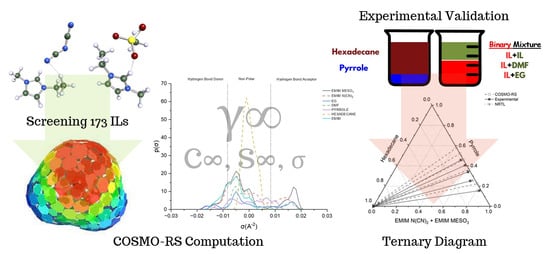
2.1. Computational Screening of ILs
The screening process for the potential solvents involved the evaluation of two criteria: capacity and selectivity at infinite dilution. A higher selectivity suggests improved separation and a reduced number of extraction stages, whereas a higher capacity indicates a larger amount of extraction. In the COSMO-RS, the maximum amount of solute that a solvent can dissolve can be represented by the capacity at infinite dilution (
C∞), which is expressed using Equation (6).
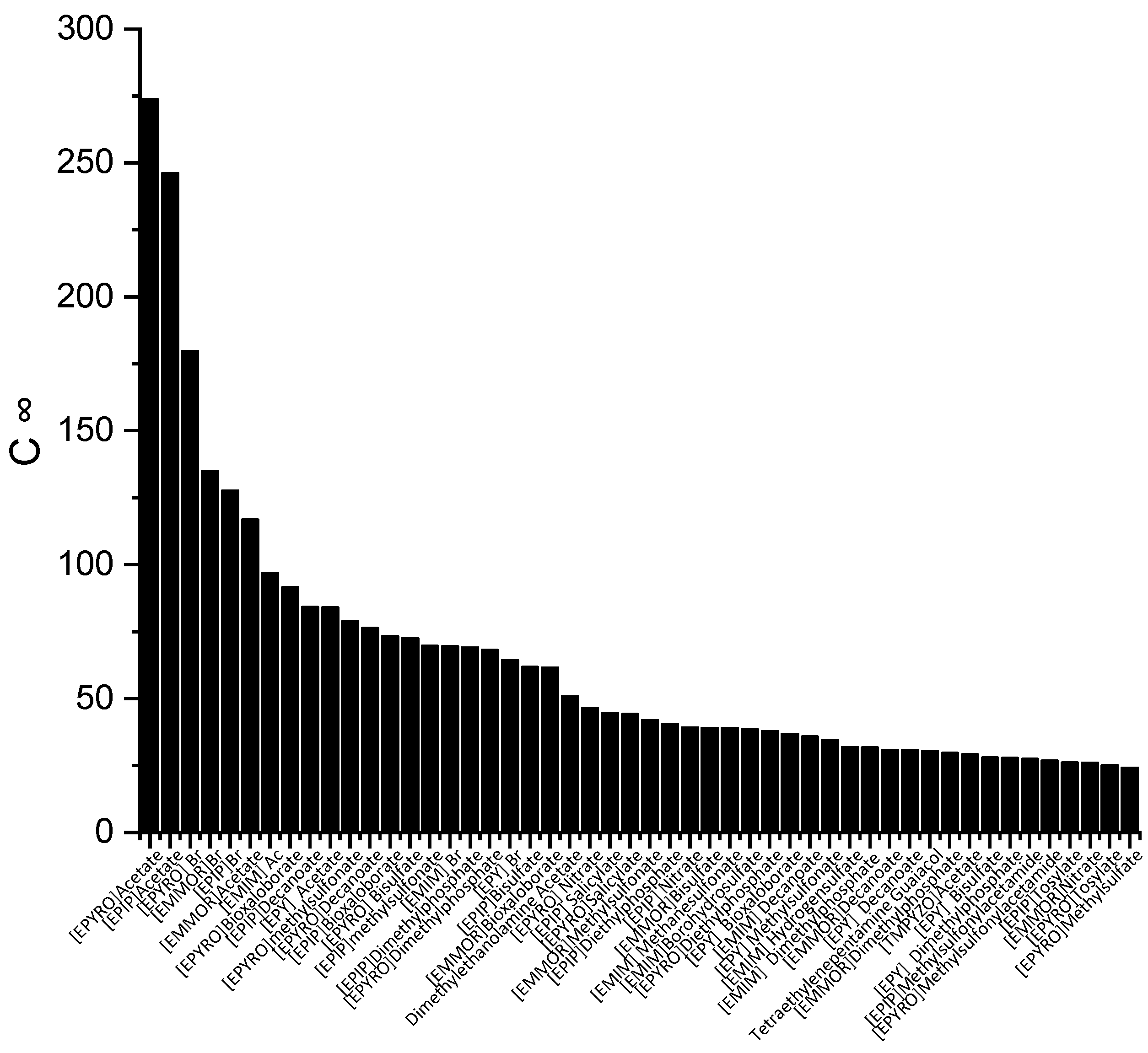
Figure 1 illustrates the top 50 ILs with respect to the
C∞ at a temperature of 298 K.
