T.V.; Rudenko, T.V.; Vashchenko, B.V.; Vashchenko, B.V.; Matvienko, V.O.; Kondratov, I.S.; Kondratov, I.S.; Tolmachev, A.A.; et al. Synthesis of Fused Pyridine Carboxylates by Reaction of β-Alkoxyvinyl Glyoxylates with Amino Heterocycles. Synthesis 2020, 52, 1915–1926. [Google Scholar] [CrossRef]
- Han, Z.G.; Zhang, G.; Jiang, B.; Ning, M.; Shi, F.; Tu, S.J. Diversity Synthesis of Pyrimido[4,5-£][l,6]Naphthyridine and Its Derivatives under Microwave Irradiation. J. Comb. Chem. 2009, 11, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.C.; Lin, W.; Wang, H.Y.; Huang, Z.B.; Shi, D.Q. Improved and Efficient Synthesis of Chromeno[4,3-d]Pyrazolo[3,4-b]Pyridin-6(3H)-Ones and Their Fluorescence Properties. J. Heterocycl. Chem. 2014, 51, 1036–1044. [Google Scholar] [CrossRef]
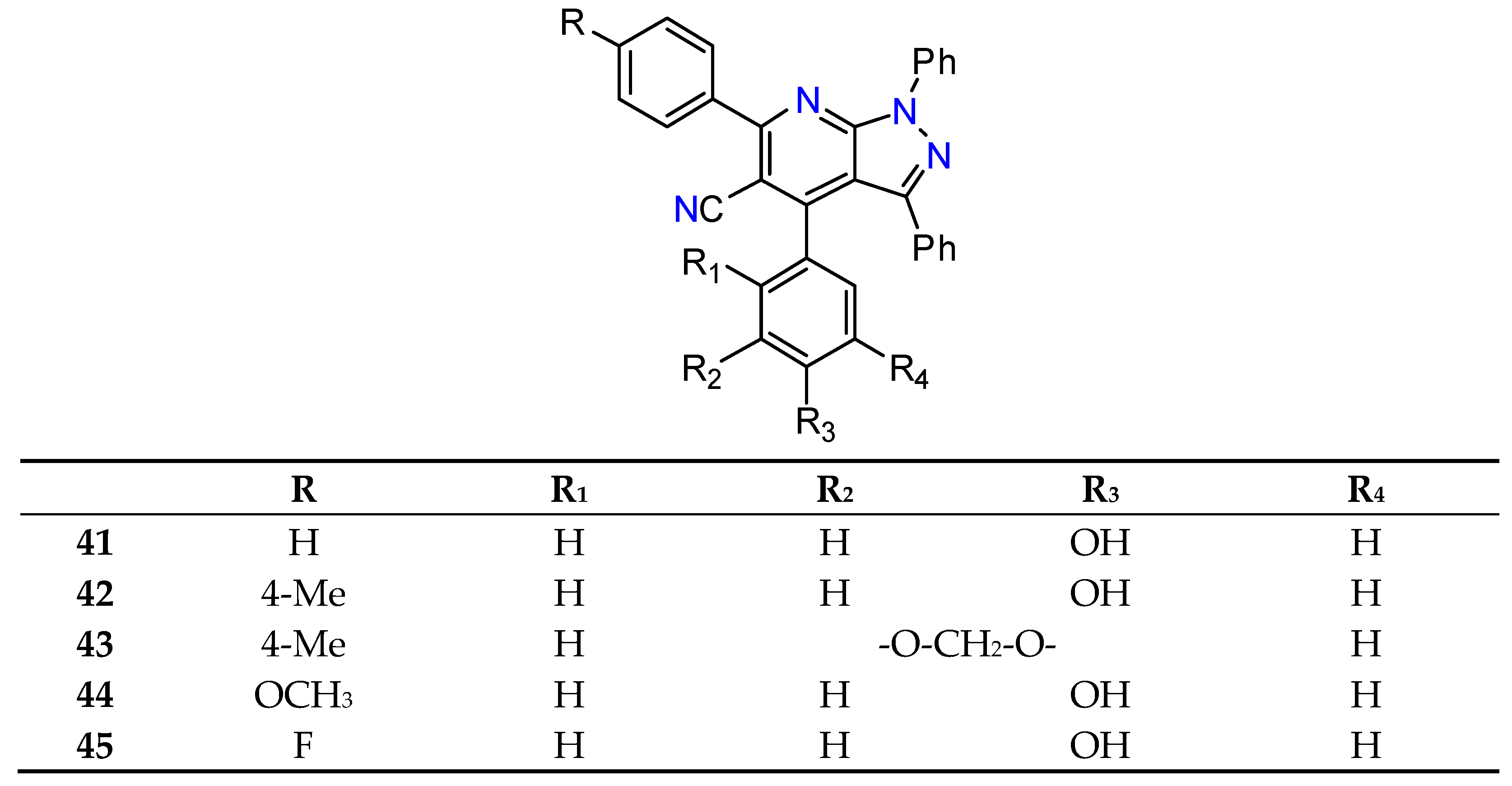
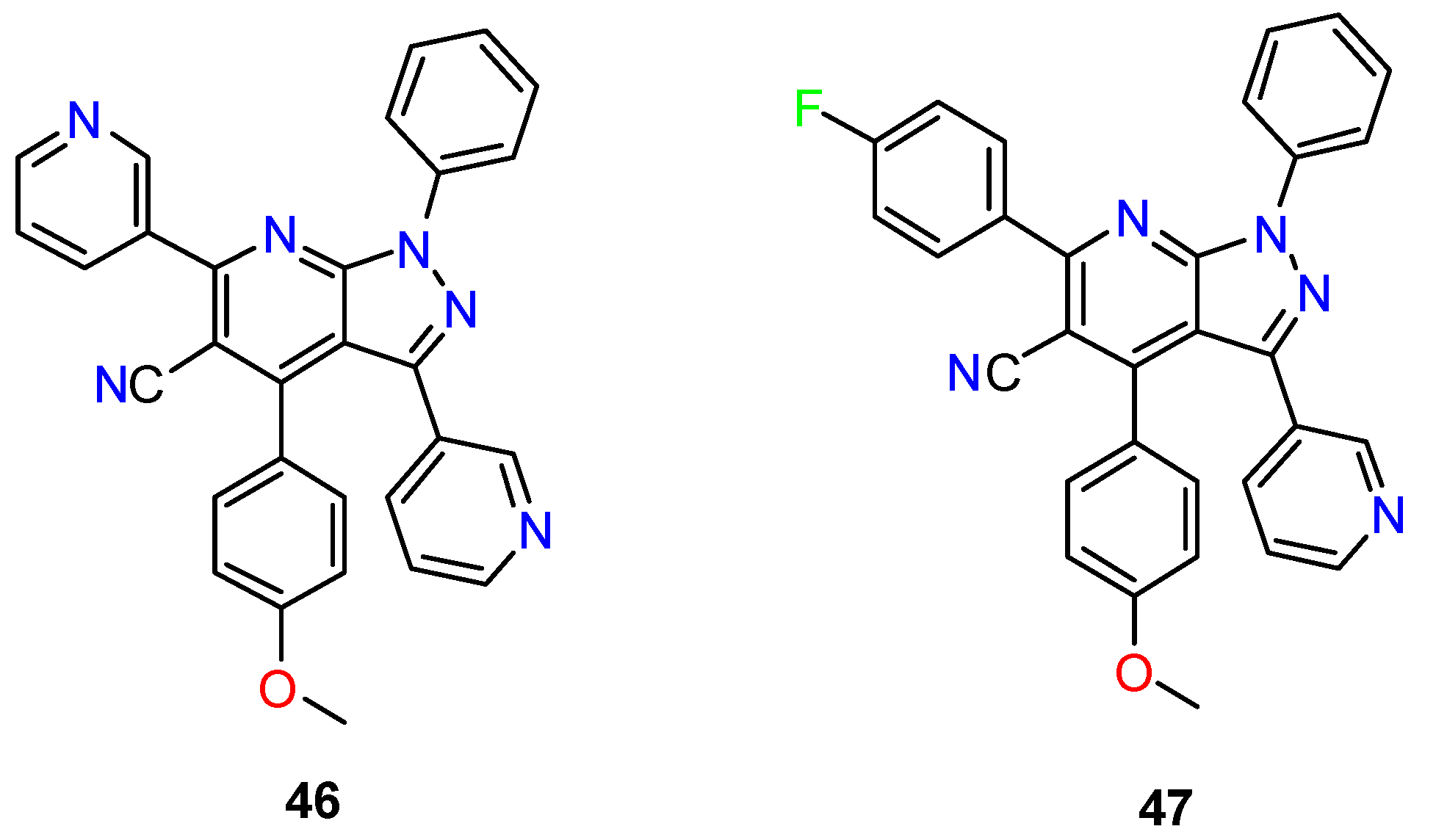
- Liu, M.; Yin, G. ZrCl 4-Catalysed Synthesis of New 4-(2-Hydroxyphenyl)Pyrazolo[3,4-b]Pyridine Derivatives. J. Chem. Res. 2015, 263, 263–266. [Google Scholar] [CrossRef]
- Zou, X.; Tu, S.; Shi, F.; Xu, J. An Efficient Synthesis of Pyrazolo[3,4-b]Pyridine Derivatives under Microwave Irradiation. Gen. Pap. ARKIVOC 2006, 2, 130–135. [Google Scholar] [CrossRef]
- Shi, D.Q.; Zhou, Y.; Liu, H. An Efficient Synthesis of Pyrazolo[3,4-b]Pyridine Derivatives in Ionic Liquid. Synth. Commun. 2010, 40, 3660–3668. [Google Scholar] [CrossRef]
- Lavrard, H.; Popowycz, F. Harnessing Cascade Suzuki-Cyclization Reactions of Pyrazolo[3,4-b]Pyridine for the Synthesis of Tetracyclic Fused Heteroaromatics. Eur. J. Org. Chem. 2017, 2017, 600–608. [Google Scholar] [CrossRef]
- Miah, A.H.; Champigny, A.C.; Graves, R.H.; Hodgson, S.T.; Percy, J.M.; Procopiou, P.A. Identification of Pyrazolopyrimidine Arylsulfonamides as CC-Chemokine Receptor 4 (CCR4) Antagonists. Bioorganic Med. Chem. 2017, 25, 5327–5340. [Google Scholar] [CrossRef] [PubMed]
- Guercio, G.; Castoldi, D.; Giubellina, N.; Lamonica, A.; Ribecai, A.; Stabile, P.; Westerduin, P.; Dams, R.; Nicoletti, A.; Rossi, S.; et al. Overall Synthesis of GSK356278: Quick Delivery of a PDE4 Inhibitor Using a Fit-for-Purpose Approach. Org. Process. Res. Dev. 2010, 14, 1153–1161. [Google Scholar] [CrossRef]
- Azevedo, A.R.; Ferreira, V.F.; de Mello, H.; Leäo-Ferreira, L.R.; Jabor, A.V.; Frugulhetti, I.C.P.P.; Pereira, H.S.; Moussatche, N.; Rolim Bernardino, A.M. Synthesis and biological evaluation of 1H-pyrazolo[3,4-b]pyridine-5-carboxylic acids against vaccinia virus. Heterocycl. Commun. 2002, 8, 427–432. [Google Scholar] [CrossRef]
- Rimland, J.; Dunne, A.; Hunjan, S.S.; Sasse, R.; Uings, I.; Montanari, D.; Caivano, M.; Shah, P.; Standing, D.; Gray, D.; et al. The Identification a Novel, Selective, Non-Steroidal, Functional Glucocorticoid Receptor Antagonist. Bioorganic Med. Chem. Lett. 2010, 20, 2340–2343. [Google Scholar] [CrossRef]
