
In Vitro Affinity Maturation of Nanobodies against Mpox Virus A29 Protein Based on Computer-Aided Design
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Phage Screening of A29
2.2. Antibody Expression, Purification, and Affinity Validation
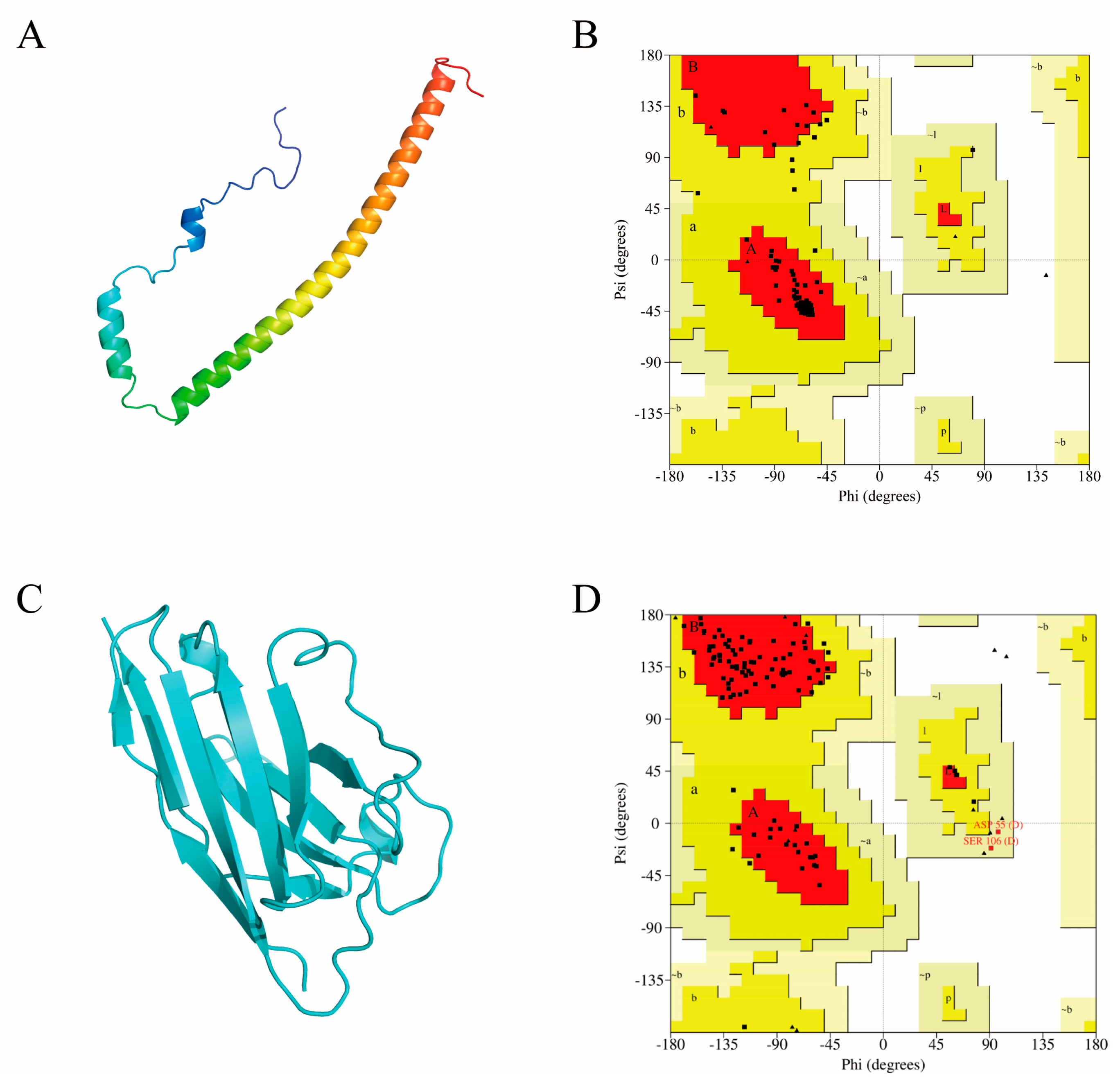
2.3. Computer-Aided Modeling and Docking of Structures
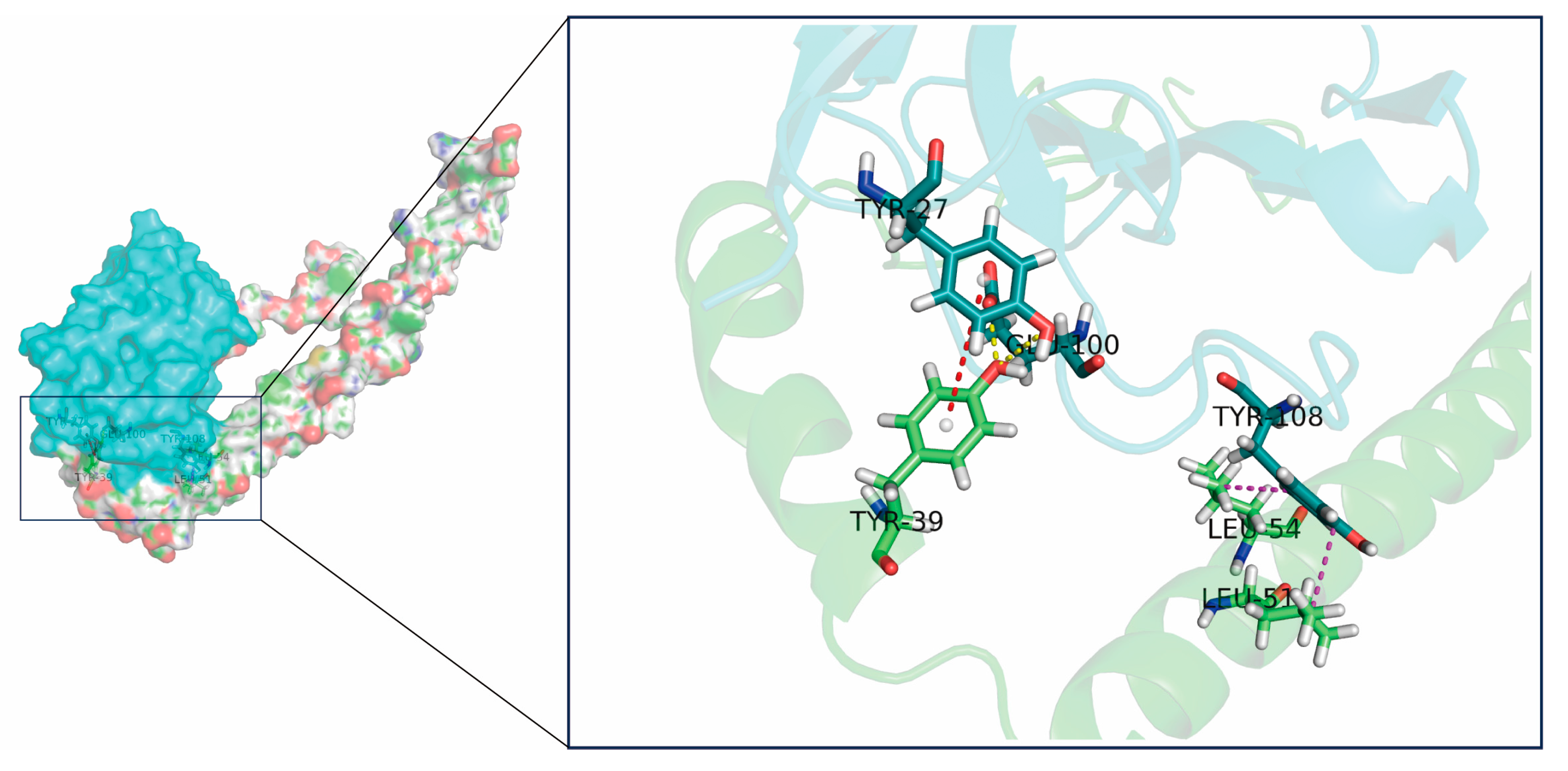
2.4. Key Residue Positions for Affinity Maturation
2.5. Mutagenesis for Affinity Maturation In Silico
2.6. Affinity and Recognition Site Validation after Mutation
3. Discussion
4. Materials and Methods
4.1. Sequencing
4.2. Phage Screening for Nanobody Selection
4.3. Phage Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. Construction, Expression, and Purification of the Nanobody
4.5. Affinity ELISA
4.6. Antigen Epitope Analysis
4.7. Homology Modeling
4.8. Molecular Docking
4.9. MD Simulation
4.10. Identifying Key Residue Positions
4.11. In Silico Affinity Maturation
4.12. Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- WHO. Multi-Country Outbreak of Monkeypox. External Situation Report 2; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- WHO. 2022–2023 Mpox Outbreak: Global Trends; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Fine, P.E.; Jezek, Z.; Grab, B.; Dixon, H. The transmission potential of monkeypox virus in human populations. Int. J. Epidemiol. 1988, 17, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Lustig, S.; Fogg, C.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H.; Moss, B. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 2005, 79, 13454–13462. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Acharya, A.; Gendelman, H.E.; Byrareddy, S.N. The 2022 outbreak and the pathobiology of the monkeypox virus. J. Autoimmun. 2022, 131, 102855. [Google Scholar] [CrossRef]
- Gilchuk, I.; Gilchuk, P.; Sapparapu, G.; Lampley, R.; Singh, V.; Kose, N.; Blum, D.L.; Hughes, L.J.; Satheshkumar, P.S.; Townsend, M.B.; et al. Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections. Cell 2016, 167, 684–694.e689. [Google Scholar] [CrossRef]
- Mucker, E.M.; Golden, J.W.; Hammerbeck, C.D.; Kishimori, J.M.; Royals, M.; Joselyn, M.D.; Ballantyne, J.; Nalca, A.; Hooper, J.W. A Nucleic Acid-Based Orthopoxvirus Vaccine Targeting the Vaccinia Virus L1, A27, B5, and A33 Proteins Protects Rabbits against Lethal Rabbitpox Virus Aerosol Challenge. J. Virol. 2022, 96, e0150421. [Google Scholar] [CrossRef]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting Nanobodies’ Singular Traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef]
