
A Combined Solution and Solid-State Study on the Tautomerism of an Azocalix[4]arene Chromoionophore


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of 2
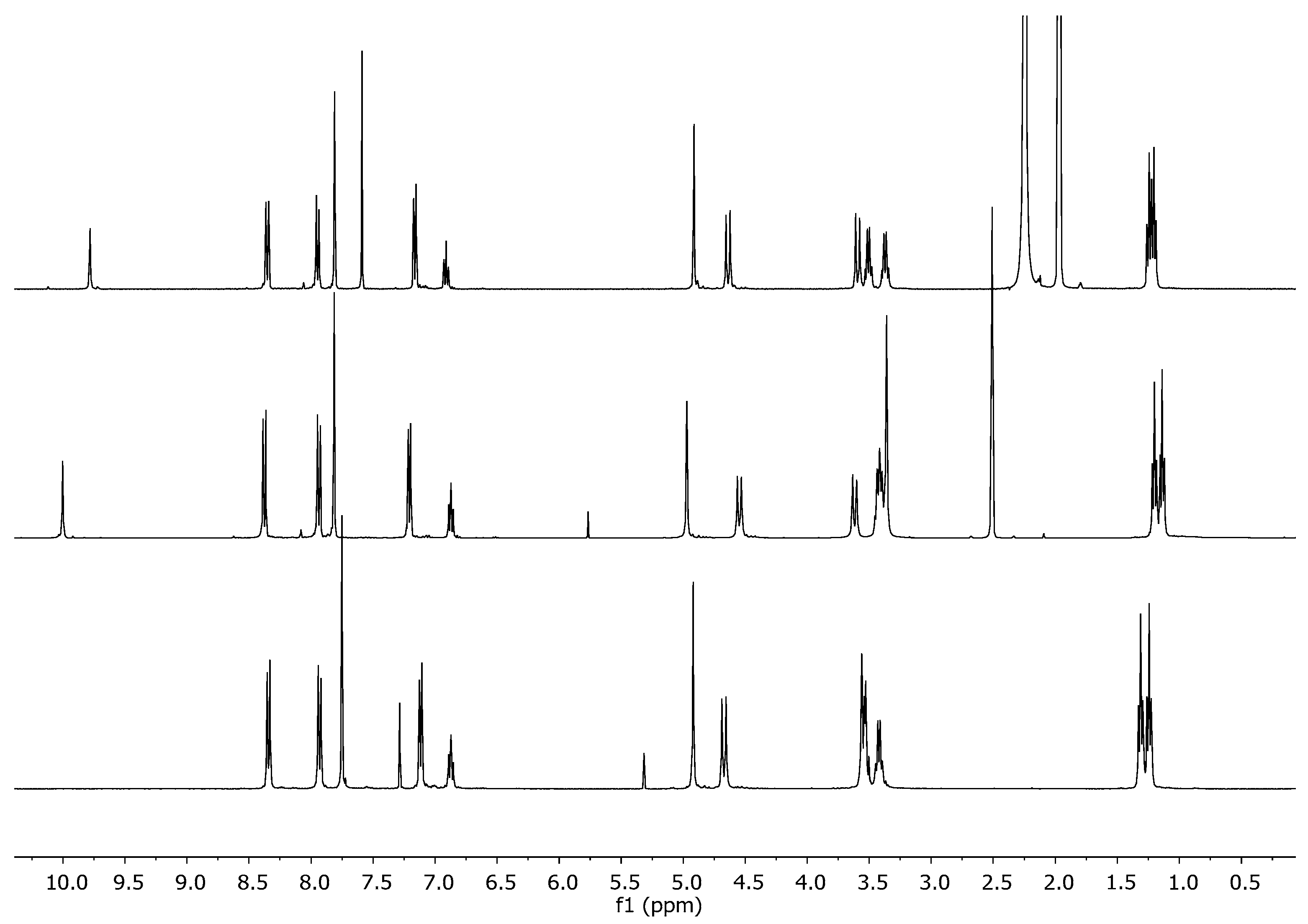
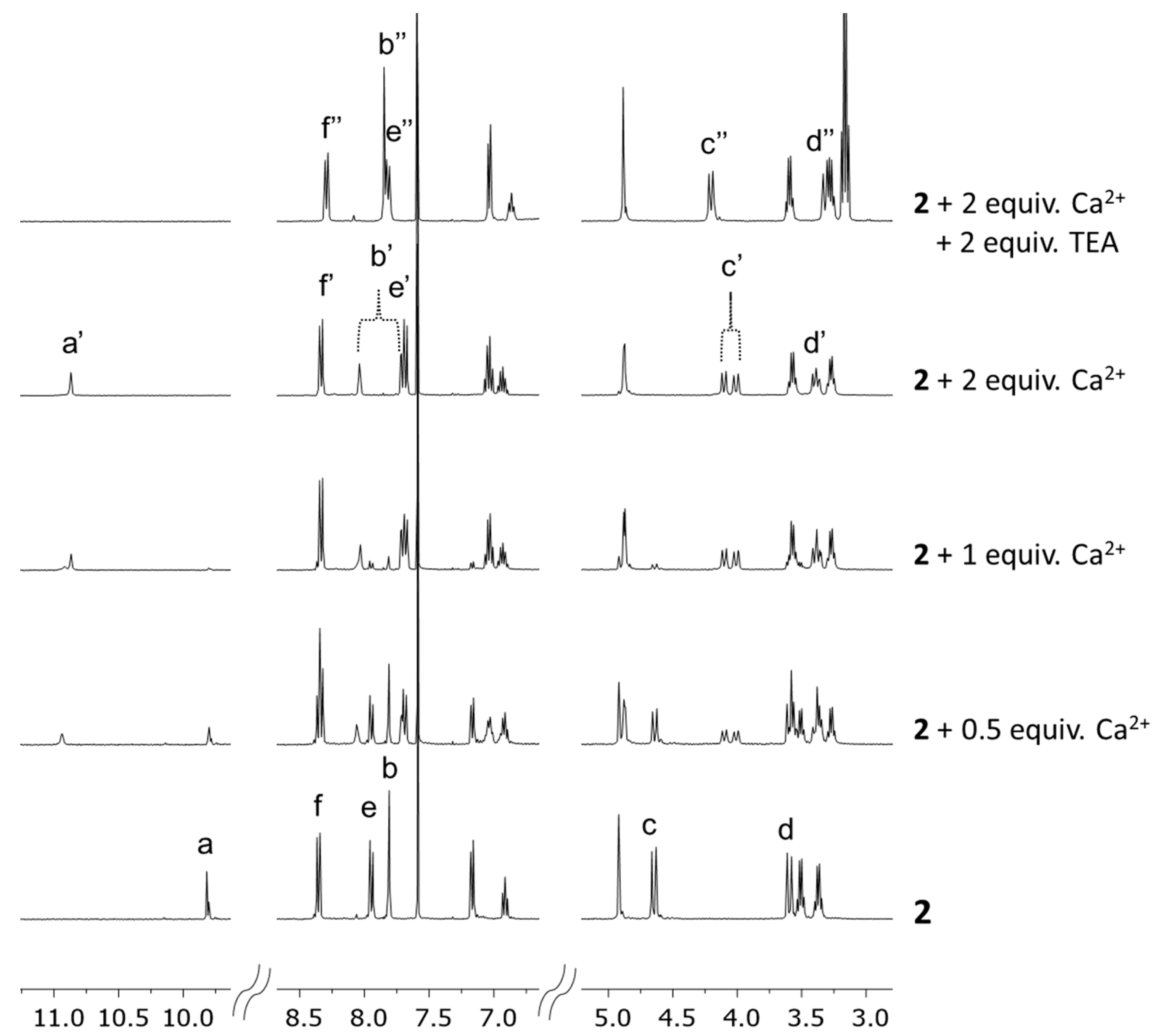
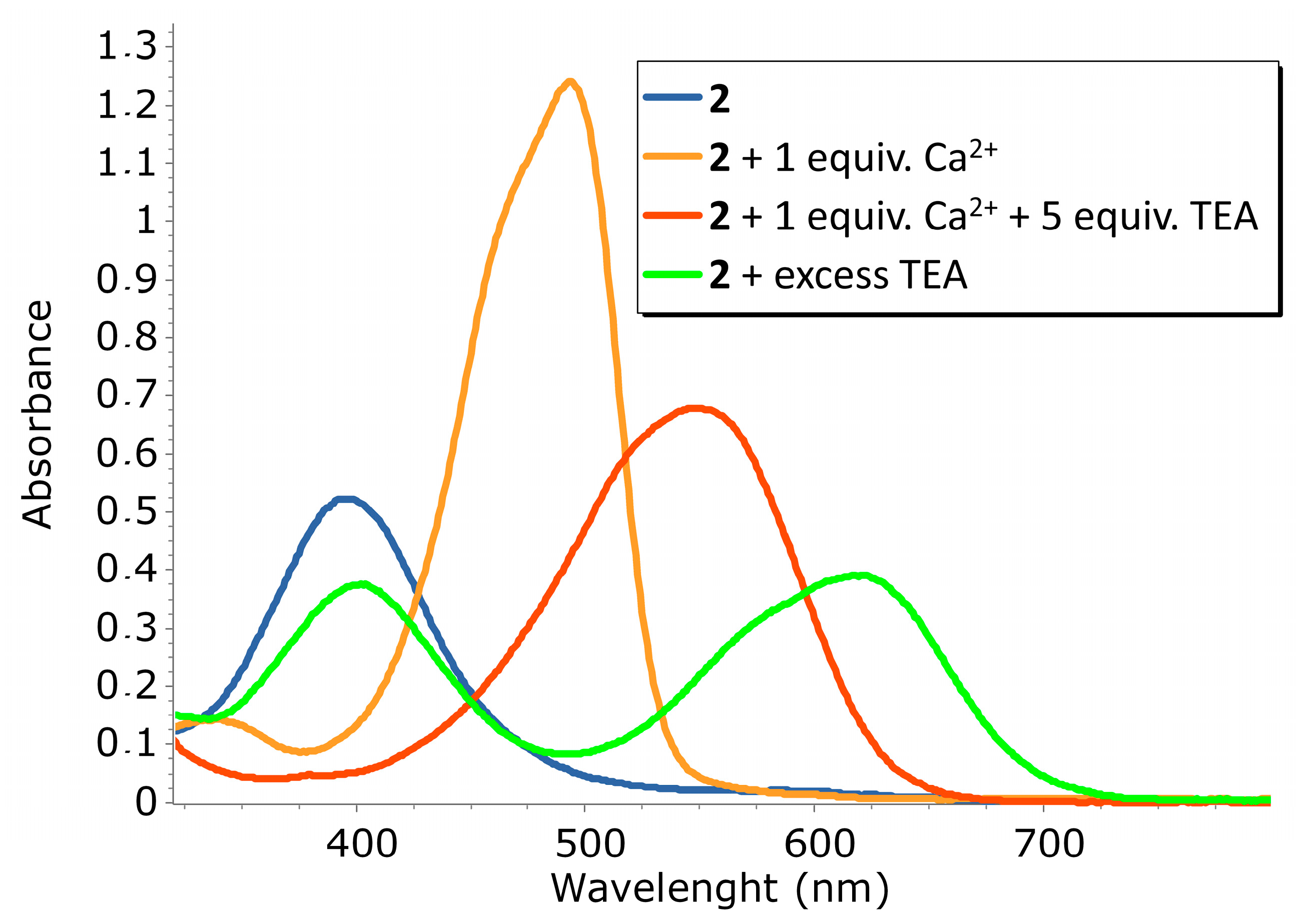
2.2. Ca2+ Complexation Studies
3. Materials and Methods
3.1. General
3.2. Synthesis of Compound 2
3.3. X-ray Data Collection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lohr, H.G.; Vogtle, F. Chromo- and Fluoroionophores. A New Class of Dye Reagents. Acc. Chem. Res. 1985, 18, 65–72. [Google Scholar] [CrossRef]
- Ludwig, R. Turning Ionophores into Chromo- and Fluoro-Ionophores. In Calixarenes 2001; Asfari, Z., Böhmer, V., Harrowfield, J., Vicens, J., Saadioui, M., Eds.; Springer: Dordrecht, The Netherlands, 2001; pp. 598–611. [Google Scholar]
- Diamond, D.; McKervey, M.A. Calixarene-Based Sensing Agents. Chem. Soc. Rev. 1996, 25, 15–24. [Google Scholar] [CrossRef]
- Na Kim, H.; Xiu Ren, W.; Seung Kim, J.; Yoon, J. Fluorescent and Colorimetric Sensors for Detection of Lead, Cadmium, and Mercury Ions. Chem. Soc. Rev. 2012, 41, 3210–3244. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.J.; Kim, M.Y.; Chang, S.K. A New Hg2+-Selective Chromoionophore Based on Calix[4]arenediazacrown Ether. Chem. Commun. 2001, 1, 1664–1665. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Choe, J.I.; Chang, S.K. Novel Ca2+-Selective Merocyanine-Type Chromoionophore Derived from Calix[4]arene-diamide. Tetrahedron Lett. 2003, 44, 5299–5302. [Google Scholar] [CrossRef]
- Chang, K.C.; Su, I.H.; Lee, G.H.; Chung, W.S. Triazole- and Azo-Coupled Calix[4]arene as a Highly Sensitive Chromogenic Sensor for Ca2+ and Pb2+ Ions. Tetrahedron Lett. 2007, 48, 7274–7278. [Google Scholar] [CrossRef]
