Abstract
A new protocol for the synthesis of N-vinyl azoles using vinyl selenones and azoles in the presence of potassium hydroxide was developed. This reaction proceeded under mild and transition metal-free conditions through an addition/elimination cascade process. Both aromatic and aliphatic vinyl selenones and various mono-, bi- and tri-cyclic azoles can be tolerated and give terminal N-vinyl azoles in moderate to high yields. A plausible mechanism is also proposed.
1. Introduction
N-vinyl azoles are common structural motifs of natural products, agrochemicals and pharmaceuticals and occupy an important place in heterocyclic chemistry, representing useful building blocks in organic synthesis and in material science [1,2]. N-vinyl imidazoles display antifungal activity [3], and diverse N-vinyl azoles are incorporated in structures of medical interest [4,5]. N-vinyl indole derivatives are useful intermediates for alkaloid synthesis [6,7,8]. They are also highly reactive monomers that generate polymeric materials with various properties. In particular, poly(N-vinyl indoles) are used as semiconductors and photosensitive materials [9,10] and the poly(N-vinyl carbazoles) are extensively studied photoconductive polymers with several applications such as light emitting diodes, capacitors or memory devices [11].
Owing to the widespread application of N-vinyl azoles, many strategies have been developed for their synthesis, and selected examples are reported in Scheme 1. A convenient route is the direct condensation of aldehydes on N-H indoles in the presence of a Brønsted or Lewis acid (Scheme 1, route a) [12]. This method requires harsh reaction conditions causing low functional group tolerance. With the vigorous growth of organometallic chemistry, the number of methods for the synthesis of N-vinyl azoles by metal-catalyzed cross-coupling reactions has increased over the years. A reliable method is copper- or palladium-catalyzed N-vinylation of azoles with vinyl bromide (Scheme 1, route b) [13,14,15]. N-vinyl indoles were also prepared through a palladium-catalyzed oxidative cross-coupling reaction of indoles with N-tosylhydrazones (Scheme 1, route c) [16,17] or by direct reaction with alkenes (Scheme 1, route d) [18]. All transition metal-catalyzed reactions have some limitations such as expensive catalysts/ligands and the preparation of the specific starting materials. Therefore, it is desirable to develop new types of coupling reactions that may circumvent these drawbacks. Base-mediated hydroamination of alkynes represents an alternative approach for the preparation of N-vinyl azoles [19,20,21] (Scheme 1, route e). Very recently, N-vinyl azoles were also obtained employing vinyl sulfonium salts in presence of a base [22,23] (Scheme 1, route f) or through a three-component reaction between aromatic aldehydes, dimethyl sulfoxide DMSO and azoles (Scheme 1, route g) [24].
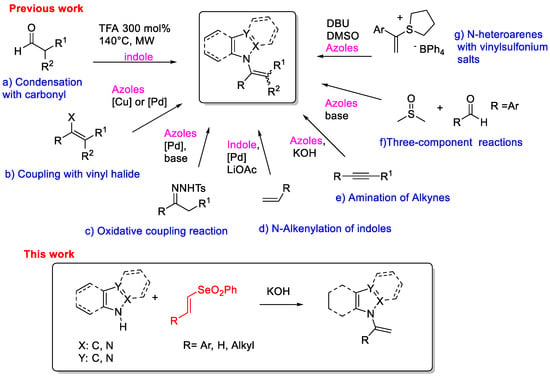
Scheme 1.
Synthetic approaches toward N-vinylazoles.
On these grounds, and inspired by renewed interest in vinyl selenones due to their useful applications in challenging fields of organic synthesis [25,26,27,28], we decided to synthesize N-vinyl azoles employing these derivatives in a domino process. Although their chemistry shows analogies with that of the vinyl sulfones, their reactivity presents marked differences. Both contain an electron-withdrawing group that activates the double bond to the conjugate nucleophilic attack, but the weak C–Se bond donates to the phenylselenonyl moiety a better leaving group character for further substitution or elimination reactions. While several Michael addition/cyclization cascade reactions using vinyl selenones are widely reported in the literature [29,30,31], also in asymmetrical versions [32,33,34,35,36,37], only sporadic examples involving addition/elimination domino processes are investigated [38,39].
Herein we report a new application of the chemistry of vinyl selenones to an addition/elimination cascade process using mono-, bi- and tri-cyclic azoles with variously substituted vinyl selenones in presence of base.
2. Results and Discussion
The vinyl phenyl selenones necessary for the present investigation were synthesized starting from the corresponding vinyl selenides using Oxone® as oxidant [40]. Initially, we explored the vinylation of indole 1a with the phenyl vinyl selenone 2a as a model reactant. As shown in Table 1, different inorganic and organic bases and different polar and apolar solvents were employed, and the best result was obtained using potassium hydroxide (2.5 equiv.) as base and N, N-dimethylformamide (DMF) as solvent (entry 5, 86% yield). Other dipolar aprotic solvents such as dimethyl sulfoxide or acetonitrile permit the progress of the reaction (entries 6–7), while the reaction did not occur in dichloromethane or in ethanol (entries 3–4). The substitution of potassium hydroxide with other inorganic bases such as cesium carbonate or sodium hydride gave the product 3a in lower yields and longer reaction times (entries 8–9). Employing organic bases, the reaction proceeded with a yield of 79% with potassium tert-butoxide, while it did not proceed with 1,5-diazabiciclo[5.4.0]undec-7-ene DBU (entries 10–11). Conducting the reaction using a lower amount of potassium hydroxide (1.5 equiv.), the desirable product 3a was isolated only in 40% yield (entry 12) and its formation was not observed in the absence of base (entry 13).

Table 1.
Optimization of the reaction conditions.
The formation of the product 3a can be explained through a one-pot reaction involving the addition/elimination process depicted in Scheme 2. An initial aza-Michael addition of indole 1a to vinyl selenone 2a forms the adduct X, which undergoes β-elimination of phenylseleninic acid to afford the product 3a. The choice of base and solvent is crucial to the success of the reaction. As expected [41], the formation of more ionic salts, such as the potassium salt, and the use of highly coordinating solvents, such as the DMF, favor the N–alkylation of indole in the aza-Michael addition. Moreover, an excess of a strong base and the presence of an aprotic polar solvent is required to assist the subsequent E2 elimination step [22]. While the elimination of selenoxides is a well-known process in organochalcogen chemistry [42], the same reaction carried out on the selenones is much less common [43,44,45], hence the interest in exploring the result.
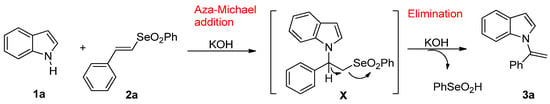
Scheme 2.