- Tu, Z.; Chen, Q.; Li, Y.; Xiong, Y.; Xu, Y.; Hu, N.; Tao, Y. Identification and characterization of species-specific nanobodies for the detection of Listeria monocytogenes in milk. Anal. Biochem. 2016, 493, 1–7. [Google Scholar] [CrossRef]
- Morales-Yanez, F.J.; Sariego, I.; Vincke, C.; Hassanzadeh-Ghassabeh, G.; Polman, K.; Muyldermans, S. An innovative approach in the detection of Toxocara canis excretory/secretory antigens using specific nanobodies. Int. J. Parasitol. 2019, 49, 635–645. [Google Scholar] [CrossRef]
- Liu, W.; Song, H.; Chen, Q.; Yu, J.; Xian, M.; Nian, R.; Feng, D. Recent advances in the selection and identification of antigen-specific nanobodies. Mol. Immunol. 2018, 96, 37–47. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Gerace, D.; Vaddinelli, D.; Allegra, A.G.; Musolino, C. Nanobodies and Cancer: Current Status and New Perspectives. Cancer Investig. 2018, 36, 221–237. [Google Scholar] [CrossRef]
- Chakravarty, R.; Goel, S.; Cai, W. Nanobody: The “magic bullet” for molecular imaging? Theranostics 2014, 4, 386–398. [Google Scholar] [CrossRef]
- Mitchell, L.S.; Colwell, L.J. Analysis of nanobody paratopes reveals greater diversity than classical antibodies. Protein Eng. Des. Sel. PEDS 2018, 31, 267–275. [Google Scholar] [CrossRef]
- Zavrtanik, U.; Lukan, J.; Loris, R.; Lah, J.; Hadži, S. Structural Basis of Epitope Recognition by Heavy-Chain Camelid Antibodies. J. Mol. Biol. 2018, 430, 4369–4386. [Google Scholar] [CrossRef]
- Ledsgaard, L.; Ljungars, A.; Rimbault, C.; Sørensen, C.V.; Tulika, T.; Wade, J.; Wouters, Y.; McCafferty, J.; Laustsen, A.H. Advances in antibody phage display technology. Drug Discov. Today 2022, 27, 2151–2169. [Google Scholar] [CrossRef]
- Deutscher, S. Phage Display to Detect and Identify Autoantibodies in Disease. New Engl. J. Med. 2019, 381, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.Z.B.; Oroojalian, F.; Eyvazi, S.; Hejazi, M.; Baradaran, B.; Pouladi, N.; Tohidkia, M.R.; Mokhtarzadeh, A.; Muyldermans, S. An overview on display systems (phage, bacterial, and yeast display) for production of anticancer antibodies; advantages and disadvantages. Int. J. Biol. Macromol. 2022, 208, 421–442. [Google Scholar] [CrossRef] [PubMed]