The ILs in
Figure 1 are ranked from the largest to smallest
C∞ values. As observed, the
C∞ is affected by the type of cation family, in the order of EPYRO > EPIP > EMMOR > EPY > EMIM > TMPYZO. This trend can be explained due to the lesser amount of steric hindrance of the first three compared to EMIM, TMPYZO, and EPY, which are encompassed by an aromatic ring. Furthermore,
Figure 1 also implies that anions with lesser numbers of heteroatomic atoms, such as acetate, bromide, and decanoate, result in a significant effect in terms of capacity. A lesser negative charge reduces the coulombic force and increases the solvent capacity.
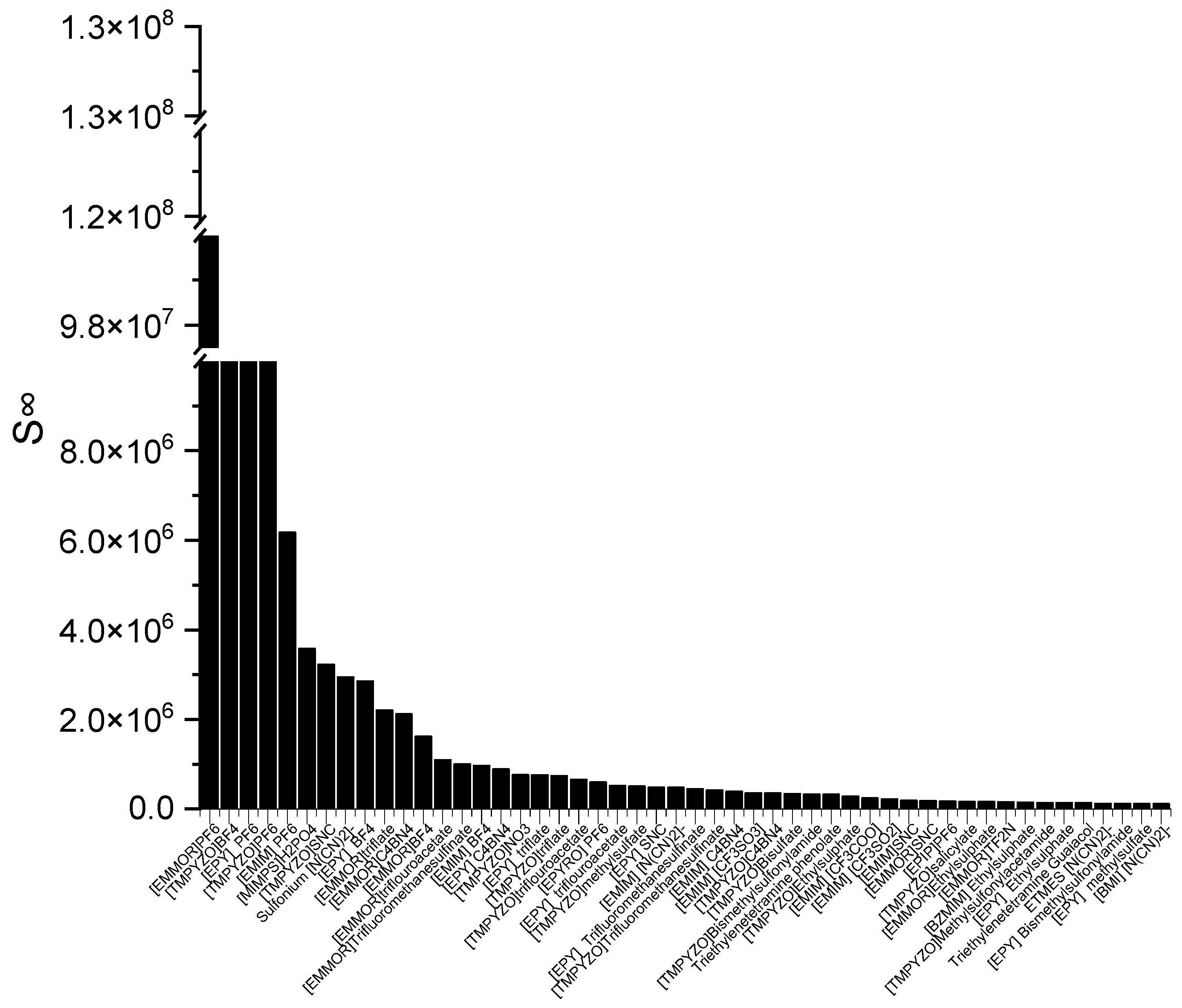
Figure 2 presents the top 50 ILs based on the calculated
S∞ at 298 K.
The corresponding ILs are listed from the largest to smaller order of their COSMO-RS reading result. The cation family was shown to have a significant influence on the S∞. The cation families EMMOR and TMPYZO showed the greatest influence on the S∞, followed by EPY and EMIM. This could be caused by the influence of the heteroatom in EMMOR, i.e., N and O, which increased the charge of the cation. In addition, the presence of an aromatic ring in TMPYZO, which is rich in electron density, resulted in a strong π-π interaction with the pyrrole compound.