- Pan, T.; Xie, S.; Zhou, Y.; Hu, J.; Luo, H.; Li, X.; Huang, L. Dual Functional Cholinesterase and PDE4D Inhibitors for the Treatment of Alzheimer’s Disease: Design, Synthesis and Evaluation of Tacrine-Pyrazolo[3,4-b]Pyridine Hybrids. Bioorganic Med. Chem. Lett. 2019, 29, 2150–2152. [Google Scholar] [CrossRef]
- Ezzati, M.; Khalafy, J.; Marjani, A.P.; Prager, R.H. The Catalyst-Free Syntheses of Pyrazolo[3,4-b]Quinolin-5-One and Pyrazolo[4′,3′:5,6]Pyrido[2,3-d]Pyrimidin-5,7-Dione Derivatives by One-Pot, Three-Component Reactions. Tetrahedron 2017, 73, 6587–6596. [Google Scholar] [CrossRef]
- Quiroga, J.; Trilleras, J.; Sánchez, A.I.; Insuasty, B.; Abonía, R.; Nogueras, M.; Cobo, J. Regioselective Three-Component Synthesis of Indolylpyrazolo[3,4-b]Pyridines Induced by Microwave and under Solvent-Free Conditions. Lett. Org. Chem. 2009, 6, 381–383. [Google Scholar] [CrossRef]
- Huang, Z.; Hu, Y.; Zhou, Y.; Shi, D. Efficient One-Pot Three-Component Synthesis of Fused Pyridine Derivatives in Ionic Liquid. ACS Comb. Sci. 2011, 13, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, A.; Khalesi, Z. Catalyst Free Synthesis of Fused Pyrido[2,3-d]Pyrimidines and Pyrazolo[3,4-b]Pyridines in Water. Chin. Chem. Lett. 2012, 23, 1149–1152. [Google Scholar] [CrossRef]
- Shi, C.L.; Shi, D.Q.; Kim, S.H.; Huang, Z.B.; Ji, S.J.; Ji, M. A Novel and Efficient One-Pot Synthesis of Furo[3′,4′:5,6]Pyrido[2,3-c]Pyrazole Derivatives Using Organocatalysts. Tetrahedron 2008, 64, 2425–2432. [Google Scholar] [CrossRef]
- Shi, D.Q.; Yang, F.; Ni, S.N. A Facile Synthesis of Furo[3,4-e]Pyrazolo[3,4-b]Pyridine-5(7H)-One Derivatives via Three-Component Reaction in Ionic Liquid without Any Catalyst. J. Heterocycl. Chem. 2009, 46, 469–476. [Google Scholar] [CrossRef]
- Hamama, W.S.; El-Gohary, H.G.; Soliman, M.; Zoorob, H.H. A Versatile Synthesis, PM3-Semiempirical, Antibacterial, and Antitumor Evaluation of Some Bioactive Pyrazoles. J. Heterocycl. Chem. 2012, 49, 543–554. [Google Scholar] [CrossRef]
- Marzouk, M.I.; Sayed, G.H.; Abd ElHalim, M.S.; Mansour, S.Y. Synthesis and Characterization of Novel Pyrazolone Derivatives. Eur. J. Chem. 2014, 5, 24–32. [Google Scholar] [CrossRef]
- Witherington, J.; Bordas, V.; Garland, S.L.; Hickey, D.M.B.; Ife, R.J.; Liddle, J.; Saunders, M.; Smith, D.G.; Ward, R.W. 5-Aryl-Pyrazolo[3,4-b]Pyridines: Potent Inhibitors of Glycogen Synthase Kinase-3 (GSK-3). Bioorganic Med. Chem. Lett. 2003, 13, 1577–1580. [Google Scholar] [CrossRef]
- Shi, J.; Xu, G.; Zhu, W.; Ye, H.; Yang, S.; Luo, Y.; Han, J.; Yang, J.; Li, R.; Wei, Y.; et al. Design and Synthesis of 1,4,5,6-Tetrahydropyrrolo[3,4-c]Pyrazoles and Pyrazolo[3,4-b]Pyridines for Aurora-A Kinase Inhibitors. Bioorganic Med. Chem. Lett. 2010, 20, 4273–4278. [Google Scholar] [CrossRef]