- Soler, M.A.; Fortuna, S.; de Marco, A.; Laio, A. Binding affinity prediction of nanobody-protein complexes by scoring of molecular dynamics trajectories. Phys. Chem. Chem. Phys. PCCP 2018, 20, 3438–3444. [Google Scholar] [CrossRef]
- Sudha, G.; Nussinov, R.; Srinivasan, N. An overview of recent advances in structural bioinformatics of protein-protein interactions and a guide to their principles. Prog. Biophys. Mol. Biol. 2014, 116, 141–150. [Google Scholar] [CrossRef]
- Cumbers, S.J.; Williams, G.T.; Davies, S.L.; Grenfell, R.L.; Takeda, S.; Batista, F.D.; Sale, J.E.; Neuberger, M.S. Generation and iterative affinity maturation of antibodies in vitro using hypermutating B-cell lines. Nat. Biotechnol. 2002, 20, 1129–1134. [Google Scholar] [CrossRef]
- Qiao, C.; Lv, M.; Li, X.; Geng, J.; Li, Y.; Zhang, J.; Lin, Z.; Feng, J.; Shen, B. Affinity maturation of antiHER2 monoclonal antibody MIL5 using an epitope-specific synthetic phage library by computational design. J. Biomol. Struct. Dyn. 2013, 31, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef] [PubMed]
- Kiyoshi, M.; Caaveiro, J.M.; Miura, E.; Nagatoishi, S.; Nakakido, M.; Soga, S.; Shirai, H.; Kawabata, S.; Tsumoto, K. Affinity improvement of a therapeutic antibody by structure-based computational design: Generation of electrostatic interactions in the transition state stabilizes the antibody-antigen complex. PLoS ONE 2014, 9, e87099. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B. mCSM-AB: A web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. 2016, 44, W469–W473. [Google Scholar] [CrossRef]
- Myung, Y.; Rodrigues, C.H.M.; Ascher, D.B.; Pires, D.E.V. mCSM-AB2: Guiding rational antibody design using graph-based signatures. Bioinformatics 2020, 36, 1453–1459. [Google Scholar] [CrossRef]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, Y.; Lu, H.; Zhao, F.; Alvarez, R.V.; Goncearenco, A.; Panchenko, A.R.; Li, M. MutaBind2: Predicting the Impacts of Single and Multiple Mutations on Protein-Protein Interactions. iScience 2020, 23, 100939. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef]