- Song, K.C.; Choi, M.G.; Ryu, D.H.; Kim, K.N.; Chang, S.K. Ratiometric Chemosensing of Mg2+ Ions by a Calix[4]arene Diamide Derivative. Tetrahedron Lett. 2007, 48, 5397–5400. [Google Scholar] [CrossRef]
- Chawla, H.M.; Sahu, S.N. Synthesis of Novel Chromogenic Azocalix[4]arenemonoquinones and Their Binding with Alkali Metal Cations. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 141–149. [Google Scholar] [CrossRef]
- Shinkai, S.; Araki, K.; Shibata, J.; Tsugawa, D.; Manabe, O. Diazo-Coupling Reactions with Calix[4]arene. pKa Determination with Chromophoric Azocalix[4]Arenes. Chem. Lett. 1989, 18, 931–934. [Google Scholar] [CrossRef]
- Oueslati, F.; Dumazet-Bonnamour, I.; Lamartine, R. New Chromogenic Azocalix[4]arene Podands Incorporating 2,2′-Bipyridyl Subunits. New J. Chem. 2003, 27, 644–647. [Google Scholar] [CrossRef]
- Rouis, A.; Mlika, R.; Dridi, C.; Davenas, J.; Ben Ouada, H.; Halouani, H.; Bonnamour, I.; Jaffrezic, N. Optical Spectroscopy Studies of the Complexation of Chromogenic Azo-Calix[4]arene with Eu3+, Ag+ and Cu2+ Ions. Mater. Sci. Eng. C 2006, 26, 247–252. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chung, W.S. Tetrazoles and Para-Substituted Phenylazo-Coupled Calix[4]arenes as Highly Sensitive Chromogenic Sensors for Ca2+. Eur. J. Org. Chem. 2009, 2009, 4770–4776. [Google Scholar] [CrossRef]
- Wang, J.; Guan, H.; Ge, C.; Fan, P.; Xing, X.; Shang, Y. Azocalix[4]arene with Three Distal Ethyl Ester Residues as a Highly Selective Chromogenic Sensor for Ca2+ Ions. Heterocycl. Commun. 2018, 24, 147–150. [Google Scholar] [CrossRef]
- Shimizu, H.; Iwamoto, K.; Fujimoto, K.; Shinkai, S. Chromogenic Calix[4]arene. Chem. Lett. 1991, 20, 2147–2150. [Google Scholar] [CrossRef]