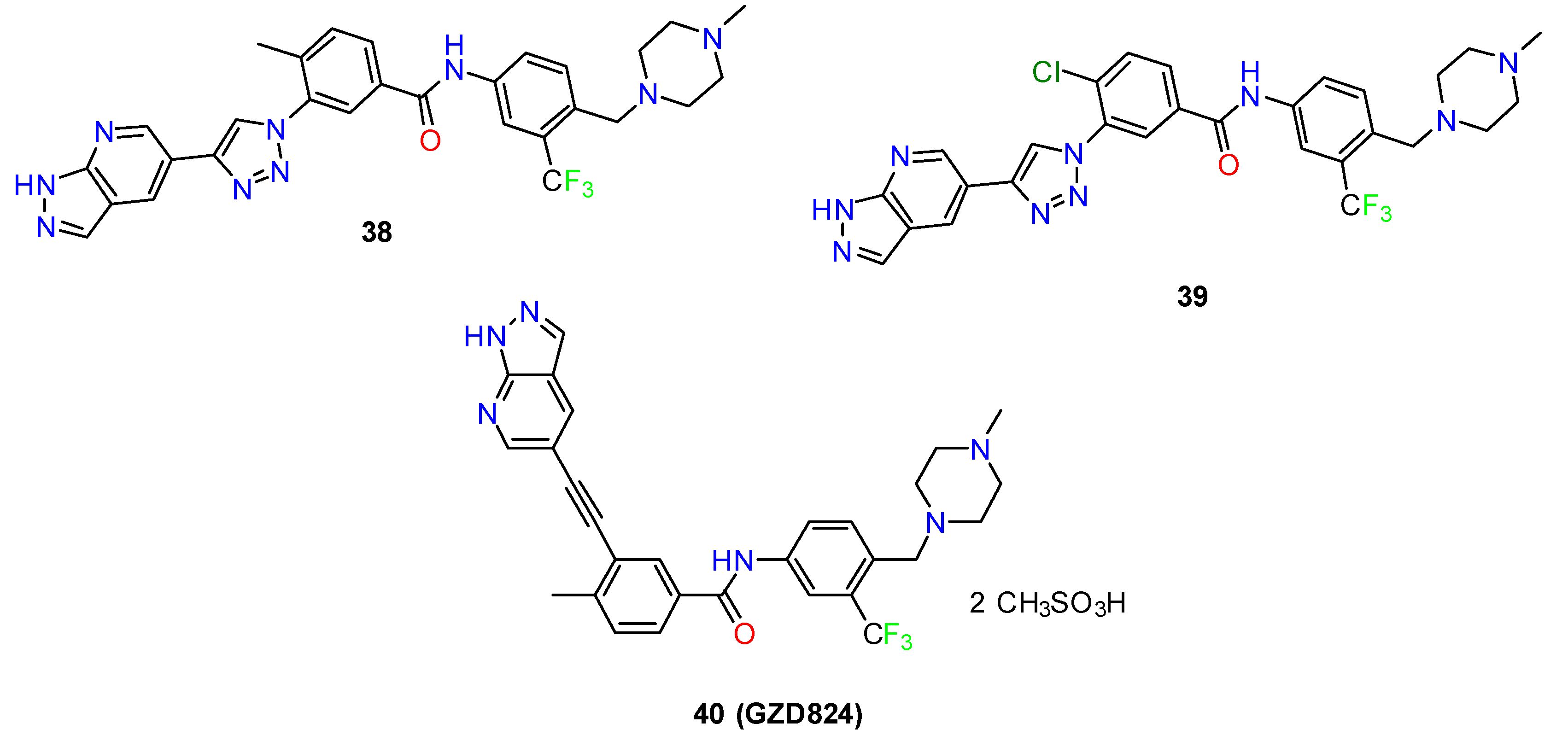
- Ren, X.; Pan, X.; Zhang, Z.; Wang, D.; Lu, X.; Li, Y.; Wen, D.; Long, H.; Luo, J.; Feng, Y.; et al. Identification of GZD824 as an Orally Bioavailable Inhibitor That Targets Phosphorylated and Nonphosphorylated Breakpoint Cluster Region-Abelson (Bcr-Abl) Kinase and Overcomes Clinically Acquired Mutation-Induced Resistance against Imatinib. J. Med. Chem. 2013, 56, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, H.; Zhang, Z.; Zhuang, X.; Luo, J.; Long, H.; Zhou, Y.; Xu, Y.; Taghipouran, R.; Li, D.; et al. N-(3-Ethynyl-2,4-Difluorophenyl)Sulfonamide Derivatives as Selective Raf Inhibitors. ACS Med. Chem. Lett. 2015, 6, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Arafa, W.A.A.; Hussein, M.F. Design, Sonosynthesis, Quantum-Chemical Calculations, and Evaluation of New Mono- and Bis-Pyridine Dicarbonitriles as Antiproliferative Agents. Chin. J. Chem. 2020, 38, 501–508. [Google Scholar] [CrossRef]
- Rateb, N.M. Synthesis and Reactions of 4-Trifluoromethyl-3-Cyano Pyridine-2(1H)-Thione/One Derivatives. J. Sulfur Chem. 2011, 32, 611–622. [Google Scholar] [CrossRef]
- Bakhite, E.A.; Abdel-Rahman, A.E.; Al-Taifi, E.A. Fluorine-Containing Heterocycles: Part III. Synthesis of Some New Furo[2,3-b]-, Pyrazolo[3,4-b]- and Thieno[2,3-b]Pyridines with Anticipated Biological Activities. Arab. J. Chem. 2014, 7, 936–946. [Google Scholar] [CrossRef]
- van Linden, O.P.J.; Farenc, C.; Zoutman, W.H.; Hameetman, L.; Wijtmans, M.; Leurs, R.; Tensen, C.P.; Siegal, G.; de Esch, I.J.P. Fragment Based Lead Discovery of Small Molecule Inhibitors for the EPHA4 Receptor Tyrosine Kinase. Eur. J. Med. Chem. 2012, 47, 493–500. [Google Scholar] [CrossRef]
- Mali, J.R.; Pratap, U.R.; Jawale, D.V.; Mane, R.A. Water-Mediated One-Pot Synthetic Route for Pyrazolo[3,4-b]Quinolines. Tetrahedron Lett. 2010, 51, 3980–3982. [Google Scholar] [CrossRef]
- Waly, M.A.; El-Hawary, I.I.; Hamama, W.S.; Zoorob, H.H. Synthesis and Antitumor Evaluation of Some New Fused and Binary Pyridines. J. Heterocycl. Chem. 2013, 50, E12–E17. [Google Scholar] [CrossRef]
- Lapa, G.B.; Bekker, O.B.; Mirchink, E.P.; Danilenko, V.N.; Preobrazhenskaya, M.N. Regioselective Acylation of Congeners of 3-Amino-1H-Pyrazolo[3,4-b] Quinolines, Their Activity on Bacterial Serine/Threonine Protein Kinases and in Vitro Antibacterial (Including Antimycobacterial) Activity. J. Enzym. Inhib. Med. Chem. 2013, 28, 1088–1093. [Google Scholar] [CrossRef]
- Masoud, M.S.; AbouEl-Enein, S.A.; Ali, A.E.; Abd Elhamed, E.H. Chelating Behavior of 3-Amino-4,6-Dimethyl-1H-Pyrazolo[3,4-b]Pyridine Ligand towards Some Metal Ions, Spectral and Thermal Measurements as Well as Molecular Modeling and Biological Studies. J. Mol. Struct. 2020, 1202, 127172. [Google Scholar] [CrossRef]