- Zhong, Z.; Yang, Y.; Chen, X.; Han, Z.; Zhou, J.; Li, B.; He, X. Positive charge in the complementarity-determining regions of synthetic nanobody prevents aggregation. Biochem. Biophys. Res. Commun. 2021, 572, 1–6. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, K.; Wu, Q.; Huang, S.Y. NLDock: A Fast Nucleic Acid-Ligand Docking Algorithm for Modeling RNA/DNA-Ligand Complexes. J. Chem. Inf. Model. 2021, 61, 4771–4782. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yan, Y.; He, J.; Tao, H.; Wu, Q.; Huang, S.Y. Docking and scoring for nucleic acid-ligand interactions: Principles and current status. Drug Discov. Today 2022, 27, 838–847. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Suganami, A.; Ishida, I.; Tamura, Y.; Maeda, Y. Affinity maturation of a CDR3-grafted VHH using in silico analysis and surface plasmon resonance. J. Biochem. 2013, 154, 325–332. [Google Scholar] [CrossRef]
- Tan, L.; Chen, H.; Yu, S.; Qiu, X.; Song, C.; Chen, D.; Zhang, S.; Zhang, F.; He, S.; Shen, X.; et al. A SOE-PCR method of introducing multiple mutations into Mycoplasma gallisepticum neuraminidase. J. Microbiol. Methods 2013, 94, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Bunge, E.M.; Hoet, B.; Chen, L.; Lienert, F.; Weidenthaler, H.; Baer, L.R.; Steffen, R. The changing epidemiology of human monkeypox-A potential threat? A systematic review. PLoS Neglected Trop. Dis. 2022, 16, e0010141. [Google Scholar] [CrossRef]
- Siegrist, E.A.; Sassine, J. Antivirals With Activity Against Mpox: A Clinically Oriented Review. Clin. Infect. Dis. An. Off. Publ. Infect. Dis. Soc. Am. 2023, 76, 155–164. [Google Scholar] [CrossRef]
- Lim, C.K.; Roberts, J.; Moso, M.; Liew, K.C.; Taouk, M.L.; Williams, E.; Tran, T.; Steinig, E.; Caly, L.; Williamson, D.A. Mpox diagnostics: Review of current and emerging technologies. J. Med. Virol. 2023, 95, e28429. [Google Scholar] [CrossRef]
- Shi, D.; He, P.; Song, Y.; Cheng, S.; Linhardt, R.J.; Dordick, J.S.; Chi, L.; Zhang, F. Kinetic and Structural Aspects of Glycosaminoglycan-Monkeypox Virus Protein A29 Interactions Using Surface Plasmon Resonance. Molecules 2022, 27, 5898. [Google Scholar] [CrossRef]