- Gordon, J.L.M.; Böhmer, V.; Vogt, W. A Calixarene-Based Chromoionophore for the Larger Alkali Metals. Tetrahedron Lett. 1995, 36, 2445–2448. [Google Scholar] [CrossRef]
- Hayashita, T.; Kunogi, K.; Yamamoto, H.; Shinkai, S. Selective Colorimetry of Sodium Ion in Acidic Aqueous Media by Calix[4]crown Chromoionophore. Anal. Sci. 1997, 13, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.Y.; Chang, S.-K. Calix[4]arenes Bearing Two Distal Azophenol Moieties: Highly Selective Chromogenic Ionophores for the Recognition of Ca2+ Ion. J. Org. Chem. 1998, 63, 2362–2364. [Google Scholar] [CrossRef]
- Van Der Veen, N.J.; Egberink, R.J.M.; Engbersen, J.F.J.; Van Veggel, F.J.C.M.; Reinhoudt, D.N. Conformationally Flexible Calix[4]arene Chromoionophores: Optical Transduction of Soft Metal Ion Complexation by Cation-π Interactions. Chem. Commun. 1999, 681–682. [Google Scholar] [CrossRef]
- Halouani, H.; Dumazet-Bonnamour, I.; Duchamp, C.; Bavoux, C.; Ehlinger, N.; Perrin, M.; Lamartine, R. Synthesis, Conformations and Extraction Properties of New Chromogenic Calix[4]arene Amide Derivatives. Eur. J. Org. Chem. 2002, 4202–4210. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, G.; Kim, C.R.; Lee, S.H.; Lee, J.H.; Kim, J.S. UV Band Splitting of Chromogenic Azo-Coupled Calix[4]crown upon Cation Complexation. J. Org. Chem. 2003, 68, 1933–1937. [Google Scholar] [CrossRef]
- Kishimoto, S.; Kitahara, S.; Manabe, O.; Hiyama, H. Tautomerism and Dissociation of 4-Arylazo-1-Naphthols in Various Solvents. J. Org. Chem. 1978, 43, 3882–3886. [Google Scholar] [CrossRef]