- Moir, M.; Lane, S.; Lai, F.; Connor, M.; Hibbs, D.E.; Kassiou, M. Strategies to Develop Selective CB2 Receptor Agonists from Indole Carboxamide Synthetic Cannabinoids. Eur. J. Med. Chem. 2019, 180, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, D.; Park, C.; So, W.; Jo, M.; Ok, T.; Kwon, J.; Kong, S.; Jo, S.; Kim, Y.; et al. Discovery of Phenylaminopyridine Derivatives as Novel HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. ACS Med. Chem. Lett. 2012, 3, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Al-Kaabi, S.S.; Elgemeie, G.E.H. Studies on Fused 2(1H)-Pyridenethiones-New Routes for the Synthesis of Fused 1H-Pyrazolo[3,4-b]Pyridines and Fused Thieno[2,3-b]Pyridines. Chem. Soc. Jpn. 1992, 65, 2241–2245. [Google Scholar] [CrossRef]
- Selvi, S.T.; Nadaraj, V.; Mohan, S.; Sasi, R.; Hema, M. Solvent Free Microwave Synthesis and Evaluation of Antimicrobial Activity of Pyrimido[4,5-b]- and Pyrazolo[3,4-b]Quinolines. Bioorganic Med. Chem. 2006, 14, 3896–3903. [Google Scholar] [CrossRef] [PubMed]
- El-Borai, M.A.; Rizk, H.F.; Beltagy, D.M.; El-Deeb, I.Y. Microwave-Assisted Synthesis of Some New Pyrazolopyridines and Their Antioxidant, Antitumor and Antimicrobial Activities. Eur. J. Med. Chem. 2013, 66, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.; Medishetti, N.; Kanugala, S.; Ganesh Kumar, C.; Atmakur, K. Na2S-Promoted Reduction of Azides in Water: Synthesis of Pyrazolopyridines in One Pot and Evaluation of Antimicrobial Activity. Org. Biomol. Chem. 2019, 17, 3186–3194. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.A.; El-Emary, T.I.; Mourad, A.F.E.; Alyan, Z.K.; Bräse, S.; Nieger, M. Synthesis of New Heterocycles from Reactions of 1-Phenyl-1H-Pyrazolo[3,4-b]Pyridine-5-Carbonyl Azides. J. Heterocycl. Chem. 2019, 56, 1369–1375. [Google Scholar] [CrossRef]
- Ghattas, A.B.A.G.; Khodairy, A.; Abd-Rahman, M.A.; Younes, S. Synthesis of Some New Pyrazolopyridines, Pyrazolothienopyridines, Pyrazolopyridothienopyrimidines and Pyrazolopyridothienotriazines. Phosphorus Sulfur Silicon Relat. Elem. 2003, 178, 1781–1794. [Google Scholar] [CrossRef]
- Girisha, K.S.; Kalluraya, B.; Vidyashree, J.H.S. Synthesis, Characterisation and Antimicrobial Activity of 3-Methyl-6-(Aryl)-1-Phenyl-1H-Pyrazolo[3,4-b]Pyridine. Indian J. Chem. 2012, 51, 1767–1770. [Google Scholar]
- Castillo, J.C.; Quiroga, J.; Abonia, R.; Rodriguez, J.; Coquerel, Y. The Aryne Aza-Diels-Alder Reaction: Flexible Syntheses of Isoquinolines. Org. Lett. 2015, 17, 3374–3377. [Google Scholar] [CrossRef]
- Xu, H.; Li, L.; Wang, Z.; Xi, J.; Rong, L. A Green Synthesis of 1,7-Dihydrodipyrazolo[3,4-b:4′,3′-e]Pyridin-3(2H)-One Derivatives from Deamination Cyclization Reactions in Aqueous Medium. Res. Chem. Intermed. 2018, 44, 3211–3226. [Google Scholar] [CrossRef]