- Hughes, L.J.; Goldstein, J.; Pohl, J.; Hooper, J.W.; Lee Pitts, R.; Townsend, M.B.; Bagarozzi, D.; Damon, I.K.; Karem, K.L. A highly specific monoclonal antibody against monkeypox virus detects the heparin binding domain of A27. Virology 2014, 464–465, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.Y.; Dubuc, G.; Li, S.; Hirama, T.; Mackenzie, C.R.; Jermutus, L.; Hall, J.C.; Tanha, J. Affinity maturation of a V(H)H by mutational hotspot randomization. J. Immunol. Methods 2005, 297, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Sulea, T.; Hussack, G.; Ryan, S.; Tanha, J.; Purisima, E.O. Application of Assisted Design of Antibody and Protein Therapeutics (ADAPT) improves efficacy of a Clostridium difficile toxin A single-domain antibody. Sci. Rep. 2018, 8, 2260. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. USA 2015, 112, 10920–10925. [Google Scholar] [CrossRef]
- Rege, N.K.; Wickramasinghe, N.P.; Tustan, A.N.; Phillips, N.F.B.; Yee, V.C.; Ismail-Beigi, F.; Weiss, M.A. Structure-based stabilization of insulin as a therapeutic protein assembly via enhanced aromatic-aromatic interactions. J. Biol. Chem. 2018, 293, 10895–10910. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Petsko, G.A. Aromatic-aromatic interaction: A mechanism of protein structure stabilization. Science 1985, 229, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Paesani, F.; Zhang, W.; Case, D.A.; Cheatham, T.E., 3rd; Voth, G.A. An accurate and simple quantum model for liquid water. J. Chem. Phys. 2006, 125, 184507. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Martonák, R.; Laio, A.; Parrinello, M. Predicting crystal structures: The Parrinello-Rahman method revisited. Phys. Rev. Lett. 2003, 90, 075503. [Google Scholar] [CrossRef]
- Li, L.; Chen, S.; Miao, Z.; Liu, Y.; Liu, X.; Xiao, Z.X.; Cao, Y. AbRSA: A robust tool for antibody numbering. Protein Sci. A Publ. Protein Soc. 2019, 28, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.S.; Schein, C.H.; Oezguen, N.; Power, T.D.; Braun, W. InterProSurf: A web server for predicting interacting sites on protein surfaces. Bioinformatics 2007, 23, 3397–3399. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interface Residues Analysis | PyMOL Analysis | ||
|---|---|---|---|
| 1–5 | - | 1–5 | - |
| 27 | ARG(R) | 27 | ARG(R) |
| 37 | PHE(F) | 37–46 | - |
| 39 | GLN(Q) | 95 | TYR(Y) |
| 42–46 | - | 99 | ALA(A) |
| 95 | TYR(Y) | 100 | MET(M) |
| 100 | MET(M) | 101 | ILE(I) |
| 101 | ILE(I) | 102 | TYR(Y) |
| 102 | TYR(Y) | 108 | GLN(Q) |
| 108 | GLN(Q) | 109 | TRP(W) |
| 109 | TRP(W) | 110 | SER(S) |
| 110 | SER(S) | 112–116 | - |
| 112–116 | - | 117 | GLY(G) |
| 124 | SER(S) | 124 | SER(S) |
| Mutants | MutaBind2 | mCSM-AB2 |