Plausible mechanism of Michael addition/elimination cascade.
With the optimized reaction conditions, we evaluated the versatility of the methodology. Firstly, we tested the reactivity of the indole with aryl vinyl selenones. As highlighted in Table 2, various indoles bearing different functional groups such as CH3, Br, I, CH2OH, CN and CO2Et were investigated. Some of these substituents may provide the possibility of further synthetic transformations.
Table 2.
Synthesis of N-vinyl indoles with different vinyl selenones.
N-vinyl indoles 3a–b were isolated in excellent yields using phenyl (E)-2-phenylvinyl selenone 2a. Different aryl vinyl selenones 2b–e bearing electron-deficient groups (R2 = 4-Cl–C6H4) and electron-rich groups (R2 = 4-CH3O–C6H4, 4-CH3–C6H4, 2-CH3–C6H4) have been successfully used, affording the corresponding N-vinyl indoles 3c–g in good yields. A better yield was obtained starting from the selenone 2b bearing an electron-deficient group in para position of the phenyl ring (3c, 87% yield), while when starting from the selenone 2e bearing a methyl group in ortho position, the lowest yield (3f, 45%) was observed, probably due to steric hindrance. Interestingly, starting from (5-bromo-1H-indol-2yl) methanol, the exclusive formation of compound 3h was achieved, demonstrating that when using a bis nucleophile there is also no trace of the Michael addition/cyclization cascade product. This result is reasonably a consequence of the high stability of the resulting conjugate system.
It is worth noting that when an unsubstituted vinyl selenone (2f, R2 = H) was used, the corresponding N-vinyl indoles 3i–l were obtained in satisfactory yields, despite the formation of a less stable alkene. However, in this case the (5-bromo-1H-indol-2yl) methanol afforded the biologically relevant 8-bromo-3,4-dihydro-1H-[1,4]oxazino [4,3-a]indole (22% yield), as a result of a domino Michael/intramolecular nucleophilic substitution pathway [30], beside the expected N-vinyl indole 3l (36% yield).
Encouraged by the results obtained with the indole scaffold, and in order to expand the substrate scope of the method, we proceeded to apply the same procedure to other azoles. As reported in Table 3, we first explored reactions with other benzo-fused mono-, di- and tri-azoles such as carbazole, benzoimidazole and benzotriazole and then with monocyclic azoles such as pyrrole, imidazole and pyrazole.
Table 3.
Vinylation of other N-heteroarenes substrates.
When the reaction was carried out on a carbazole nucleus employing aryl and unsubstituted vinyl selenones, the N-vinyl carbazoles 5a–b were obtained in acceptable yields. N-vinyl benzimidazoles 5c–d were obtained in good yields using benzoimidazole derivatives with aryl vinyl selenones. Employing benzotriazole with unsubstituted vinyl selenone, we observed the formation of N-vinyl benzotriazole 5e, even if with a lower yield. The low reactivity of benzotriazole can be a consequence of its poor nucleophilicity as reported in the literature [13]. Switching to monocyclic azoles, N-vinyl pyrroles 5f–g, N-vinyl imidazoles 5h–j and N-vinyl pyrazoles 5k–l were isolated in good yields using different aromatic and aliphatic vinyl selenones.
As shown in Scheme 3, the N-vinylation reaction of unsymmetrical benzo-fused or monocyclic diazoles can lead to two regioisomers due to the functionalization at the NH position of the two tautomers. The N-vinyl benzoimidazole regioisomers 5m and 5m′ were isolated when the 5-bromo benzoimidazole 4g was employed as nucleophile. These compounds were separated in pure form by column chromatography in an almost 1:1 ratio. Structures were assigned according to the coupling constants and the shielding effects observed in their 1H NMR spectra. In both cases, the 4-chloro phenyl group causes a shielding effect on the benzoimidazole proton falling into its shielding cone. In particular, HA appears as a doublet at 6.81 ppm (d, 3J = 8.6 Hz) in the compound 5m and at 7.63 ppm (d, 3J = 8.6 Hz) in the compound 5m′, while the proton HB absorbs at 7.92 ppm (d, 4J = 1.1 Hz) in the compound 5m and 7.17 ppm (d, 4J = 1.9 Hz) in the compound 5m’. The structural assignment was confirmed by NOESY experiments (see supporting information). Similarly, the N-vinylation of the 4-bromo-3-methyl-1H-pyrazole 4h led to the two regioisomers 5n and 5n′ in a 2.5:1 ratio. The formation of 5n’ as minor isomer suggests that the steric hindrance of the methyl group plays a significant role in the N-functionalization.
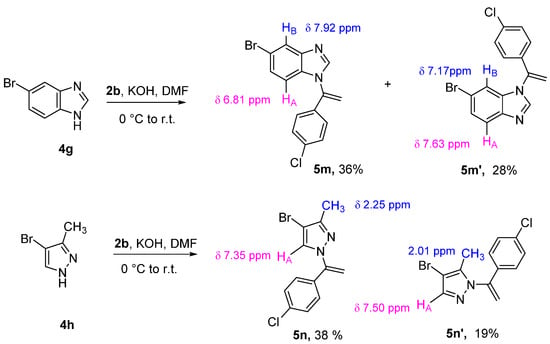
Scheme 3.
Vinylation of unsymmetrical N-diazoles.
3. Materials and Methods
3.1. General Information
Solvents and reagents were used as received unless otherwise noted. Thin-layer chromatography (TLC) was performed on silica gel 60 F254 (Merck, KGaA, Darmstadt, Germany). The products of the reactions were purified by normal chromatography column using Silica Gel Kiesegel 60 (70–230 mesh). Yields corresponded to isolated compounds. Melting points were determined in Kofler melting apparatus and values are uncorrected. All synthesized compounds were characterized by 1H NMR and 13C NMR spectroscopy. NMR experiments were obtained at 25 °C on a Bruker Avance at 400 MHz spectrometer, a Bruker Avance NEO 600 MHz spectrometer or a Bruker DPX 200 MHz spectrometer (Bruker, Billerica, MA, USA). Chemical shifts (δ) are reported in parts per million (ppm) in CDCl3 solution, if not otherwise specified. The following abbreviations are used to indicate multiplicity: s—singlet; d—doublet; t—triplet; q—quartet; quin—quintet; m—multiplet. Exact mass analyses were obtained by mass spectrometer Ion-Mobility QTof Agilent 6560 coupled with UHPLC 1290 Infinity II Agilent (UHPLC Agilent Technologies, Santa Clara, CA, USA).