- Cacciari, B.; Spalluto, G. Facile and Versatile Route to the Synthesis of Fused 2-Pyridones: Useful Intermediates for Polycyclic Sytems. Synth. Commun. 2006, 36, 1173–1183. [Google Scholar] [CrossRef]
- Witherington, J.; Bordas, V.; Gaiba, A.; Garton, N.S.; Naylor, A.; Rawlings, A.D.; Slingsby, B.P.; Smith, D.G.; Takle, A.K.; Ward, R.W. 6-Aryl-Pyrazolo[3,4-b]Pyridines: Potent Inhibitors of Glycogen Synthase Kinase-3 (GSK-3). Bioorganic Med. Chem. Lett. 2003, 13, 3055–3057. [Google Scholar] [CrossRef]
- Misra, R.N.; Xiao, H.Y.; Rawlins, D.B.; Shan, W.; Kellar, K.A.; Mulheron, J.G.; Sack, J.S.; Tokarski, J.S.; Kimball, S.D.; Webster, K.R. 1H-Pyrazolo[3,4-b]Pyridine Inhibitors of Cyclin-Dependent Kinases: Highly Potent 2,6-Difluorophenacyl Analogues. Bioorganic Med. Chem. Lett. 2003, 13, 2405–2408. [Google Scholar] [CrossRef]
- Huang, S.; Lin, R.; Yu, Y.; Lu, Y.; Connolly, P.J.; Chiu, G.; Li, S.; Emanuel, S.L.; Middleton, S.A. Synthesis of 3-(1H-Benzimidazol-2-Yl)-5-Isoquinolin-4-Ylpyrazolo[1,2-b]Pyridine, a Potent Cyclin Dependent Kinase 1 (CDK1) Inhibitor. Bioorganic Med. Chem. Lett. 2007, 17, 1243–1245. [Google Scholar] [CrossRef]
- Jing, L.; Tang, Y.; Xiao, Z. Discovery of Novel CDK Inhibitors via Scaffold Hopping from CAN508. Bioorganic Med. Chem. Lett. 2018, 28, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Connolly, P.J.; Lu, Y.; Chiu, G.; Li, S.; Yu, Y.; Huang, S.; Li, X.; Emanuel, S.L.; Middleton, S.A.; et al. Synthesis and Evaluation of Pyrazolo[3,4-b]Pyridine CDK1 Inhibitors as Anti-Tumor Agents. Bioorganic Med. Chem. Lett. 2007, 17, 4297–4302. [Google Scholar] [CrossRef]
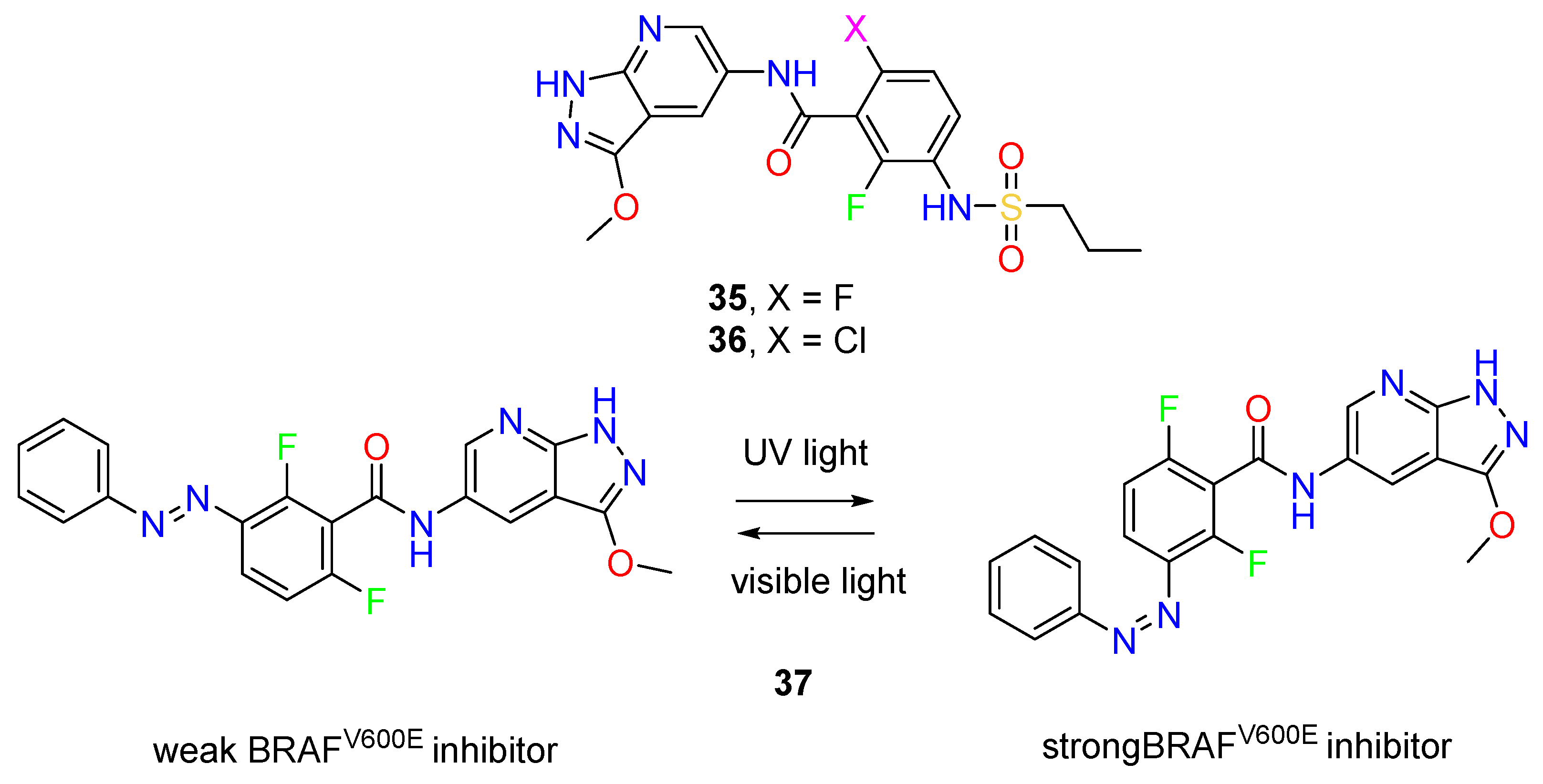
- Hoorens, M.W.H.; Ourailidou, M.E.; Rodat, T.; van der Wouden, P.E.; Kobauri, P.; Kriegs, M.; Peifer, C.; Feringa, B.L.; Dekker, F.J.; Szymanski, W. Light-Controlled Inhibition of BRAFV600E Kinase. Eur. J. Med. Chem. 2019, 179, 133–146. [Google Scholar] [CrossRef]
- Li, Y.; Shen, M.; Zhang, Z.; Luo, J.; Pan, X.; Lu, X.; Long, H.; Wen, D.; Zhang, F.; Leng, F.; et al. Design, Synthesis, and Biological Evaluation of 3-(1H-1,2,3-Triazol-1-Yl) Benzamide Derivatives as Potent Pan Bcr-Abl Inhibitors Including the Threonine315→isoleucine315 Mutant. J. Med. Chem. 2012, 55, 10033–10046. [Google Scholar] [CrossRef]
- Barghash, R.F.; Eldehna, W.M.; Kovalová, M.; Vojáčková, V.; Kryštof, V.; Abdel-Aziz, H.A. One-Pot Three-Component Synthesis of Novel Pyrazolo[3,4-b]Pyridines as Potent Antileukemic Agents. Eur. J. Med. Chem. 2022, 227, 113952. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Bair, K.W.; Bauer, P.; Baumeister, T.; Bowman, K.K.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Feng, Y.; Han, B.; et al. Identification of Amides Derived from 1H-Pyrazolo[3,4-b]Pyridine-5-Carboxylic Acid as Potent Inhibitors of Human Nicotinamide Phosphoribosyltransferase (NAMPT). Bioorganic Med. Chem. Lett. 2013, 23, 5488–5497. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.V.M.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Göktürk, C.; Sanjiv, K.; Strömberg, K.; et al. Stereospecific Targeting of MTH1 by (S)-Crizotinib as an Anticancer Strategy. Nature 2014, 508, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.; Liu, S.; Gao, M.; Wang, L.; Sun, J.; Du, J.; Guan, Q.; Bao, K.; Zuo, D.; Wu, Y.; et al. 3,5-Diaryl-1H-Pyrazolo[3,4-b]Pyridines as Potent Tubulin Polymerization Inhibitors: Rational Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2019, 168, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Hwang, D.; Kim, N.; Seo, H.S.; Selim, K.B.; Sim, T. Identification of 1H-Pyrazolo[3,4-b]Pyridine Derivatives as Potent ALK-L1196M Inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 1426–1438. [Google Scholar] [CrossRef]
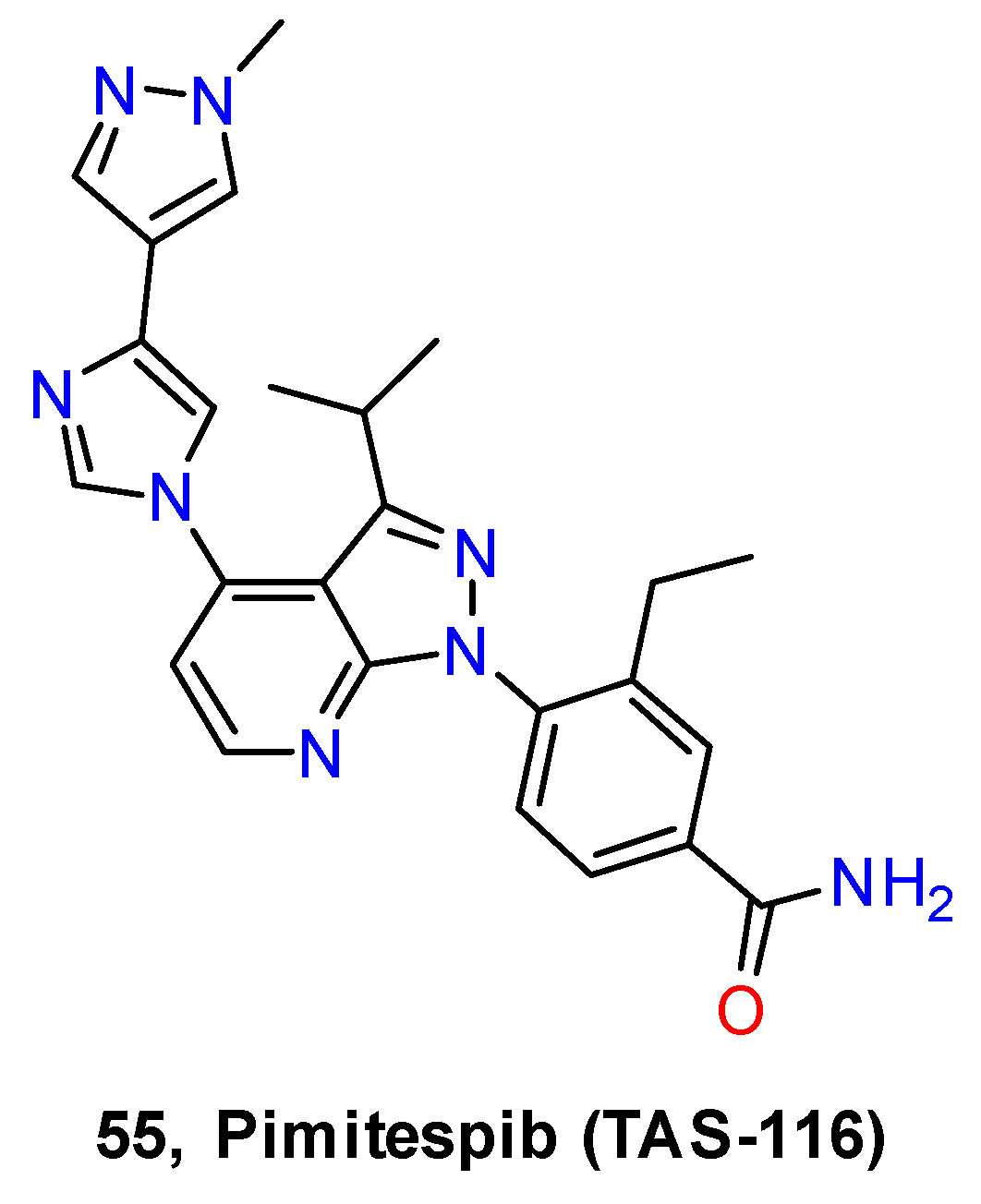
- Kihara, T.; Yuan, J.; Watabe, T.; Kitajima, K.; Kimura, N.; Ohkouchi, M.; Hashikura, Y.; Ohkubo, S.; Takahashi, T.; Hirota, S. Pimitespib Is Effective on Cecal GIST in a Mouse Model of Familial GISTs with KIT-Asp820Tyr Mutation through KIT Signaling Inhibition. Exp. Mol. Pathol. 2021, 123, 104692. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, Y.; Dong, G.; Zhang, W.; Liu, N.; Sheng, C. Identification of Pyrazolopyridine Derivatives as Novel Spleen Tyrosine Kinase Inhibitors. Arch. Pharm. 2018, 351, 1800083. [Google Scholar] [CrossRef]
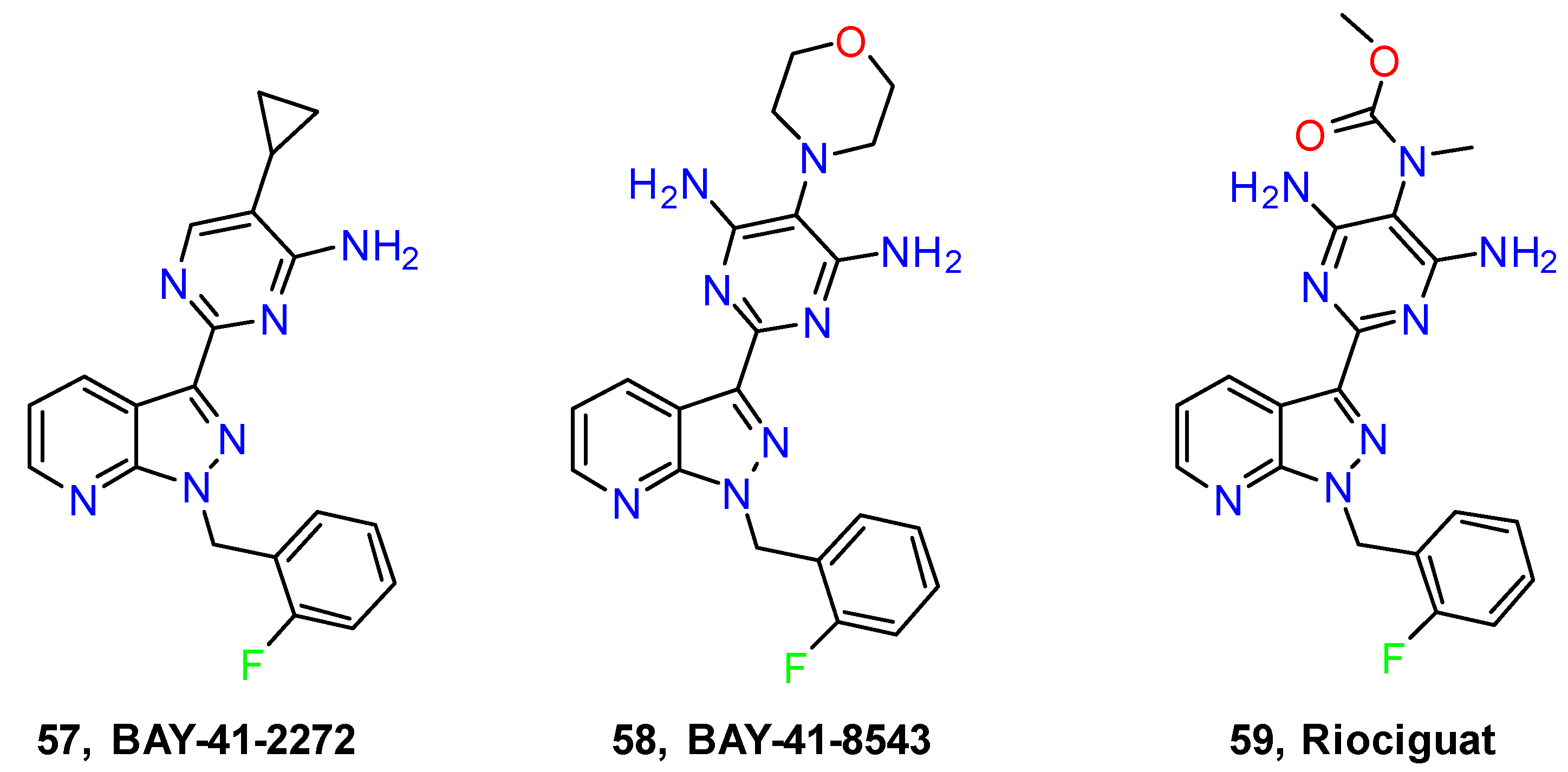
- Goldenberg, N.M.; Rabinovitch, M.; Steinberg, B.E. Inflammatory Basis of Pulmonary Arterial HypertensionImplications for Perioperative and Critical Care Medicine. Anesthesiology 2019, 131, 898–907. [Google Scholar] [CrossRef]
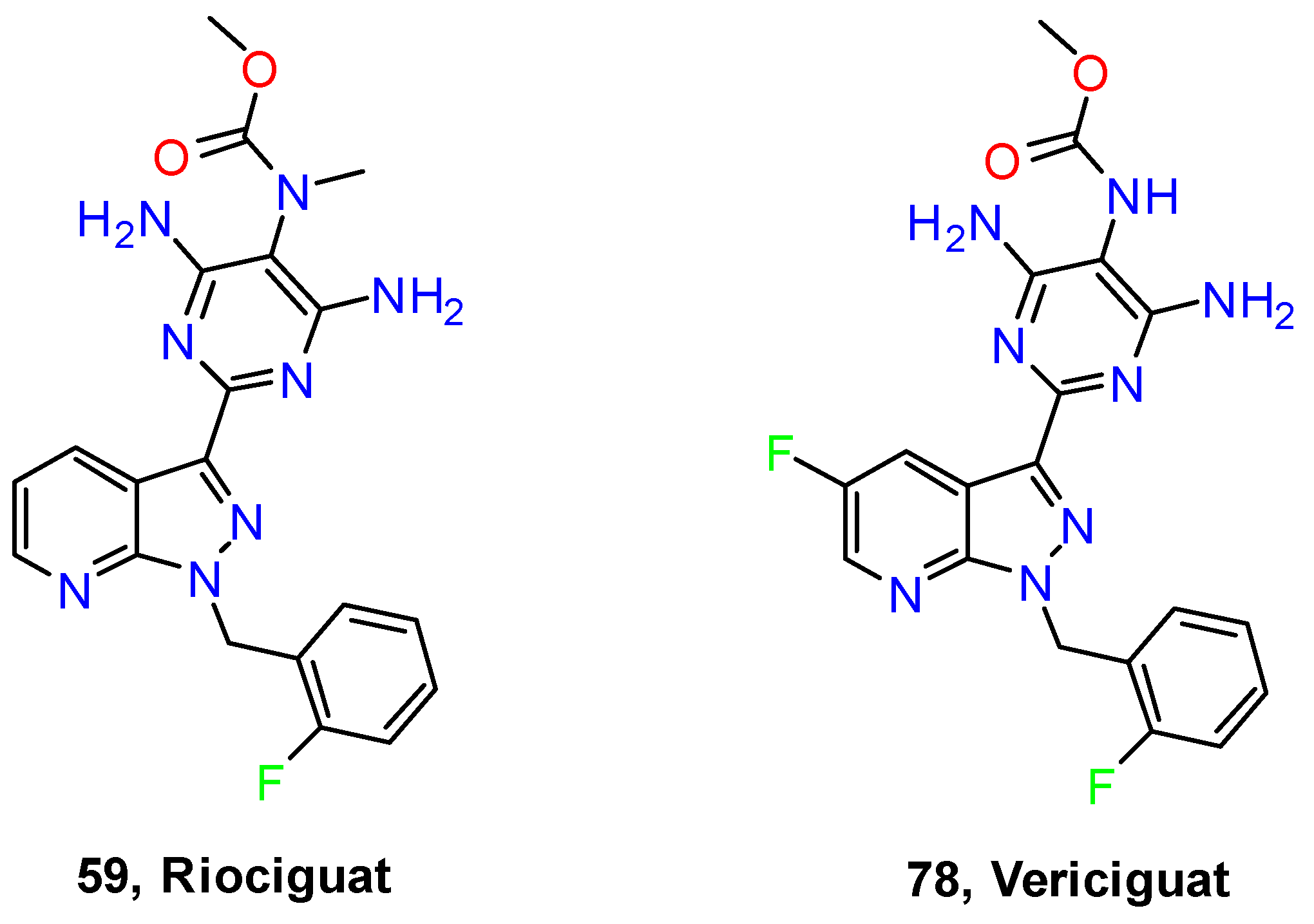
- Mittendorf, J.; Weigand, S.; Alonso-Alija, C.; Bischoff, E.; Feurer, A.; Gerisch, M.; Kern, A.; Knorr, A.; Lang, D.; Muenter, K.; et al. Discovery of Riociguat (BAY 63-2521): A Potent, Oral Stimulator of Soluble Guanylate Cyclase for the Treatment of Pulmonary Hypertension. ChemMedChem 2009, 4, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Mahajan, T.R.; Gole, Y.R.; Nambiar, M.; Matan, T.T.; Kulkarni-Almeida, A.; Balachandran, S.; Junjappa, H.; Balakrishnan, A.; Vishwakarma, R.A. Synthesis and Evaluation of Pyrazolo[3,4-b]Pyridines and Its Structural Analogues as TNF-α and IL-6 Inhibitors. Bioorganic Med. Chem. 2008, 16, 7167–7176. [Google Scholar] [CrossRef]
- Revesz, L.; Blum, E.; di Padova, F.E.; Buhl, T.; Feifel, R.; Gram, H.; Hiestand, P.; Manning, U.; Neumann, U.; Rucklin, G. Pyrazoloheteroaryls: Novel P38α MAP Kinase Inhibiting Scaffolds with Oral Activity. Bioorganic Med. Chem. Lett. 2006, 16, 262–266. [Google Scholar] [CrossRef]
