
Dynamic Phenomena and Complexation Effects in the α-Lithiation and Asymmetric Functionalization of Azetidines

 ,
,  , ,
, ,
Abstract
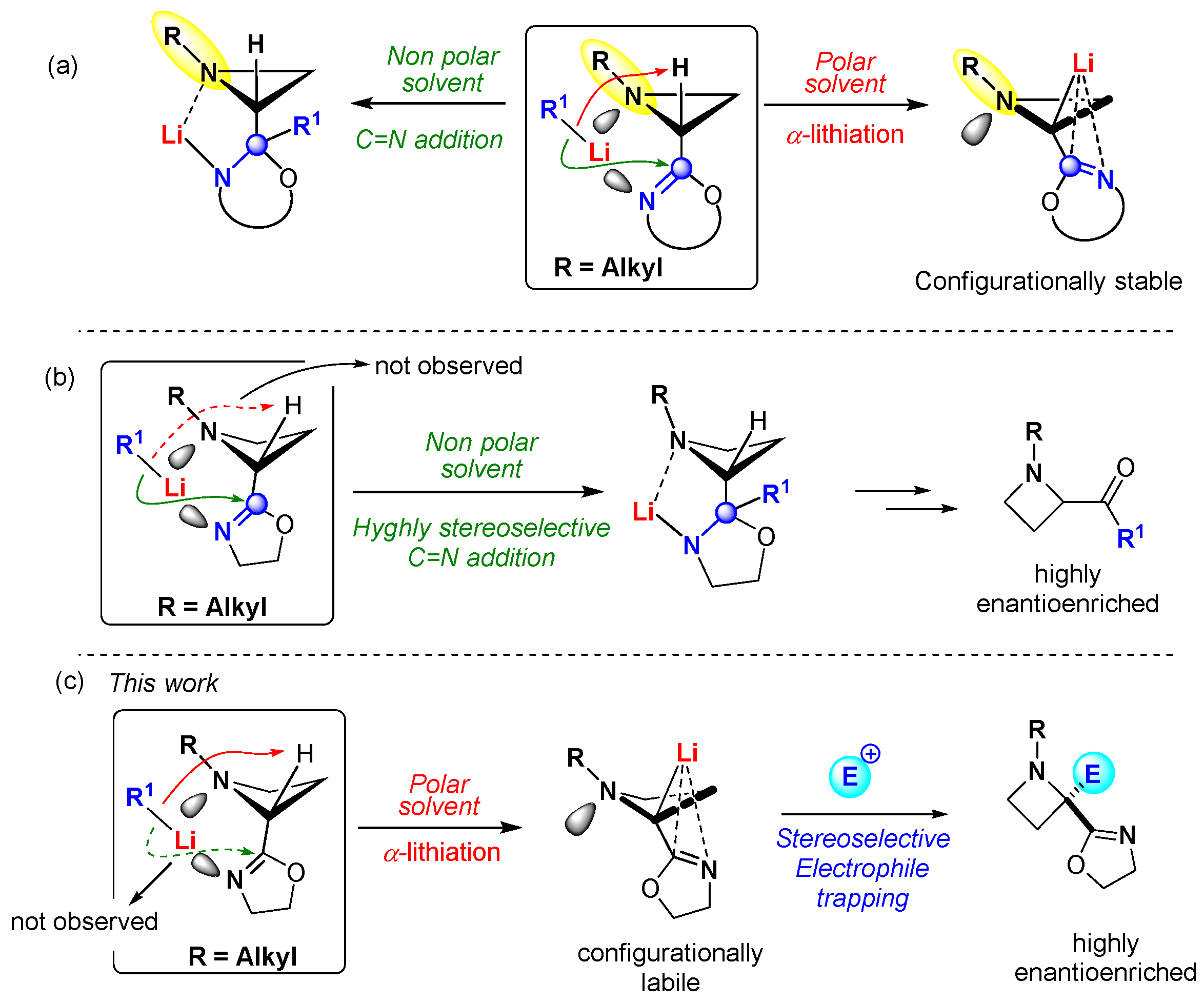
:1. Introduction
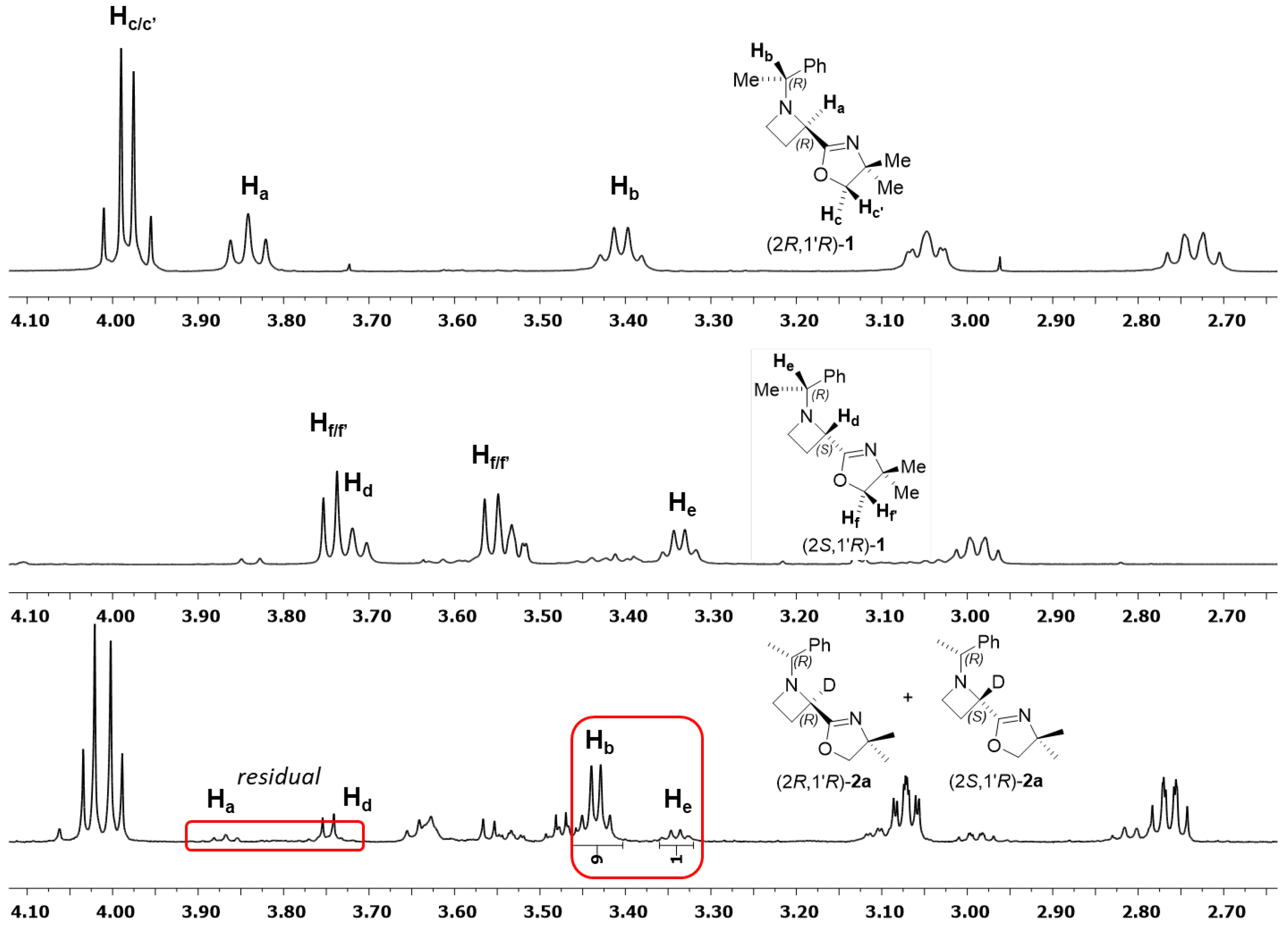
2. Results
3. In Situ FT-IR Investigation
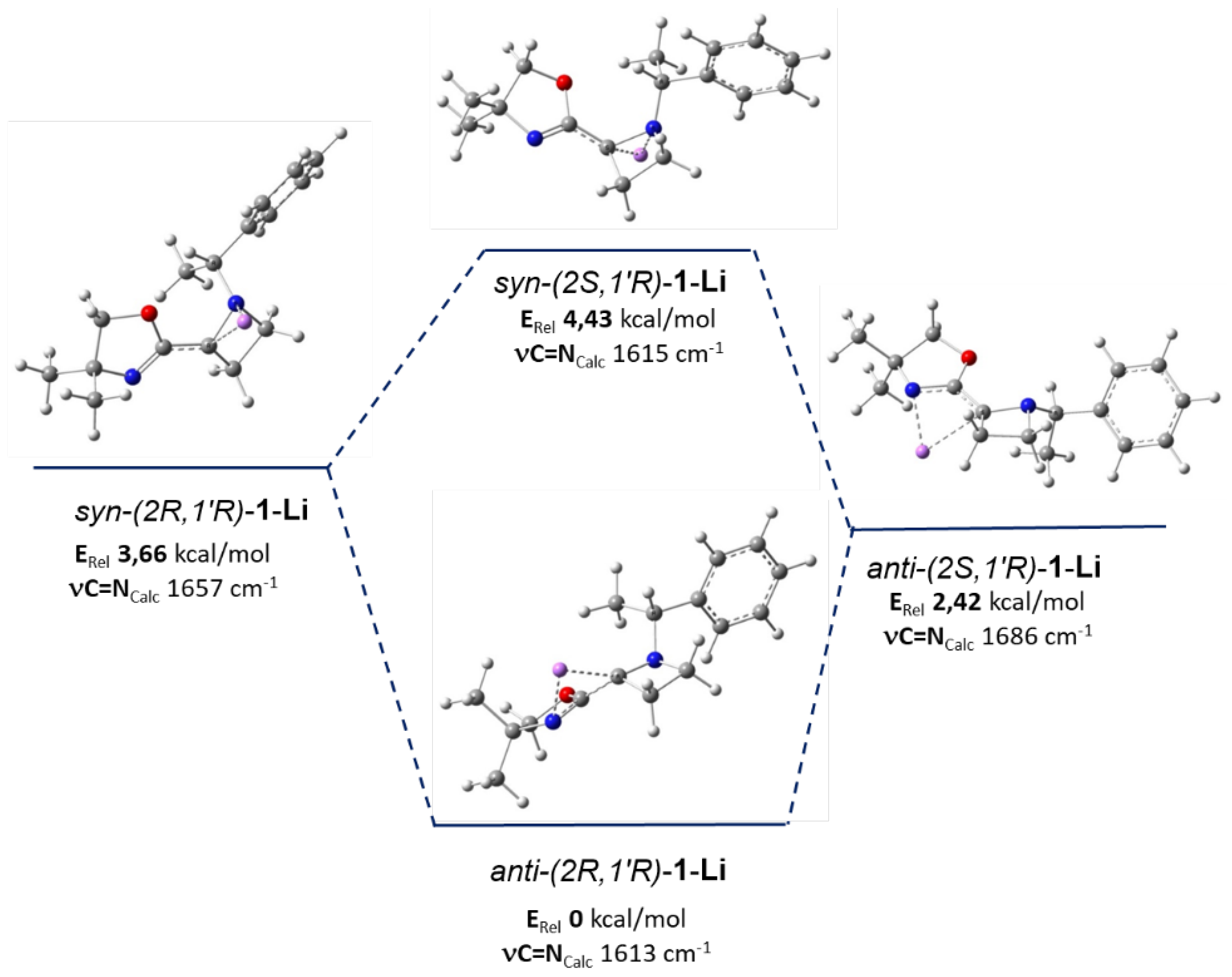
4. DFT Studies
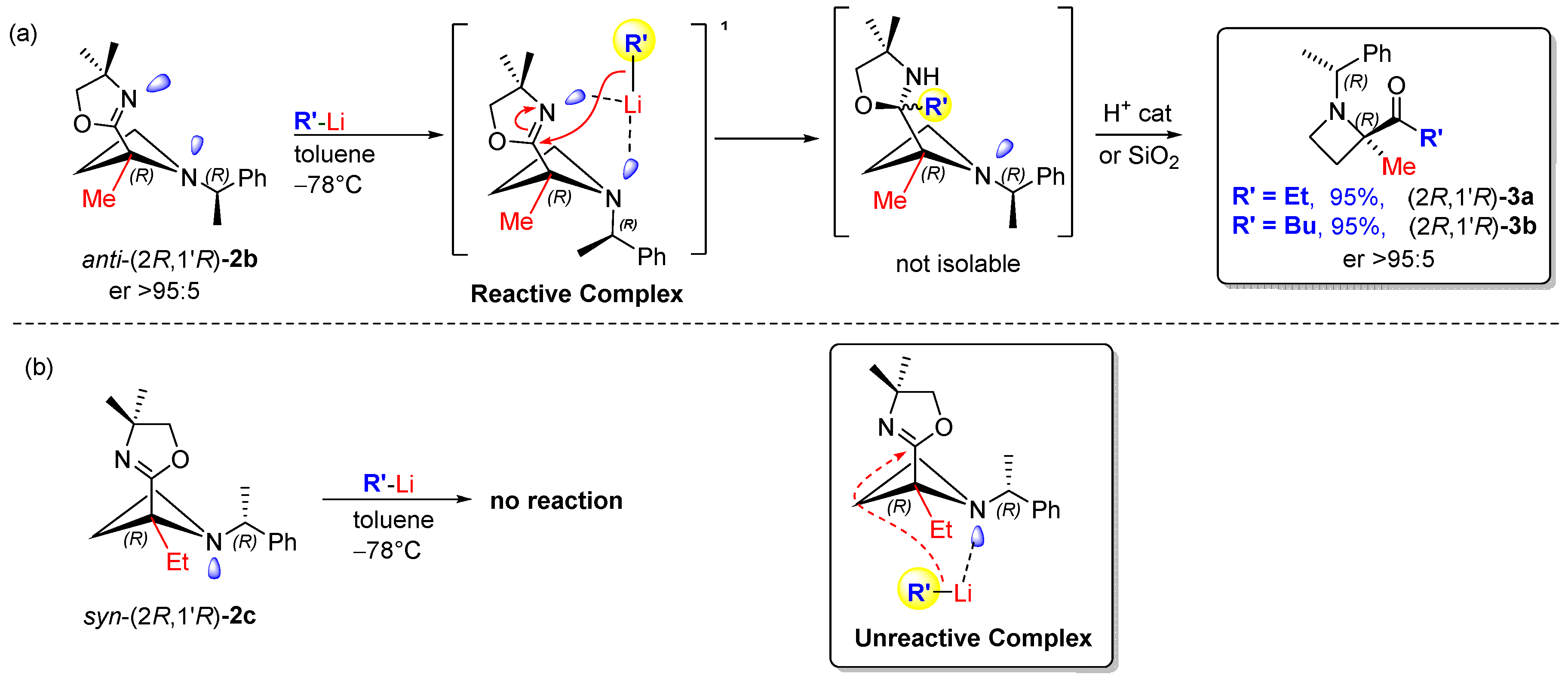
5. Reaction Scope
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References and Notes
- Andresini, M.; Degennaro, L.; Luisi, R. Azetidines, Azetines and Azetes: Monocyclic. In Comprehensive Heterocyclic Chemistry IV; Black, D., St, C., Cossy, J., Stevens, C.V., Eds.; Elsevier: Oxford, UK, 2022; Volume 2, pp. 1–115. [Google Scholar]
- Mughal, H.; Szostak, M. Recent advances in the synthesis and reactivity of azetidines: Strain-driven character of the four-membered heterocycle. Org. Biomol. Chem. 2021, 19, 3274–3286. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.D.; Becker, M.R.; Schindler, C.S. Synthesis of azetidines by aza Paternò–Büchi reactions. Chem. Sci. 2020, 11, 7553–7561. [Google Scholar]
- Becker, M.R.; Wearing, E.R.; Schindler, C.S. Synthesis of azetidines via visible-light-mediated intermolecular [2+2] photocycloadditions. Nat. Chem. 2020, 12, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Andresini, M.; Degennaro, L.; Luisi, R. The renaissance of strained 1-azabicyclo [1.1. 0] butanes as useful reagents for the synthesis of functionalized azetidines. Org. Biomol. Chem. 2020, 18, 5798–5810. [Google Scholar] [CrossRef]