3.2. Starting Materials
The indoles 1a–f and other azoles 4a–h used as starting products are commercially available, except for (5-bromo-1H-indol-2yl) methanol 1d that was prepared following the method reported in the literature [30]. According to the literature procedures, starting vinyl selenones 2a–g were prepared from the corresponding vinyl selenides by oxidation with an excess of Oxone [40].
3.3. General Procedure for the Synthesis of N-Vinyl Azoles
A stirred solution of N-indoles 1a–f or other azoles 4a–h (1 mmol) in DMF (2 mL) was treated with potassium hydroxide (2.5 equivalents) at 0 °C under argon atmosphere. After 10 min, a solution of the vinyl selenones 2a–g (1 mmol) in DMF (2 mL) was added at 0 °C and the reaction mixtures were allowed to warm to room temperature. The progress of the reaction was monitored by TLC (petroleum ether/ethyl acetate 80:20), verifying the disappearance of the starting product. The reaction mixture was extracted with ethyl acetate (3 × 5 mL) and organic phase was then washed with H2O (3 × 5 mL). After drying with Na2SO4, the organic extracts were filtered and evaporated under reduced pressure. The products were purified using column chromatography on silica gel affording the N-vinyl indoles 3a–l and other N-vinyl azoles 5a–n, 5m′–5n′.
1-(1-Phenylvinyl)-1H-indole, 3a [24]: The crude product was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to afford 3a as a white solid (m.p. 92–94 °C) in 82% yield. R.f. 0.80 (petroleum ether: ethyl acetate 80:20). 1H NMR (200 MHz, CDCl3, 25 °C, TMS): δ 7.71–7.66 (m, 1H), 7.42–7.32 (m, 5H), 7.21 (d, J = 3.3 Hz, 1H), 7.17–7.11 (m, 3H), 6.64 (d, J = 3.3 Hz, 1H), 5.61 (br s, 1H, CHH=), 5.40 (br s, 1H, CHH=).
3-Methyl-1-(1-phenylvinyl)-1H-indole, 3b [24]: The crude product was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to afford 3b as a yellow oil in 86% yield. R.f. 0.75 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 7.62 (d, J = 7.3 Hz, 1H), 7.41–7.38 (m, 5H), 7.17–7.14 (m, 3H), 6.98 (s, 1H), 5.52 (br s, 1H, CHH=), 5.35 (br s, 1H, CHH=), 2.38 (s, 3H, CH3).
1-[1-(4-Chlorophenyl)vinyl]-1H-indole, 3c [16]: The crude product was purified by silica gel chromatography (petroleum ether: ethyl acetate 98:2 as eluent mixture:) to afford 3c as a white solid (m.p. 68–70 °C) in 87% yield. R.f. 0.83 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.61–7.54 (m, 1H), 7.28–7.21 (m, 2H), 7.20–7.14 (m, 2H), 7.08 (d, J = 3.3 Hz, 1H), 7.07–7.01 (m, 3H), 6.55 (dd, J = 3.3, 0.6 Hz, 1H), 5.50 (br s, 1H, CHH=), 5.31 (br s, 1H, CHH=).
1-[1-(4-Methoxyphenyl)vinyl]-1H-indole, 3d [24]: The crude product was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to afford 3d as a yellow solid (m.p. 51–53 °C) in 60% yield. R.f. 0.72 (petroleum ether: ethyl acetate 80:20). 1H NMR (200 MHz, CDCl3, 25 °C, TMS): δ 7.64–7.58 (m, 1H), 7.24–7.04 (m, 6H), 6.86–6.79 (m, 2H), 6.57 (d, J = 3.25 Hz, 1H), 5.44 (br s, 1H, CHH=), 5.22 (br s, 1H, CHH=), 3.78 (s, 3H, CH3O).
1-[1-(4-Methylphenyl)vinyl]-1H-indole, 3e [17]: The crude product was purified by silica gel chromatography (petroleum ether: ethyl acetate 97:3 as eluent mixture) to obtain 3e as a dark brown solid (m.p. 52–55 °C) in 65% yield. R.f. 0.88 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.61–7.53 (m, 1H), 7.16–6.98 (m, 8H), 6.53 (d, J = 3.4 Hz, 1H), 5.46 (br s, 1H, CHH=), 5.23 (br s, 1H, CHH=), 2.29 (s, 3H, CH3).
1-[1-(2-Methylphenyl)vinyl]-1H-indole, 3f [24]: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 97:3 as eluent mixture) to obtain 3f as a yellow oil in 45% yield. R.f. 0.86 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.58–7.50 (m, 1H), 7.36 (d, J = 8.7 Hz, 1H), 7.29–7.19 (m, 3H), 7.10–7.01 (m, 3H), 6.97–6.93 (m, 1H), 6.50–6.46 (m, 1H), 5.42 (br s, 1H, CHH=), 5.12 (br s, 1H, CHH=), 1.76 (s, 3H, CH3).
1-[1-(2-Methylphenyl)vinyl]-1H-indole-4-carbonitrile, 3g: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to obtain 3g as a greenish oil in 71% yield. R.f. 0.60 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.42–7.15 (m, 6H), 7.12–7.02 (m, 2H), 6.72 (d, J = 3.4, 0.9 Hz, 1H), 5.44 (br s, 1H, CHH=), 5.23 (br s, 1H, CHH=), 1.73 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 144.7, 136.7, 136.5, 135.3, 130.9, 130.1, 130.3, 130.2, 129.7, 126.5, 125.8, 122.1, 118.6, 116.4, 108.6, 103.7, 102.5, 19.3. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C18H15N2 259.1230; found 259.1225.
{5-Bromo-1-[1-(4-chlorophenyl)vinyl]-1H-indol-2-yl}methanol, 3h: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 75:25 as eluent mixture) to obtain 3h as a yellow oil in 50% yield. R.f. 0.25 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.68 (d, J = 1.9 Hz, 1H), 7.23–7.16 (m, 2H), 7.13 (dd, J = 8.7, 1.9 Hz, 1H), 7.01–6.91 (m, 2H), 6.89 (d, J = 8.7 Hz, 1H), 6.52 (s, 1H), 5.97 (br s, 1H, CHH=), 5.45 (br s, 1H, CHH=), 4.53 (br s, 2H, CH2). 13C NMR (100 MHz, CDCl3): δ 141.6, 140.8, 137.0, 135.5, 135.0, 129.4, 129.3 (2C), 127.1 (2C), 125.6, 123.4, 114.6, 113.8, 112.5, 102.5, 57.5. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C17H14BrClNO 361.9942; found 361.9924.