- Hamdy, N.A.; Gamal-Eldeen, A.M. New Pyridone, Thioxopyridine, Pyrazolopyridine and Pyridine Derivatives That Modulate Inflammatory Mediators in Stimulated RAW 264.7 Murine Macrophage. Eur. J. Med. Chem. 2009, 44, 4547–4556. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, H.; Ishida, A.; Ohtani, T.; Kusumi, K.; Kishikawa, K.; Yamamoto, S.; Takeda, H.; Obata, T.; Nakai, H.; Toda, M. New Orally Active PDE4 Inhibitors with Therapeutic Potential. Bioorganic Med. Chem. 2004, 12, 4089–4100. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, J.N.; Angell, T.D.R.; Ballantine, S.P.; Cook, C.M.; Cooper, A.W.J.; Dawson, J.; Delves, C.J.; Jones, P.S.; Lindvall, M.; Lucas, F.S.; et al. Pyrazolopyridines as a Novel Structural Class of Potent and Selective PDE4 Inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 4237–4241. [Google Scholar] [CrossRef]
- Mitchell, C.J.; Ballantine, S.P.; Coe, D.M.; Cook, C.M.; Delves, C.J.; Dowle, M.D.; Edlin, C.D.; Hamblin, J.N.; Holman, S.; Johnson, M.R.; et al. Pyrazolopyridines as Potent PDE4B Inhibitors: 5-Heterocycle SAR. Bioorganic Med. Chem. Lett. 2010, 20, 5803–5806. [Google Scholar] [CrossRef]
- Jimenez, J.M.; Boyall, D.; Brenchley, G.; Collier, P.N.; Davis, C.J.; Fraysse, D.; Keily, S.B.; Henderson, J.; Miller, A.; Pierard, F.; et al. Design and Optimization of Selective Protein Kinase C θ (PKCθ) Inhibitors for the Treatment of Autoimmune Diseases. J. Med. Chem. 2013, 56, 1799–1810. [Google Scholar] [CrossRef]
- Schenone, S.; Bruno, O.; Fossa, P.; Ranise, A.; Menozzi, G.; Mosti, L.; Bondavalli, F.; Martini, C.; Trincavelli, L. Synthesis and Biological Data of 4-Amino-1-(2-Chloro-2-Phenylethyl)-1H-Pyrazolo[3,4-b]Pyridine-5-Carboxylic Acid Ethyl Esters, a New Series of A 1-Adenosine Receptor (A 1 AR) Ligands. Bioorganic Med. Chem. Lett. 2001, 11, 2529–2531. [Google Scholar] [CrossRef]
- Menegatti, R.; Silva, G.M.S.; Zapata-Sudo, G.; Raimundo, J.M.; Sudo, R.T.; Barreiro, E.J.; Fraga, C.A.M. Design, Synthesis, and Pharmacological Evaluation of New Neuroactive Pyrazolo[3,4-b]Pyrrolo[3,4-d]Pyridine Derivatives with in Vivo Hypnotic and Analgesic Profile. Bioorganic Med. Chem. 2006, 14, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Dyck, B.; Grigoriadis, D.E.; Gross, R.S.; Guo, Z.; Haddach, M.; Marinkovic, D.; McCarthy, J.R.; Moorjani, M.; Regan, C.F.; Saunders, J.; et al. Potent, Orally Active Corticotropin-Releasing Factor Receptor-1 Antagonists Containing a Tricyclic Pyrrolopyridine or Pyrazolopyridine Core. J. Med. Chem. 2005, 48, 4100–4110. [Google Scholar] [CrossRef]