- Antermite, D.; Degennaro, L.; Luisi, R. Recent advances in the chemistry of metallated azetidines. Org. Biomol. Chem. 2017, 15, 34. [Google Scholar] [CrossRef]
- Parmar, D.; Henkel, L.; Dib, J.; Rueping, M. Iron catalysed cross-couplings of azetidines–application to the formal synthesis of a pharmacologically active molecule. Chem. Commun. 2015, 51, 2111–2113. [Google Scholar] [CrossRef]
- Hodgson, D.M.; Kloesges, J. Lithiation–Electrophilic Substitution of N-Thiopivaloylazetidine. Angew. Chem. Int. Ed. 2010, 49, 2900. [Google Scholar] [CrossRef]
- Jackson, K.E.; Mortimer, C.L.; Odell, B.; McKenna, J.M.; Claridge, T.D.W.; Paton, R.S.; Hodgson, D.M. α- and α’-Lithiation–Electrophile Trapping of N-Thiopivaloyl and N-tert-Butoxythiocarbonyl α-Substituted Azetidines: Rationalization of the Regiodivergence Using NMR and Computation. J. Org. Chem. 2015, 80, 9838–9846. [Google Scholar] [CrossRef]
- Delany, P.K.; Mortimer, C.L.; Hodgson, D.M. Electrophile dependent mechanisms in the asymmetric trapping of α-lithio-N-(tert-butoxythiocarbonyl) azetidine. Chem. Commun. 2020, 56, 12174–12177. [Google Scholar] [CrossRef]
- Colella, M.; Musci, P.; Cannillo, D.; Spennacchio, M.; Aramini, A.; Degennaro, L.; Luisi, R. Development of a Continuous Flow Synthesis of 2-Substituted Azetines and 3-Substituted Azetidines by Using a Common Synthetic Precursor. J. Org. Chem. 2021, 86, 13943–13954. [Google Scholar] [CrossRef]