1-Vinyl-1H-indole, 3i [9]: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 97:3 as eluent mixture) to obtain 3i as whitish crystalline solid (m. p. 34–36 °C) in 60% yield. R.f. 0.75 (petroleum ether: ethyl acetate 80:20). 1H NMR (600MHz, Chloroform-d): δ 7.54 (d, J = 7.9 Hz, 1H), 7.40 (dd, J = 8.3, 1.0 Hz, 1H), 7.36 (d, J = 3.4 Hz, 1H), 7.19–7.13 (m, 2H, Har, CH=), 7.10–7.06 (m, 1H), 6.56 (d, J = 3.4 Hz, 1H), 5.12 (dd, J = 15.7, 1.4 Hz, 1H, CHH=), 4.70 (dd, J = 8.9, 1.4 Hz, 1H, CHH=).
Ethyl 5-bromo-1-vinyl-1H-indole-2-carboxylate, 3j: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 97:3 as eluent mixture) to obtain 3j as a yellowish oil in 35% yield. R.f. 0.77 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.47 (d, J = 1.9 Hz, 1H), 7.69–7.59 (m, 2H, Har, CH=), 7.45 (dd, J = 9.0, 1.9 Hz, 1H), 7.03 (s, 1H), 5.40 (dd, J = 15.8, 1.2 Hz, 1H, CHH=), 5.31 (dd, J = 8.7, 1.2 Hz, 1H, CHH=), 7.01 (quart, J = 7.1Hz, 2H, CH2O), 1.38 (t, J = 7.1 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 161.5, 136.7, 132.1, 129.0, 128.7, 128.6, 125.1, 114.7, 114.1, 111.1, 108.2, 60.7, 13.2. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C13H13BrNO2 294.0124; found 294.0114.
5-Iodo-1-vinyl-1H-indole, 3k: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to obtain 3k as a white powder (m.p. 62–67 °C) in 50% yield. R.f. 0.80 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.87 (d, J = 1.7 Hz, 1H), 7.43 (dd, J = 8.6, 1.7 Hz, 1H), 7.30 (d, J = 3.4 Hz, 1H), 7.20–7.13 (m, 1H), 7.07 (dd, J = 15.7, 8.9 Hz, 1H, CH=), 6.47 (d, J = 3.4 Hz, 1H), 5.13 (dd, J = 15.7, 1.5 Hz, 1H, CHH=), 4.74 (dd, J = 9.0, 1.5 Hz, 1H, CHH=). 13C NMR (100 MHz, CDCl3): δ 134.7, 131.6, 131.1, 130.1, 129.4, 124.3, 111.6, 104.2, 97.7, 84.3. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C10H9IN 269.9774; found 269.9772.
(5-Bromo-1-vinyl-1H-indol-2-yl)methanol, 3l: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 70:30 as eluent mixture) to obtain 3l as a yellow solid (m.p.88–93 °C) in 36% yield. R.f. 0.23 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.63 (d, J = 2.0 Hz, 1H), 7.41 (d, J = 8.7 Hz, 1H), 7.25 (dd, J = 8.8, 2.0 Hz, 1H), 7.11 (dd, J = 15.9, 9.1 Hz, 1H, CH=), 6.41 (s, 1H), 5.43 (dd, J = 15.9, 1.0 Hz, 1H, CHH=), 5.09 (dd, J = 9.1, 0.9 Hz, 1H, CHH=), 4.74 (d, J = 4.0 Hz, 2H, CH2O). 13C NMR (100 MHz, CDCl3): δ 139.4, 135.5, 130.1, 129.9, 126.0, 123.6, 114.1, 112.9, 105.4, 103.7, 57.6 HRMS (ESI Q-TOF): m/z [M]+ calculated for C11H10BrNO 250.9940; found 250.9942.
8-Bromo-3,4-dihydro-1H-[1,4]oxazino [4,3-a]indole [30]: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 80:20 as eluent mixture) to obtain a white solid (m.p. 158–159 °C) in 22% yield. R.f. 0,46 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.61 (d, J = 1.9 Hz, 1H), 7.20–7.16 (m, 1H), 7.07 (d, J = 8.6 Hz, 1H), 6.08 (s, 1H), 4.90 (s, 2H), 4.11–3.96 (m, 4H).
9-[1-(2-Methylphenyl)vinyl]-9H-carbazole, 5a: The crude was purified by silica gel chromatography (petroleum ether:ethyl acetate 90:10 as eluent mixture) to obtain 5a as a white solid (m.p. 76–80 °C) in 40% yield. R.f. 0.93 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 8.01 (dt, J = 7.7, 1.0 Hz, 2H), 7.43 (dd, J = 6.9, 2.3 Hz, 1H), 7.28–7.12 (m, 8H), 7.00 (dd, J = 6.4, 2.4 Hz, 1H), 5.63 (br s, 1H, CHH=), 5.60 (br s, 1H, CHH=), 1.68 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 143.6, 140.3 (2C), 137.5, 136.6, 131.1, 129.9, 129.1, 126.4, 126.0 (2C), 123.8 (2C), 120.2 (2C), 120.1 (2C), 113.3, 111.3 (2C), 20.1. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C21H18N 284.1434; found 284.1434.
9-Vinyl-9H-carbazole, 5b [19]: The crude was purified by silica gel chromatography (petroleum ether:ethyl acetate 90:10 as eluent mixture) to obtain 5b as a white solid (m.p. 64–65 °C) in 50% yield. R.f. 0.74 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 8.00 (dt, J = 7.7, 1.0 Hz, 2H), 7.59 (dt, J = 8.3, 0.9 Hz, 2H), 7.40 (ddd, J = 8.3, 7.2, 1.3 Hz, 2H), 7.29–7.17 (m, 3H, 2Har, CH=), 5.48 (dd, J = 15.9, 0.9 Hz, 1H, CHH=), 5.09 (dd, J = 9.2, 0.9 Hz, 1H, CHH=).
1-(1-Phenylvinyl)-1H-benzimidazolo, 5c [24]: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 50:50 as eluent mixture) to obtain 5c as a yellow solid (m.p. 57–58 °C) in 77% yield. R.f. 0.58 (petroleum ether: ethyl acetate 40:60). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 8.01 (s, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.45–7.21 (m, 7H), 7.10 (d, J = 8.1 Hz, 1H), 5.72 (s, 1H), 5.50 (s, 1H).