- Stoyanov, S.; Antonov, L.; Soloveytchik, B.; Petrova, V. Quantitative Analysis of Tautomeric Equilibrium in 1-Phenylazo-4-Naphthols-a New Approach. Dye. Pigment. 1994, 26, 149–158. [Google Scholar] [CrossRef]
- Chapoteau, E.; Czech, B.P.; Gebauer, C.R.; Kumar, A.; Leong, K.; Mytych, D.T.; Zazulak, W.; Desai, D.H.; Luboch, E.; Krzykawski, J.; et al. Phenylazophenol-Quinone Phenylhydrazone Tautomerism in Chromogenic Cryptands and Corands with Inward-Facing Phenolic Units and Their Acyclic Analogues. J. Org. Chem. 1991, 56, 2575–2579. [Google Scholar] [CrossRef]
- Joshi, H.; Kamounah, F.S.; van der Zwan, G.; Gooijer, C.; Antonov, L. Temperature Dependent Absorption Spectroscopy of Some Tautomeric Azo Dyes and Schiff Bases. J. Chem. Soc. Perkin Trans. 2 2001, 12, 2303–2308. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, S.H.; Van Tan, L.; Dong, Y.; Kim, H.; Kim, J.S. Diazo-Coupled Calix[4]arenes for Qualitative Analytical Screening of Metal Ions. Talanta 2008, 74, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H. Spectrophotometric and Electrochemical Study for Metal Ion Binding of Azocalix[4]arene Bearing p-Ethylester Group. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 178, 8–13. [Google Scholar] [CrossRef]
- Beer, P.D.; Drew, M.G.B.; Leeson, P.B.; Ogden, M.I. Metal Complexes of a Calix[4]arene Diamide: Syntheses, Crystal Structures and Molecular Mechanics Calculations on [Fe(L1-2H)][FeCl4] and [Er(L1-2H)(Picrate)] (L1 = 5,11,17,23-Tetra-Tert-Butyl-25,27-Bis(Diethylcarbamoylmethoxy)Calix[4]Arene). Inorg. Chim. Acta 1996, 246, 133–141. [Google Scholar] [CrossRef]
- Beer, P.D.; Drew, M.G.B.; Kan, M.; Leeson, P.B.; Ogden, M.I.; Williams, G. Lanthanide Structures, Coordination, and Extraction Investigations of a 1,3-Bis(Diethyl Amide)-Substituted Calix[4]arene Ligand. Inorg. Chem. 1996, 35, 2202–2211. [Google Scholar] [CrossRef]