- Musci, P.; Keutz, T.; Belaj, F.; Degennaro, L.; Cantillo, D.; Kappe, C.O.; Luisi, R. Flow Technology for Telescoped Generation, Lithiation and Electrophilic (C3) Functionalization of Highly Strained 1-Azabicyclo[1.1.0]butanes. Angew. Chem. Int. Ed. 2021, 60, 6395–6399. [Google Scholar] [CrossRef]
- Degennaro, L.; Musio, B.; Luisi, R. Nitrogen-Bearing Lithium Compounds in Modern Synthesis. In Lithium Compounds in Organic Synthesis: From Fundamentals to Applications; Wiley Blackwell: Weinheim, Germany, 2014; ISBN 9783527333431. pp. 191–224. [Google Scholar]
- Andresini, M.; De Angelis, S.; Uricchio Visaggio, A.; Romanazzi, G.; Ciriaco, F.; Corriero, N.; Degennaro, L.; Luisi, R. Azetidine–borane complexes: Synthesis, reactivity, and stereoselective functionalization. J. Org. Chem. 2018, 83, 10221–10230. [Google Scholar]
- Azzena, U.; Dettori, G.; Pisano, L.; Musio, B.; Luisi, R. BH3-Promoted Stereoselective β-Lithiation of N-Alkyl-2-phenylaziridines. J. Org. Chem. 2011, 76, 2291–2295. [Google Scholar] [CrossRef]
- Parisi, G.; Zenzola, M.; Capitanelli, E.; Carlucci, C.; Romanazzi, G.; Pisano, L.; Degennaro, L.; Luisi, R. Exploiting structural and conformational effects for a site-selective lithiation of azetidines. Pure Appl. Chem. 2016, 88, 631–648. [Google Scholar] [CrossRef] [Green Version]
- Parisi, G.; Capitanelli, E.; Pierro, A.; Romanazzi, G.; Clarkson, G.J.; Degennaro, L.; Luisi, R. Easy access to constrained peptidomimetics and 2, 2-disubstituted azetidines by the unexpected reactivity profile of α-lithiated N-Boc-azetidines. Chem. Commun. 2015, 51, 15588–15591. [Google Scholar] [CrossRef]