1-[1-(4-Chlorophenyl)vinyl]-1H-1,3-benzimidazole, 5d: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 50:50 as eluent mixture) to obtain 5d as a white solid (m.p. 78–81 °C) in 66% yield. R.f. 0.38 (petroleum ether: ethyl acetate 70:30), 1H NMR (400 MHz, Chloroform-d): δ 7.94 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.31–7.09 (m, 6H), 6.96 (d, J = 8.1 Hz, 1H), 5.61 (d, J = 1.1 Hz, 1H, CHH=), 5.41 (d, J = 1.1 Hz, 1H, CHH=). 13C NMR (100 MHz, CDCl3): δ 144.1, 143.1, 141.4, 136.0, 133.9, 133.7, 129.3 (2C), 128.2 (2C), 123.7, 123.0, 120.7, 111.8, 110.1 HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C15H12ClN2 255.0684; found 255.0679.
1-Vinyl-1H-1,2,3-benzotriazole, 5e: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 95:5 as eluent mixture) to obtain 5e as a yellowish oil in 30% yield. R.f. 0.48 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 8.04 (d, J = 8.2 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 7.57–7.47 (m, 2H, Har, CH=), 7.36 (ddd, J = 8.1, 7.0, 1.0 Hz, 1H), 5.90 (dd, J = 16.0, 1.5 Hz, 1H, CHH=), 5.21 (dd, J = 9.2, 1.6 Hz, 1H, CHH=). 13C NMR (100 MHz, CDCl3): δ 146.5, 131.6, 129.5, 128.5, 124.7, 120.6, 110.3, 104.2. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C8H8N3 146.0713; found 145.0713.
1-(1-Phenylvinyl)-1H-pyrrole, 5f [14]: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to obtain 5f as a yellowish oil in 53% yield. R.f. 0.78 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 7.43–7.41 (m, 5H), 6.84 (t, J = 2.15 Hz, 2H), 6.28 (t, J = 2.15 Hz, 2H), 5.20 (br s, 1H, CHH=), 5.12 (br s, 1H, CHH=).
1-[1-(2-Methylphenyl)vinyl]-1H-pyrrole, 5g: The crude was purified by silica gel chromatography (petroleum ether:ethyl acetate 90:10 as eluent mixture) to obtain 5g as a colorless oil in 53% yield. R.f. 0.81 (petroleum ether: ethyl acetate 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.24 (td, J = 8.8, 1.7 Hz, 2H), 7.18–7.09 (m, 2H), 6.63 (t, J = 2.2 Hz, 2H), 6.13 (t, J = 2.3 Hz, 2H), 5.23 (br s, 1H, CHH=), 4.66 (br s, 1H, CHH=), 1.97 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 145.8, 137.3, 136.9, 130.4, 130.3, 129.1, 125.9, 119.6 (2C), 109.9 (2C), 101.2, 19.1. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C13H14N 184.1121; found 184.1116.
1-(1-Phenylvinyl)-1H-imidazole, 5h [14]: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 20:80 as eluent mixture) to obtain 5h as a yellow oil in 63% yield. R.f. 0.58 (petroleum ether: ethyl acetate 20:80). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 7.74 (s, 1H), 7.45–7.38 (m, 5H), 7.16–7.13 (m, 1H), 7.06–7.03 (m, 1H), 5.35 (br s, 1H, CHH=), 5.32 (br s, 1H, CHH=).
1-[1-(2-Methylphenyl)vinyl]-1H-imidazole, 5i: The crude was purified by silica gel chromatography (dichloromethane: methanol 95:5 as eluent mixture) to obtain 5i as a colorless oil with a 75% yield. R.f. 0.36 (dichloromethane: methanol 98:2). 1H NMR (400 MHz, Chloroform-d): δ 7.39 (s, 1H), 7.31–7.22 (m, 2H), 7.22–7.12 (m, 2H), 7.02 (br s, 1Har), 6.93 (br s, 1Har), 5.37 (br s, 1H, CHH=), 4.87 (br s, 1H, CHH=), 1.98 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 142.9, 136.9, 136.2, 135.4, 130.8, 130.3, 130.2, 129.7, 126.3, 117.6, 104.7, 19.2. HRMS (ESI Q-TOF): m/z [M+H]+ calculated for C12H13N2 185.1073; found 185.1071.
1-(oct-1-en-2-yl)-1H-imidazolo, 5j: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 20:80 as eluent mixture) to obtain 5j as a yellow oil in 61% yield. R.f. 0.54 (petroleum ether: ethyl acetate 20:80). 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 7.71 (s, 1H), 7.10–7.15 (m, 2H), 5.08 (br s, 1H, CHH=), 4.82 (br s, 1H, CHH=), 2.51 (t, J = 7.1 Hz, 2H, CH2), 1.51 (quint, J = 7.1 Hz, 2H, CH2), 1.39–1.23 (m, 6H, 3CH2), 0.9 (t, J = 6.8 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 142.9, 129.6 (2C), 116.9, 102.7, 33.8, 31.4, 28.5, 26.8, 22.4, 13.9. HRMS (ESI Q-TOF): m/z [M+H+] calculated for C11H19N2 179.1543; found 179.1550.
1-(1-(4-Chlorophenyl)vinyl)-1H-pyrazole, 5k [46]: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate from 90:10 to 80:20 as eluent mixture) to obtain 5k as a yellow oil in 74% yield. R.f. 0,66 (petroleum ether: ethyl acetate from 80:20). 1H NMR (200 MHz, CDCl3, 25 °C, TMS): δ 7.72 (s, 1H), 7.54 (d, J = 2.15 Hz, 1H), 7.45–7.29 (m, 4H), 6.41–6.39 (m, 1H), 5.59 (br s, 1H, CHH=), 5.23 (br s, 1H, CHH=).
1-[1-(2-Methylphenyl)vinyl]-1H-pyrazole, 5l: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to obtain 5l as a brownish oil in 73% yield. R.f. 0.75 (petroleum ether: ethyl acetate from 80:20). 1H NMR (400 MHz, Chloroform-d): δ 7.59 (d, J = 1.7 Hz, 1H), 7.27 (t, J = 7.4 Hz, 2H), 7.22–7.13 (m, 2H), 7.11 (dd, J = 2.5, 0.7 Hz, 1H), 6.24–6.19 (m, 1H), 5.79 (br s, 1H, CHH=), 4.82 (br s, 1H, CHH=), 1.99 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 144.9, 141.2, 137.3, 135.7, 130.5, 130.5, 129.5, 128.7, 126.1, 106.9, 103.5, 19.2. HRMS (ESI Q-TOF): m/z [M+H+] calculated for C12H13N2 185.1073; found 185.1077.