- Nascimento-Júnior, N.M.; Mendes, T.C.F.; Leal, D.M.; Corrêa, C.M.N.; Sudo, R.T.; Zapata-Sudo, G.; Barreiro, E.J.; Fraga, C.A.M. Microwave-Assisted Synthesis and Structure-Activity Relationships of Neuroactive Pyrazolo[3,4-b]Pyrrolo[3,4-d]Pyridine Derivatives. Bioorganic Med. Chem. Lett. 2010, 20, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.D.; Yang, S.W.; Smotryski, J.; Bercovici, A.; Nechuta, T.; Smith, E.M.; McElroy, W.; Tan, Z.; Tulshian, D.; McKittrick, B.; et al. The Discovery of Potent, Selective, and Orally Active Pyrazoloquinolines as PDE10A Inhibitors for the Treatment of Schizophrenia. Bioorganic Med. Chem. Lett. 2012, 22, 1019–1022. [Google Scholar] [CrossRef]
- Goudey, B.; Fung, B.J.; Schieber, C.; Faux, N.G.; Weiner, M.W.; Aisen, P.; Petersen, R.; Jack, C.R.; Jagust, W.; Trojanowki, J.Q.; et al. A Blood-Based Signature of Cerebrospinal Fluid Aβ 1–42 Status. Sci. Rep. 2019, 9, 4163. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.; Chioua, M.; Samadi, A.; Agostinho, P.; Garção, P.; Lajarín-Cuesta, R.; de Los Ríos, C.; Iriepa, I.; Moraleda, I.; Gonzalez-Lafuente, L.; et al. Synthesis, Pharmacological Assessment, and Molecular Modeling of Acetylcholinesterase/Butyrylcholinesterase Inhibitors: Effect against Amyloid-β-Induced Neurotoxicity. ACS Chem. Neurosci. 2013, 4, 547–565. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Duggan, S. Vericiguat: First Approval. Drugs 2021, 81, 721–726. [Google Scholar] [CrossRef] [PubMed]



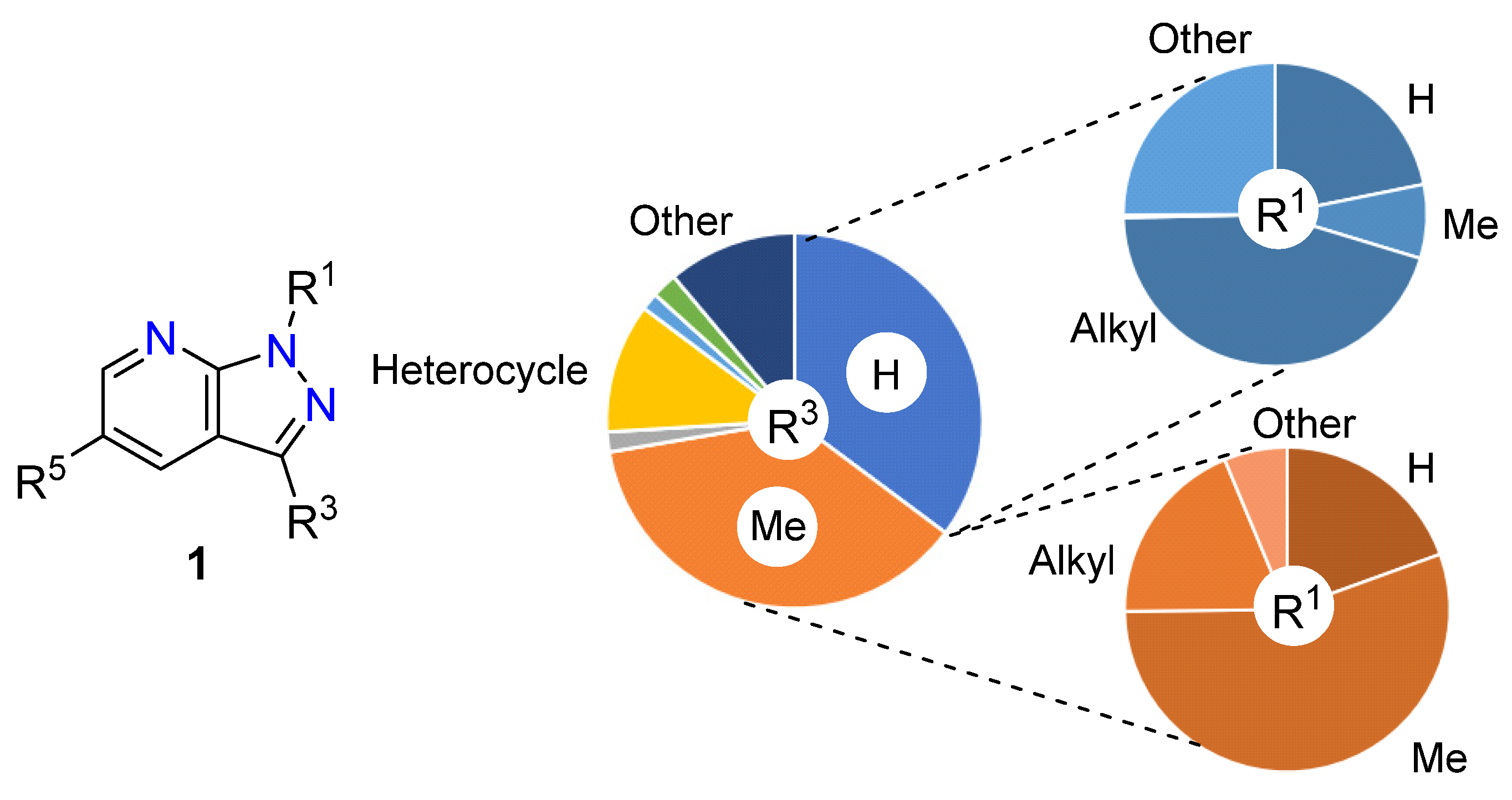






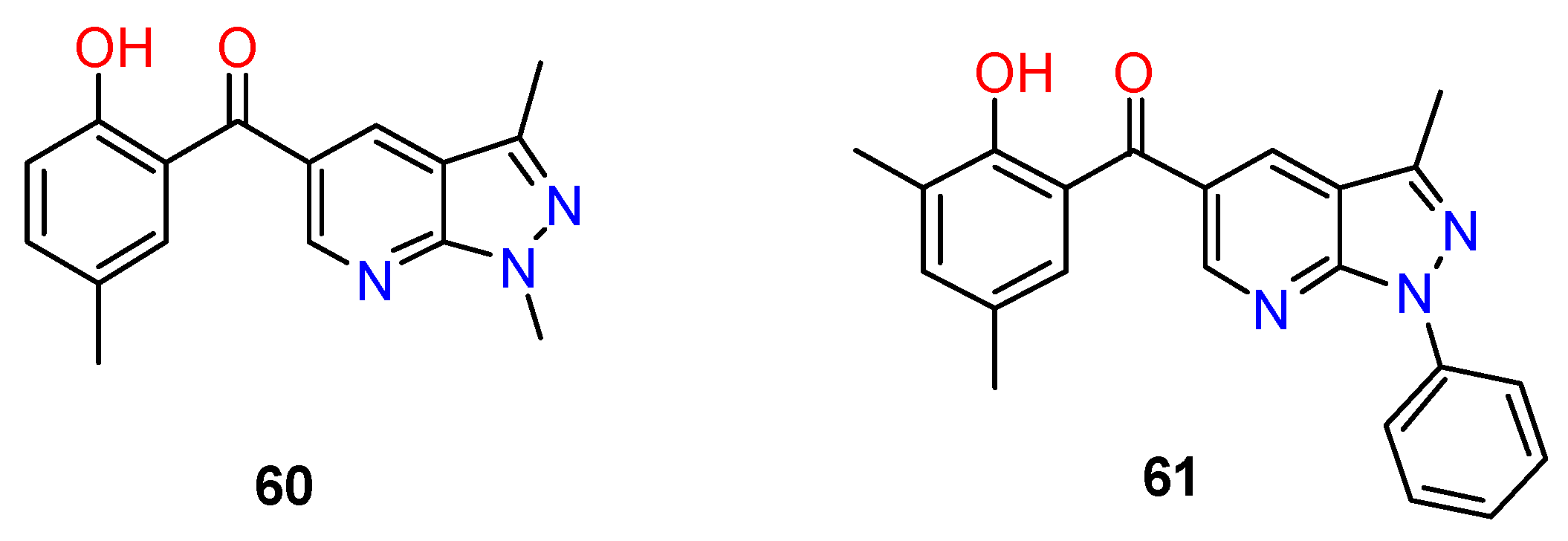

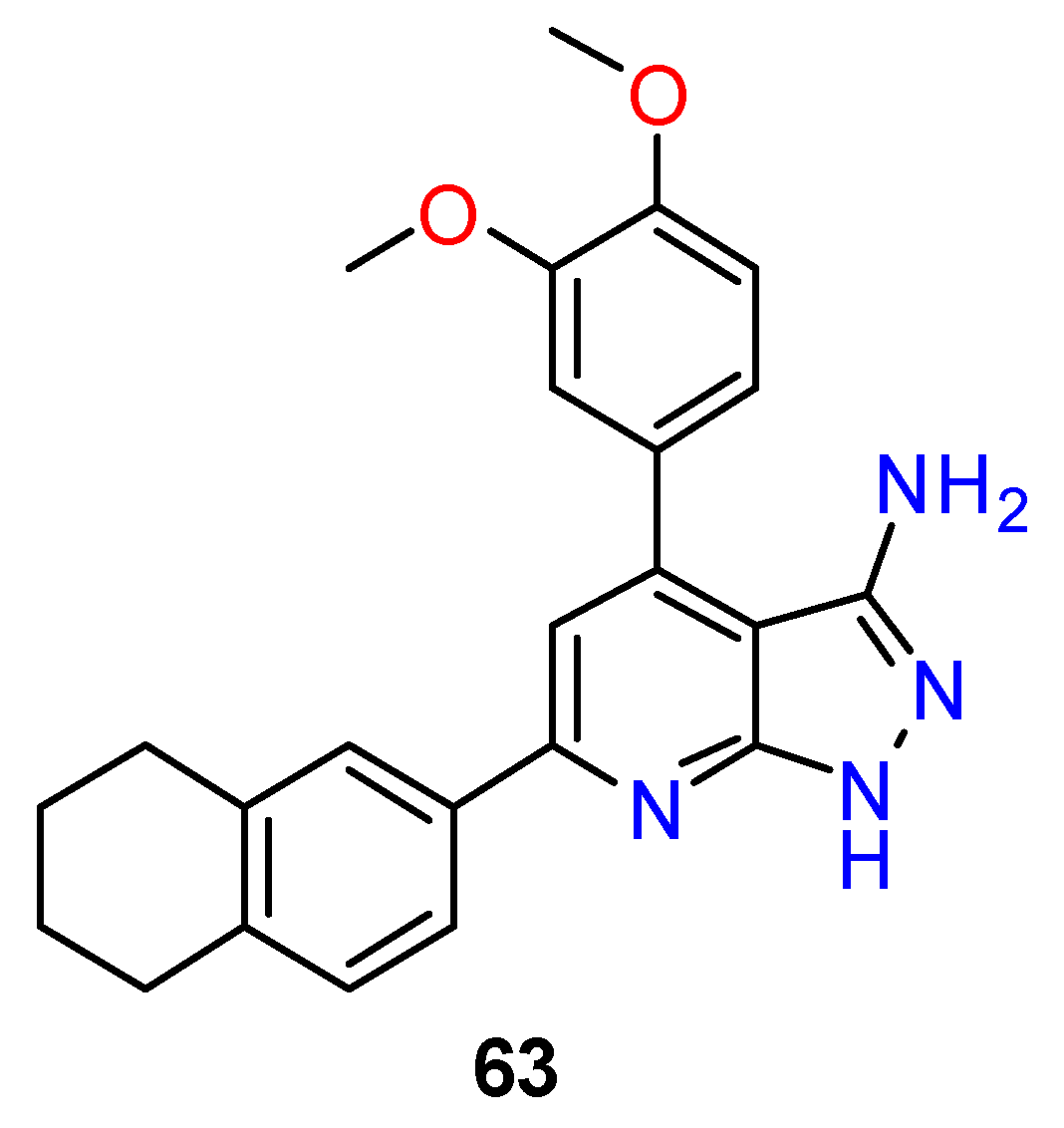
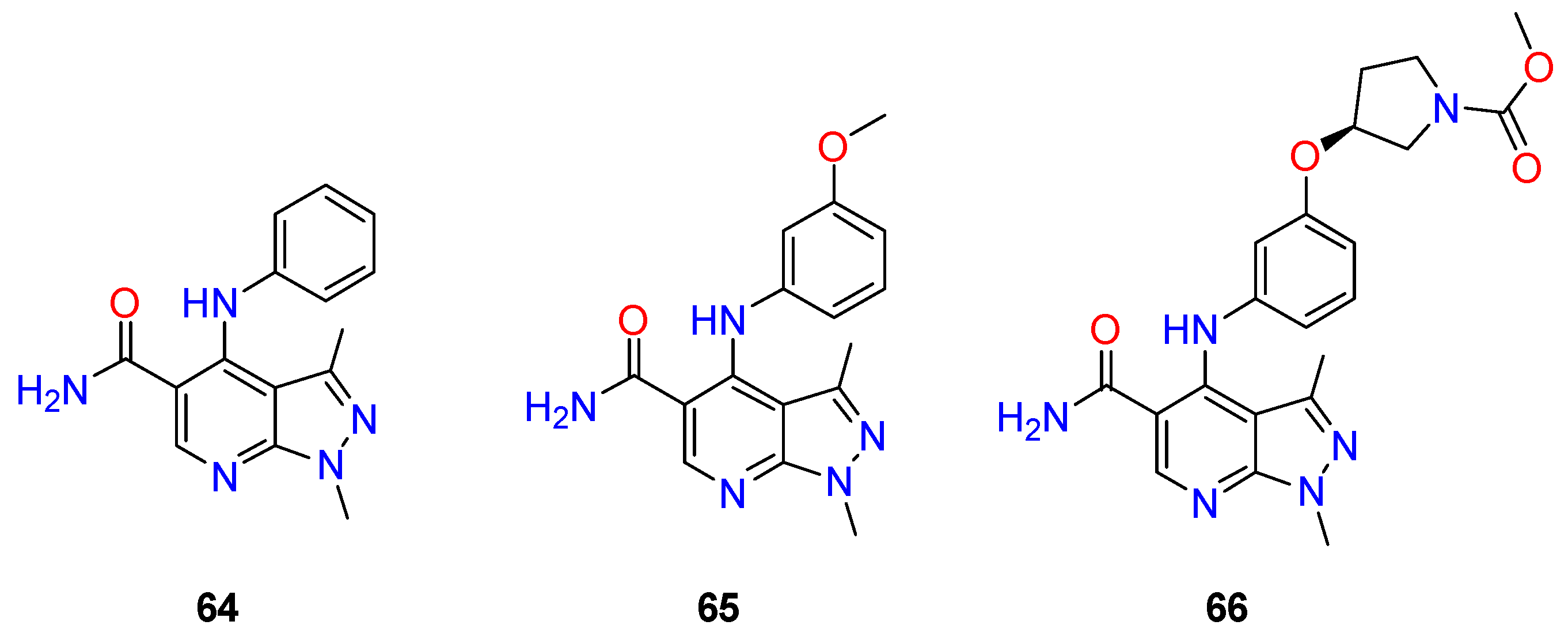
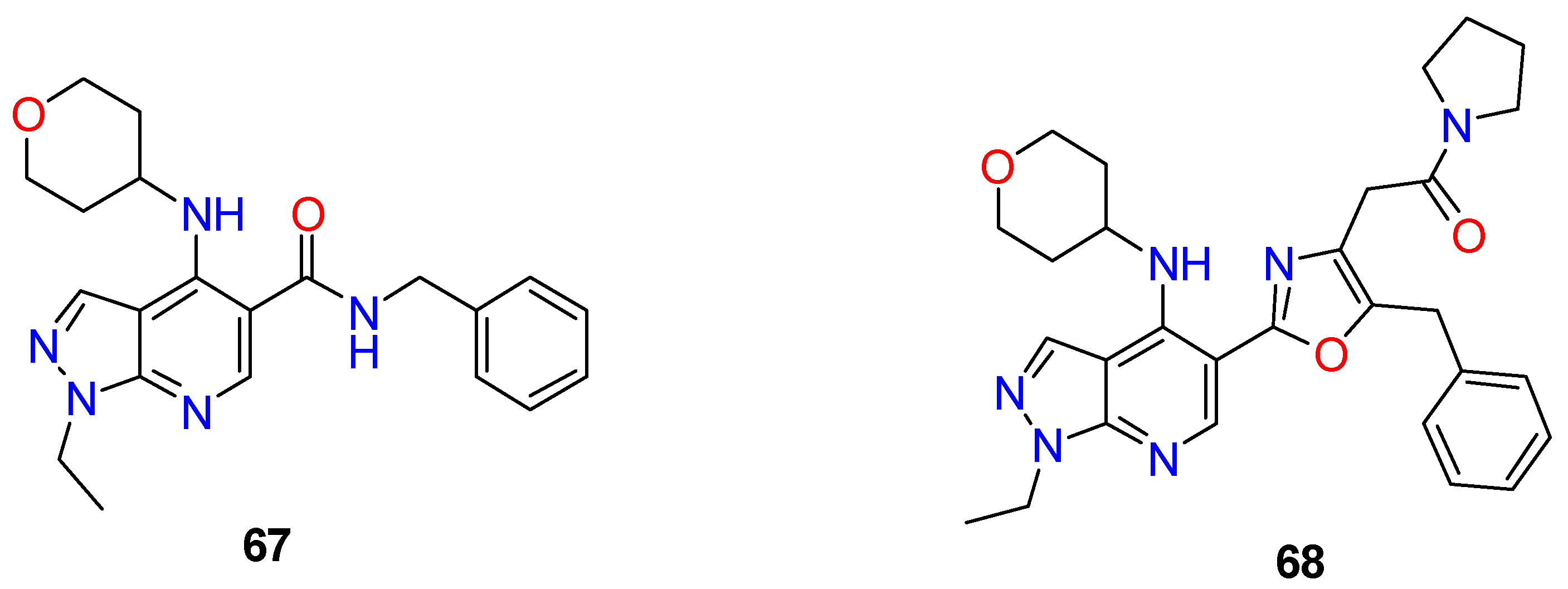



| R1 | Structures 1 (%) | Number of References | Selected References |
|---|---|---|---|
| H | 19.70 | 2520 | [18,19] |
| Me | 31.78 | 986 | [20,21] |
| Alkyl | 23.27 | 823 | [22,23] |
| Cycloalkyl | 0.70 | 47 | [24,25] |
| Ph | 15.17 | 1077 | [26,27] |
| Heterocycle | 2.33 | 284 | [27,28] |
| Other | 7.05 | - | - |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donaire-Arias, A.; Montagut, A.M.; Puig de la Bellacasa, R.; Estrada-Tejedor, R.; Teixidó, J.; Borrell, J.I. 1H-Pyrazolo[3,4-b]pyridines: Synthesis and Biomedical Applications. Molecules 2022, 27, 2237. https://doi.org/10.3390/molecules27072237
Donaire-Arias A, Montagut AM, Puig de la Bellacasa R, Estrada-Tejedor R, Teixidó J, Borrell JI. 1H-Pyrazolo[3,4-b]pyridines: Synthesis and Biomedical Applications. Molecules. 2022; 27(7):2237. https://doi.org/10.3390/molecules27072237
Chicago/Turabian StyleDonaire-Arias, Ana, Ana Maria Montagut, Raimon Puig de la Bellacasa, Roger Estrada-Tejedor, Jordi Teixidó, and José I. Borrell. 2022. "1H-Pyrazolo[3,4-b]pyridines: Synthesis and Biomedical Applications" Molecules 27, no. 7: 2237. https://doi.org/10.3390/molecules27072237
APA StyleDonaire-Arias, A., Montagut, A. M., Puig de la Bellacasa, R., Estrada-Tejedor, R., Teixidó, J., & Borrell, J. I. (2022). 1H-Pyrazolo[3,4-b]pyridines: Synthesis and Biomedical Applications. Molecules, 27(7), 2237. https://doi.org/10.3390/molecules27072237