- Degennaro, L.; Zenzola, M.; Trinchera, P.; Carroccia, L.; Giovine, A.; Romanazzi, G.; Falcicchio, A.; Luisi, R. Regioselective functionalization of 2-arylazetidines: Evaluating the ortho-directing ability of the azetidinyl ring and the α-directing ability of the N-substituent. Chem. Commun. 2014, 50, 1698–1700. [Google Scholar] [CrossRef]
- Zenzola, M.; Degennaro, L.; Trinchera, P.; Carroccia, L.; Giovine, A.; Romanazzi, G.; Mastrorilli, P.; Rizzi, R.; Pisano, L.; Luisi, R. Harnessing the ortho-Directing Ability of the Azetidine Ring for the Regioselective and Exhaustive Functionalization of Arenes. Chem. Eur. J. 2014, 20, 12190–12200. [Google Scholar] [CrossRef]
- Affortunato, F.; Florio, S.; Luisi, R.; Musio, B. α- vs. Ortho-Lithiation of N-Alkylarylaziridines: Probing the Role of the Nitrogen Inversion Process. J. Org. Chem. 2008, 73, 9214–9220. [Google Scholar] [CrossRef]
- Dammacco, M.; Degennaro, L.; Florio, S.; Luisi, R.; Musio, B.; Altomare, A. Lithiation of N-Alkyl-(o-tolyl)aziridine: Stereoselective Synthesis of Isochromans. J. Org. Chem. 2009, 74, 6319–6322. [Google Scholar] [CrossRef]
- De Ceglie, M.C.; Musio, B.; Affortunato, F.; Moliterni, A.; Altomare, A.; Florio, S.; Luisi, R. Solvent- and Temperature- Dependent Functionalisation of Enantioenriched Aziridines. Chem. Eur. J. 2011, 17, 286–296. [Google Scholar] [CrossRef]
- Degennaro, L.; Pisano, L.; Parisi, G.; Mansueto, R.; Clarkson, G.J.; Shipman, M.; Luisi, R. Nitrogen Stereodynamics and Complexation Phenomena as Key Factors in the Deprotonative Dynamic Resolution of Alkylideneaziridines: A Spectroscopic and Computational Study. J. Org. Chem. 2015, 80, 6411–6418. [Google Scholar] [CrossRef]