5-Bromo-1-[1-(4-chlorophenyl)vinyl]-1H-1,3-benzimidazole, 5m: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 60:40 as eluent mixture) to obtain 5m as a brownish oil in 30% yield. R.f. 0.33 (petroleum ether: ethyl acetate 70:30). 1H NMR (400 MHz, Chloroform-d): δ 7.93 (s, 1HC), 7.92 (d, J= 1.1 Hz, 1HB), 7.32–7.26 (m, 2Har, AA’BB’ system), 7.24 (dd, J = 8.6, 1.8 Hz, 1HD), 7.18–7.11 (m, 2Har, AA’BB’ system), 6.81 (d, J = 8.6 Hz, 1HA), 5.63 (d, J = 1.2 Hz, 1HE, CHH=), 5.41 (d, J = 1.2 Hz, 1HF, CHH=). 13C NMR (100 MHz, CDCl3): δ 145.4 (C), 144.0 (CH), 141.1 (C), 136.3 (C), 133.5 (C), 132.7 (C), 129.4 (2CH), 128.1 (2CH), 126.9 (CH), 123.6 (CH), 116.1 (C), 113.0 (CH), 110.5 (CH2=). HRMS (ESI Q-TOF): m/z [M+H+] calculated for C15H11BrClN2 332.9789; found 332.9803.
6-Bromo-1-[1-(4-chlorophenyl)vinyl]-1H-1,3-benzimidazole, 5m′: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 60:40 as eluent mixture) to obtain 5m’ as a yellowish oil in 30% yield. R.f. 0.28 (petroleum ether: ethyl acetate 70:30). 1H NMR (400 MHz, Chloroform-d): δ 7.89 (s, 1HC), 7.63 (d, J = 8.6 Hz, 1HA), 7.34 (dd, J = 8.6, 1.9 Hz, 1HD), 7.32–7.28 (m, 2Har, AA′BB′ system), 7.17 (d, J = 1.4Hz, 1HB), 7.15- 7.12 (m, 2Har, AA’BB’ system), 5.66 (d, J = 1.2 Hz, 1HE, CHH=), 5.42 (d, J = 1.2 Hz, 1HF, CHH=). 13C NMR (100 MHz, CDCl3): δ 143.6 (CH), 143.0 (C), 141.8 (C), 140.9 (C), 136.3 (C), 133.5 (C), 129.5 (2CH), 128.0 (2CH), 126.5 (CH), 122.0 (CH), 117.2 (C), 114.6 (CH), 110.9 (CH2=). HRMS (ESI Q-TOF): m/z [M+H+] calculated for C15H11BrClN2 332.9789; found 332.9810.
4-Bromo-1-[1-(4-chlorophenyl)vinyl]-3-methyl-1H-pyrazole, 5n: The crude was purified by silica gel chromatography (petroleum ether:ethyl acetate from 85:15 to 80:20 as eluent mixture) to obtain 5n as a whitish oil with a 30% yield. R.f. 0.88 (petroleum ether: ethyl acetate 70:30). 1H NMR (400 MHz, chloroform-d): δ 7.35 (s,1HA), 7.32–7.28 (m, 2Har, AA’BB’ system), 7.24–7.19 (m, 2Har, AA’BB’ system), 5.45 (br s, 1HF, CHH=), 5.07 (br s, 1HE, CHH=, 2.25 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 149.6 (C), 144.6 (C), 135.7 (C), 133.9 (C), 130.1 (CH), 129.5 (2CH), 129.0 (2CH), 105.2 (C), 96.1 (CH2=), 12.3 (CH3). HRMS (ESI Q-TOF): m/z [M+H+] calculated for C12H11BrClN2 296.9789; found 296.9776.
4-Bromo-1-[1-(4-chlorophenyl)vinyl]-5-methyl-1H-pyrazole, 5n′: The crude was purified by silica gel chromatography (petroleum ether: ethyl acetate 90:10 as eluent mixture) to obtain 5n’ as a yellowish oil with a 30% yield. R.f. 0.81 (petroleum ether: ethyl acetate 70:30). 1H NMR (400 MHz, Chloroform-d): δ 7.50 (s, 1H, HA), 7.28–7.22 (m, 2Har, AA’BB’ system), 7.07–7.01 (m, 2Har, AA′BB′ system), 5.70 (d, J = 1.0 Hz, 1HE, CHH=), 5.35 (d, J = 1.0 Hz, 1HF, CHH=), 2.01 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 144.5 (C), 140.3 (CH), 138.3 (C), 135.5 (C), 134.4 (C), 129.1 (2CH), 127.6 (2CH), 113.0 (CH2=), 95.0 (C), 10.9 (CH3). HRMS (ESI Q-TOF): m/z [M+H+] calculated for C12H11BrClN2 296.9789; found 296.9776.
4. Conclusions
In summary, we developed a novel method for the synthesis of N-vinyl azoles through a domino process. The selenonyl group plays a dual role by promoting the Michael addition and then acting as a leaving group in the one-pot elimination. This simple and metal-free approach employs easily accessible starting materials such as commercially available azoles, potassium hydroxide and bench-stable vinyl selenones. This protocol confirms the synthetic versatility of the vinyl selenones, opens the way to further studies concerning addition/elimination cascades and represents a simple and general way to synthesize a variety of particularly attractive N-vinyl heterocycles, making it a valuable addition to existing methods for their synthesis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28166026/s1, Figures S1–S45: Copies of 1H and 13C NMR Spectra of Compounds 3a–l, 5a–n, 5m′–n′ [9,14,16,17,19,24,30,46]; Figures S46–S49: NOESY experiments of compounds 5m–5m′ and 5n–5n′.
Author Contributions
Conceptualization, F.M. and L.B.; methodology M.P. and L.B.; validation, F.M. and L.B.; formal analysis, M.C., M.P., I.F.C.D. and L.B.; investigation, M.P. and M.C.; resources, C.S.; data curation M.P. and L.B.; writing—original draft preparation, M.P., I.F.C.D., M.C. and L.B.; writing—review and editing, F.M., C.S. and L.B.; supervision, F.M. and L.B.; funding acquisition, L.B. and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
University of Perugia “Fondo per la Ricerca di Base”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and in Supplementary Materials.