|---|---|---|
| R27F | −0.86 | 0.74 |
| R27Y | −1.07 | 0.45 |
| M100E | −0.39 | 1.07 |
| M100D | −0.18 | 0.51 |
| I101N | −0.03 | 0.61 |
| I101S | −0.37 | 0.14 |
| Q108F | −0.72 | 0.21 |
| Q108W | −0.41 | 0.49 |
| Q108Y | −0.7 | 0.71 |
| S110Y | −0.23 | 1.34 |
| Double Mutants | MutaBind2 | Double Mutants | MutaBind2 |
|---|---|---|---|
| R27F M100E | −0.37 | M100E Q108F | −0.91 |
| R27F M100D | −0.2 | M100E Q108W | −0.38 |
| R27F I101N | −0.59 | M100E Q108Y | −1.21 |
| R27F I101S | −0.48 | M100D Q108F | −0.64 |
| R27F Q108F | −1.44 | M100D Q108Y | −0.68 |
| R27F Q108W | −1.51 | I101N Q108F | −1.58 |
| R27F Q108Y | −1.5 | I101N Q108W | −0.54 |
| R27F S110Y | −1.05 | I101N Q108Y | −1.45 |
| R27Y M100E | −0.53 | I101N S110Y | −1.07 |
| R27Y M100D | −0.14 | I101S Q108F | −1.51 |
| R27Y I101N | −0.34 | I101S Q108W | −1.01 |
| R27Y I101S | −0.24 | I101S Q108Y | −1.76 |
| R27Y Q108F | −1.41 | I101S S110Y | −0.52 |
| R27Y Q108W | −1.74 | Q108F S110Y | −0.94 |
| R27Y Q108Y | −2.11 | Q108W S110Y | −1.27 |
| R27Y S110Y | −0.95 | Q108Y S110Y | −0.95 |
| Triple Mutants | MutaBind2 |
|---|---|
| R27F Q108W I101S | −1.77 |
| R27F Q108W S110Y | −1.66 |
| R27Y I101S Q108W | −0.84 |
| R27Y Q108W S110Y | −1.87 |
| R27Y M100E Q108Y | −2.31 |
| R27Y I101N Q108Y | −1.79 |
| R27Y I101S Q108Y | −1.57 |
| M100E I101N Q108Y | −2.02 |
| M100E I101S Q108Y | −1.98 |
| I101N Q108W S110Y | −0.89 |
| I101S Q108W S110Y | −0.83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Mao, G.; Pei, Z.; Cen, J.; Meng, W.; Wang, Y.; Zhang, S.; Li, S.; Xu, Q.; Sun, M.; et al. In Vitro Affinity Maturation of Nanobodies against Mpox Virus A29 Protein Based on Computer-Aided Design. Molecules 2023, 28, 6838. https://doi.org/10.3390/molecules28196838
Yu H, Mao G, Pei Z, Cen J, Meng W, Wang Y, Zhang S, Li S, Xu Q, Sun M, et al. In Vitro Affinity Maturation of Nanobodies against Mpox Virus A29 Protein Based on Computer-Aided Design. Molecules. 2023; 28(19):6838. https://doi.org/10.3390/molecules28196838
Chicago/Turabian StyleYu, Haiyang, Guanchao Mao, Zhipeng Pei, Jinfeng Cen, Wenqi Meng, Yunqin Wang, Shanshan Zhang, Songling Li, Qingqiang Xu, Mingxue Sun, and et al. 2023. "In Vitro Affinity Maturation of Nanobodies against Mpox Virus A29 Protein Based on Computer-Aided Design" Molecules 28, no. 19: 6838. https://doi.org/10.3390/molecules28196838