- Degennaro, L.; Mansueto, R.; Carenza, E.; Rizzi, R.; Florio, S.; Pratt, L.M.; Luisi, R. Nitrogen dynamics and reactivity of chiral aziridines: Generation of configurationally stable aziridinyllithium compounds. Chem. Eur. J. 2011, 17, 4992–5003. [Google Scholar] [CrossRef]
- Musci, P.; Colella, M.; Fanelli, F.; Altomare, A.; Pisano, L.; Carlucci, C.; Luisi, R.; Degennaro, L. Stereo-and Enantioselective Addition of Organolithiums to 2-Oxazolinylazetidines as a Synthetic route to 2-Acylazetidines. Front. Chem. 2019, 7, 614. [Google Scholar] [CrossRef] [Green Version]
- Rayner, P.J.; Smith, J.C.; Denneval, C.; O’Brien, P.; Clarke, P.A.; Horan, R.A.J. Mechanistic interrogation of the asymmetric lithiation-trapping of N-thiopivaloyl azetidine and pyrrolidine. Chem. Commun. 2016, 52, 1354–1357. [Google Scholar] [CrossRef]
- Wolf, C. Dynamic Stereochemistry of Chiral Compounds Principles and Application; RSC Publishing: London, UK, 2008. [Google Scholar]
- Pisano, L.; Degennaro, L.; Carraro, M.; Azzena, U.; Fanelli, F.; Mastrorilli, P.; Luisi, R. Computational NMR as Useful Tool for Predicting Structure and Stereochemistry of Four-Membered Sulfur Heterocycles. Eur. J. Org. Chem. 2016, 3252–3258. [Google Scholar] [CrossRef]