Acknowledgments
This manuscript is part of the scientific activity of the International Network Selenium, Sulfur, Redox and Catalysis (SeSRedCat) and National Interuniversities Consortium C.I.N.M.P.I.S.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Silva, V.L.M.; Silva, A.M.S. Revisiting the Chemistry of Vinylpyrazoles: Properties, Synthesis, and Reactivity. Molecules 2022, 27, 3493. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Matsumoto, H.; Shimizu, S.; Kida, S.; Shiro, M.; Tawara, K. Synthesis and Antifungal Activity of New 1-Vinylimidazoles. J. Med. Chem. 1987, 30, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Dreassi, E.; Brullo, C.; Crespan, E.; Tintori, C.; Bernardo, V.; Valoti, M.; Zamperini, C.; Daigl, H.; Musumeci, F.; et al. Design, Synthesis, Biological Activity, and ADME Properties of Pyrazolo[3,4-d]pyrimidines Active in Hypoxic Human Leukemia Cells: A Lead Optimization Study. J. Med. Chem. 2011, 54, 2610–2626. [Google Scholar] [CrossRef]
- Huang, W.-S.; Zhu, X.; Wang, Y.; Azam, M.; Wen, D.; Sundaramoorthi, R.; Thomas, R.M.; Liu, S.; Banda, G.; Lentini, S.P.; et al. 9-(Arenethenyl)purines as Dual Src/Abl Kinase Inhibitors Targeting the Inactive Conformation: Design, Synthesis, and Biological Evaluation. J. Med. Chem. 2009, 52, 4743–4756. [Google Scholar] [CrossRef]
- Ghost, A.; Stanley, L.M. Enantioselective hydroacylation of N-vinylindole-2-carboxaldehydes. Chem. Commun. 2014, 50, 2765–2768. [Google Scholar] [CrossRef]
- Ghosh, A.; Bainbridge, D.T.; Stanley, L.M. Enantioselective model synthesis and progress toward the Putative structure of Yuremamine. J. Org. Chem. 2016, 81, 7945–7951. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Boonnak, N.; Padwa, A. N-Alkenyl Indoles as Useful Intermediates for Alkaloid Synthesis. J. Org. Chem. 2011, 76, 9488–9496. [Google Scholar] [CrossRef]
- Maki, Y.; Mori, H.; Endo, T. Xanthate-Mediated Controlled Radical Polymerization of N-Vinylindole Derivatives. Macromolecules 2007, 40, 6119–6130. [Google Scholar] [CrossRef]
- Brustolin, F.; Castelvetro, V.; Ciardelli, F.; Ruggeri, G.; Colligiani, A. Synthesis and Characterization of Different Poly(1-vinylindole)s for Photorefractive Materials. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 253–262. [Google Scholar] [CrossRef]
- Bekkar, F.; Bettahar, F.; Moreno, I.; Meghabar, R.; Hamadouche, M.; Hernáez, E.; Vilas-Vilela, J.L.; Ruiz-Rubio, L. Polycarbazole and Its Derivatives: Synthesis and Applications. A Review of the Last 10 Years. Polymers 2020, 12, 2227. [Google Scholar] [CrossRef] [PubMed]
- Fridkin, G.; Boutard, N.; Lubell, W.D. β,β-Disubstituted C- and N-vinylindoles from One-Step Condensations of Aldehydes and Indole Derivatives. J. Org. Chem. 2009, 74, 5603–6603. [Google Scholar] [CrossRef] [PubMed]
- Taillefer, M.; Ouali, A.; Renard, B.; Spindler, J.-F. Mild Copper-Catalyzed Vinylation Reactions of Azoles and Phenols with Vinyl bromides. Chem. Eur. J. 2006, 12, 5301–5313. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Wang, Y.; Zhang, L.; Xi, C. A General Copper—Catalyzed Coupling of azoles with Vinyl Bromide. J. Org. Chem. 2009, 74, 6371–6373. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.Y.; Izmer, V.V.; Kazyul’kin, D.N.; Beletskaya, I.P.; Voskoboynikov, A.Z. Palladium-Catalyzed Stereocontrolled Vinylation of Azoles and Phenothiazine. Org. Lett. 2002, 4, 623–626. [Google Scholar] [CrossRef]
- Roche, M.; Frison, G.; Brion, J.-D.; Provot, O.; Hamze, A.; Alami, M. Csp2-N bond Formation via Ligand-Free Pd-Catalyzed Oxidative Coupling Reaction of N-Tosylhydrazones and Indole derivatives. J. Org. Chem. 2013, 78, 8485–8495. [Google Scholar] [CrossRef]
- Zeng, X.; Cheng, G.; Shen, J.; Cui, X. Palladium–catalyzed Oxidative Cross-Coupling of N-Tosylhydrazones with Indoles: Synthesis of N-Vinylindoles. Org. Lett. 2013, 15, 3022–3025. [Google Scholar] [CrossRef]
- Wu, G.; Su, W. Regio- and Stereoselective Direct N-Alkenylation of Indoles via Pd-Catalyzed Aerobic Oxidation. Org. Lett. 2013, 15, 5278–5281. [Google Scholar] [CrossRef]
- Rodygin, K.R.; Bogachenkov, A.S.; Ananikov, V.P. Vinylation of a Secondary Amine Core with Calcium Carbide for Efficient Post-Modification and Access to Polymeric Materials. Molecules 2018, 23, 648. [Google Scholar] [CrossRef]
- Verma, A.K.; Joshi, M.; Singh, V.P. Base–Mediated Regio– and Stereoselective Intermolecular Addition of Alkynes to N-Heterocycles. Org. Lett. 2011, 13, 1630–1633. [Google Scholar] [CrossRef]
- Garg, V.; Kumar, P.; Verma, A.K. Substrate-Controlled Regio- and Stereoselective Synthesis of (Z)- and (E)-N- Styrylated Carbazoles, Aza-carbazoles, and γ-Carbolines via Hydroamination of Alkynes. J. Org. Chem. 2018, 83, 11686–11702. [Google Scholar] [CrossRef]
- Zhou, M.; Tan, X.; Hu, Y.; Shen, H.C.; Qian, X. Highly Chemo- and Regioselective Vinylation of N-Heteroarenes with Vinylsulfonium salts. J. Org. Chem. 2018, 83, 8627–8635. [Google Scholar] [CrossRef]
- Zou, L.-H.; Liu, B.; Wang, C.; Shao, Z.; Zhou, J.; Shao, A.; Wen, J. Selective synthesis of alkyl amines and N- vinylazoles from vinyl sulfonium salts with N-nucleophiles. Org. Chem. Front. 2022, 9, 3231–3236. [Google Scholar] [CrossRef]
- Nie, Z.; Lv, H.; Li, H.; Su, M.; Yang, T.; Luo, W.; Liu, Q.; Guo, C. Synthesis of terminal N-Vinylazoles from Aromatic Aldehydes, DMSO, and Azoles based DMSO as Terminal Carbon Synthon. Adv. Synth Catal. 2021, 363, 4621–4626. [Google Scholar] [CrossRef]