- Casnati, A.; Fischer, C.; Guardigli, M.; Isernia, A.; Manet, I.; Sabbatini, N.; Ungaro, R. Synthesis of Calix[4]arene Receptors Incorporating (2,2′-Bipyridin-6-Yl)Methyl and (9-Methyl-1,10-Phenanthrolin-2-Yl)Methyl Chromophores and Luminescence of Their Eu3+ and Tb3+ Complexes. J. Chem. Soc. Perkin Trans. 2 1996, 3, 395–399. [Google Scholar] [CrossRef]
- Chawla, H.M.; Sahu, S.N.; Shrivastava, R. Synthesis and Binding Characteristics of Novel Calix[4]arene(amidocrown) Diquinones. Can. J. Chem. 2009, 87, 523–531. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CrysAlis Pro Software; Version 42.49; Agilent Technologies Ltd.: Oxfordshire, UK, 2013.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldini, L.; Balestri, D.; Marchiò, L.; Casnati, A. A Combined Solution and Solid-State Study on the Tautomerism of an Azocalix[4]arene Chromoionophore. Molecules 2023, 28, 4704. https://doi.org/10.3390/molecules28124704
Baldini L, Balestri D, Marchiò L, Casnati A. A Combined Solution and Solid-State Study on the Tautomerism of an Azocalix[4]arene Chromoionophore. Molecules. 2023; 28(12):4704. https://doi.org/10.3390/molecules28124704
Chicago/Turabian StyleBaldini, Laura, Davide Balestri, Luciano Marchiò, and Alessandro Casnati. 2023. "A Combined Solution and Solid-State Study on the Tautomerism of an Azocalix[4]arene Chromoionophore" Molecules 28, no. 12: 4704. https://doi.org/10.3390/molecules28124704
APA StyleBaldini, L., Balestri, D., Marchiò, L., & Casnati, A. (2023). A Combined Solution and Solid-State Study on the Tautomerism of an Azocalix[4]arene Chromoionophore. Molecules, 28(12), 4704. https://doi.org/10.3390/molecules28124704