- Sheikh, N.S.; Leonori, D.; Barker, G.; Firth, J.D.; Campos, K.R.; Meijer, A.J.H.M.; O’Brien, P.; Coldham, I. An Experimental and in Situ IR Spectroscopic Study of the Lithiation–Substitution of N-Boc-2-phenylpyrrolidine and -piperidine: Controlling the Formation of Quaternary Stereocenters. J. Am. Chem. Soc. 2012, 134, 5300–5308. [Google Scholar] [CrossRef]
- Castagnolo, D.; Degennaro, L.; Luisi, R.; Clayden, J. Enantioselective carbolithiation of S-alkenyl-N-aryl thiocarbamates: Kinetic and thermodynamic control. Org. Biomol. Chem. 2015, 13, 2330–2340. [Google Scholar] [CrossRef]
- Mansueto, R.; Degennaro, L.; Brière, J.-F.; Griffin, K.; Shipman, M.; Florio, S.; Luisi, R. A convenient enantioselective CBS-reduction of arylketones in flow-microreactor systems. Org. Biomol. Chem. 2014, 12, 8505–8511. [Google Scholar] [CrossRef] [PubMed]
- Stead, D.; Carbone, G.; O’Brien, P.; Campos, K.R.; Coldham, I.; Sanderson, A.J. Asymmetric Deprotonation of N-Boc Piperidine: React IR Monitoring and Mechanistic Aspects. Am. Chem. Soc. 2010, 132, 7260–7261. [Google Scholar] [CrossRef] [PubMed]
- Absolute stereochemistry was assigned by analogy according to the results observed in the lithiation/deuteration sequence, and on the basis of the X-ray analysis of 2i. It is assumed that reactions with the electrophile accurred with retention of configuration at the lithiated carbon.
- CCDC Deposition Number 2144109.
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Denningtom, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, non-covalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Bhaskaruni, S.V.H.S.; Maddila, S.N.; Singh, P.; Jonnalagadda, S.B. A comparison between observed and DFT calculations on structure of 5-(4-chlorophenyl)-2-amino-1, 3, 4-thiadiazole. Sci. Rep. 2019, 9, 19280. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Houk, K.N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R-Li | Equiv | Time | (R,R)-2a % [a] | (S,R)-2a % [a] | Conversion % [b] |
|---|---|---|---|---|---|---|
| 1 | n-HexLi | 1.2 | 30′ | 10 | <5 | 20 |
| 2 | n-HexLi | 2.2 | 30′ | 20 | <5 | 30 |
| 3 | n-HexLi | 2.8 | 45′ | 40 | <5 | 40 |
| 4 | n-HexLi | 3.5 | 20′ | 90 | 10 | 99 |
| 5 | s-BuLi | 2 | 20′ | 90 | 10 | 99 |
| 6 [c] | s-BuLi | 2 | 20′ | 90 | 10 | 99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musci, P.; Colella, M.; Altomare, A.; Romanazzi, G.; Sheikh, N.S.; Degennaro, L.; Luisi, R. Dynamic Phenomena and Complexation Effects in the α-Lithiation and Asymmetric Functionalization of Azetidines. Molecules 2022, 27, 2847. https://doi.org/10.3390/molecules27092847
Musci P, Colella M, Altomare A, Romanazzi G, Sheikh NS, Degennaro L, Luisi R. Dynamic Phenomena and Complexation Effects in the α-Lithiation and Asymmetric Functionalization of Azetidines. Molecules. 2022; 27(9):2847. https://doi.org/10.3390/molecules27092847
Chicago/Turabian StyleMusci, Pantaleo, Marco Colella, Angela Altomare, Giuseppe Romanazzi, Nadeem S. Sheikh, Leonardo Degennaro, and Renzo Luisi. 2022. "Dynamic Phenomena and Complexation Effects in the α-Lithiation and Asymmetric Functionalization of Azetidines" Molecules 27, no. 9: 2847. https://doi.org/10.3390/molecules27092847