- Bagnoli, L.; Santi, C. Organoselenium compound as chiral building blocks. In Chiral Building Blocks in Asymmetric Synthesis: Synthesis and Application, 1st ed.; WILEY–VCH: Hoboken, NJ, USA, 2022; Chapter 14; pp. 463–488. ISBN 978-3-527-34946-3. [Google Scholar]
- Torres-Ochoa, R.O.; Buyck, T.; Wang, Q.; Zhu, J. Heteroannulation of arynes with α-amino imides: Synthesis of 2,2-disubstituted indolin-3-ones and application to the enantioselective total synthesis of (+)-Hinckdentine A. Angew. Chem. Int. Ed. 2018, 57, 5679–5683. [Google Scholar] [CrossRef]
- Palomba, M.; De Monte, E.; Mambrini, A.; Bagnoli, L.; Santi, C.; Marini, F. A three-component [3 + 2]-cycloaddition/ elimination cascade for the synthesis of spirooxindole-pyrrolizines. Org. Biomol. Chem. 2021, 19, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, A.; Samanta, S.; Pathak, T. Enantiopure 1,4,5-trisubstituted 1,2,3-triazoles from carbohydrates: Applications of organoselenium chemistry. J. Org. Chem. 2014, 79, 6895–6904. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, L.; Casini, S.; Marini, F.; Santi, C.; Testaferri, L. Vinyl selenones: Annulation agents for the synthesis of six-membered benzo-1,4-heterocyclic compounds. Tetrahedron 2013, 69, 481–486. [Google Scholar] [CrossRef]
- Palomba, M.; Vinti, E.; Marini, F.; Santi, C.; Bagnoli, L. Synthesis of oxazino[4,3-a]indoles by domino addition-cyclization reactions of 1H-indol-2-yl)methanols and vinyl selenones in the presence of 18-crown-6. Tetrahedron 2016, 72, 7059–7064. [Google Scholar] [CrossRef]
- Palomba, M.; Sancineto, L.; Marini, F.; Santi, C.; Bagnoli, L. A domino approach to pyrazino-indoles and pyrroles using vinyl selenones. Tetrahedron 2018, 74, 7156–7163. [Google Scholar] [CrossRef]
- Buyck, T.; Wang, Q.; Zhu, J. Catalytic enantioselective Michael addition of α-aryl-α-isocyanoacetates to vinyl selenone: Synthesis of α,α-disubstituted α-amino acids and (+)- and (–)-trigonoliimine A. Angew. Chem. Int. Ed. 2013, 52, 12714–12718. [Google Scholar] [CrossRef]
- Buyck, T.; Wang, Q.; Zhu, J. Triple role of phenylselenonyl group enabled a one-pot synthesis of 1,3-oxazinan-2-ones from α-isocyanoacetates, phenyl vinyl selenones, and water. J. Am. Chem. Soc. 2014, 136, 11524–11528. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, L.; Scarponi, C.; Testaferri, L.; Tiecco, M. Preparation of both enantiomers of cyclopropane derivatives from the reaction of vinyl selenones with di-(-)-bornyl malonate. Tetrahedron Asymmetry 2009, 20, 1506–1514. [Google Scholar] [CrossRef]
- Zhang, T.; Cheng, L.; Hameed, S.; Liu, L.; Wang, D.; Chen, Y.-J. Highly enantioselective Michael addition of 2-oxindoles to vinyl selenone in RTILs catalyzed by a Cinchona alkaloid-based thiourea. Chem. Commun. 2011, 47, 6644–6646. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, L.; Scarponi, C.; Rossi, M.G.; Testaferri, L.; Tiecco, M. Synthesis of enantiopure 1,4-dioxanes, morpholines, and piperazines from the reaction of chiral 1,2-diols, amino alcohols, and diamines with vinyl selenones. Chem. Eur. J. 2011, 17, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Sternativo, S.; Walczak, O.; Battistelli, B.; Testaferri, L.; Marini, F. Organocatalytic Michael addition of indanone carboxylates to vinyl selenone for the asymmetric synthesis of polycyclic pyrrolidines. Tetrahedron 2012, 68, 10536–10541. [Google Scholar] [CrossRef]
- Bhaumik, A.; Das, A.; Pathak, T. Vinyl selenones derived from d-Fructose: A new platform for fructochemistry. Asian J. Org. Chem. 2016, 5, 1048–1062. [Google Scholar] [CrossRef]
- Bhaumik, A.; Pathak, T. Methyl-α D 2-selenonyl Pent-2-enofuranoside: A reactive selenosugar for the diversity-oriented synthesis of enantiomerically pure heterocycles, carbocycles, and isonucleosides. J. Org. Chem. 2015, 80, 11057–11064. [Google Scholar] [CrossRef]
- Palomba, M.; Trappetti, F.; Bagnoli, L.; Santi, C.; Marini, F. Oxone mediated oxidation of vinyl selenides in water. Eur. J. Org. Chem. 2018, 2018, 3914–3919. [Google Scholar] [CrossRef]
- Bandini, M.; Melloni, A.; Umani-Ronchi, A. New versatile Pd-Catalyzed Alkylation of Indoles via Nucleophilic Allylic Substitution: Controlling the Regioselectivity. Org. Lett. 2004, 6, 3199–3202. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Young, M.W.; Lauer, R.F. Reactions of Selenoxides: Thermal Syn-Elimination and H218O Exchange. Tetrahedron Lett. 1973, 14, 1979–1982. [Google Scholar] [CrossRef]
- Palomba, M.; Dias, I.F.C.; Rosati, O.; Marini, F. Modern Synthetic Strategies with Organoselenium Reagents. Molecules 2021, 26, 3148. [Google Scholar] [CrossRef]
- Azaz, T.; Mourya, H.; Singh, V.; Ram, B.; Tiwari, B. Reductive Alkenylation of Ketimines via Hydride Transfer from Aldehydes. J. Org. Chem. 2023, 88, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, A.; Azaz, T.; Singh, V.; Khatana, A.K.; Tiwari, B. Carbene/Base–Mediated Redox Alkenylation of Isatins using β-Substituted Organoselenenones and Aldehydes. J. Org. Chem. 2019, 84, 14898–14903. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, T.; Aoki, K.; Wagatsuma, T.; Suzuki, Y. Lewis Acid Catalyzed Addition of Pyrazoles to Alkynes: Selective Synthesis of Double and Single Addition Products. Eur. J. Org. Chem. 2008, 4035–4040. [Google Scholar] [CrossRef]






























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).