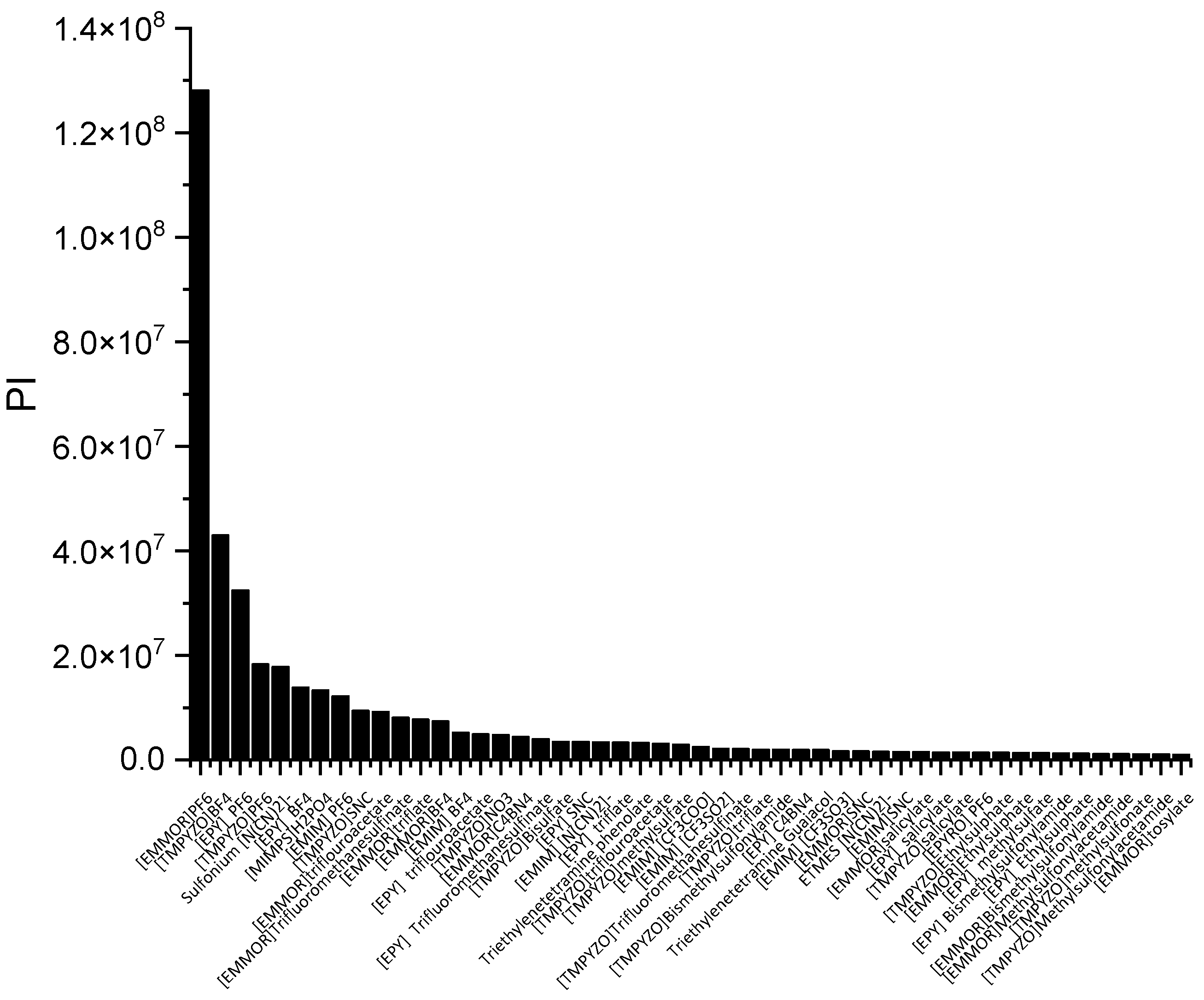
In a liquid–liquid extraction, an ideal IL would be one possessing high capacity and selectivity. However, the actual extraction process typically found that low selectivity resulted in high capacity, and vice versa. While the capacity determines the flow rate of the circulating solvent and, consequently, the size of the reactor, it is also a crucial factor in the solvent selection process. The selectivity needs to be properly assessed as well. As a result, there ought to be criteria that can consider both qualities. The performance index (PI) is one of the techniques used to complement the inverse proportionality between capacity and selectivity.
In
Figure 3, [EMMOR][PF
6] is shown to be the best candidate among the other ILs studied. However, for its application on an industrial scale, the cost and toxicity of the IL must be considered. For instance, the presence of a halide anion may result in equipment corrosiveness [
28] In this work, we considered the IL ranking after the screening, and we selected two relatively cheaper ILs that fell into the categories of high capacity, [EMIM][MeSO
3], and high selectivity, [EMIM][N(CN)
2)]. In addition, dimethyl formamide (DMF) and ethylene glycol (EG) were chosen to represent conventional solvents with high capacity and selectivity, respectively.
Table 1 summarizes the top 10 ILs based on the PI.
2.1.1. Effect of Cation Alkyl Chain Length
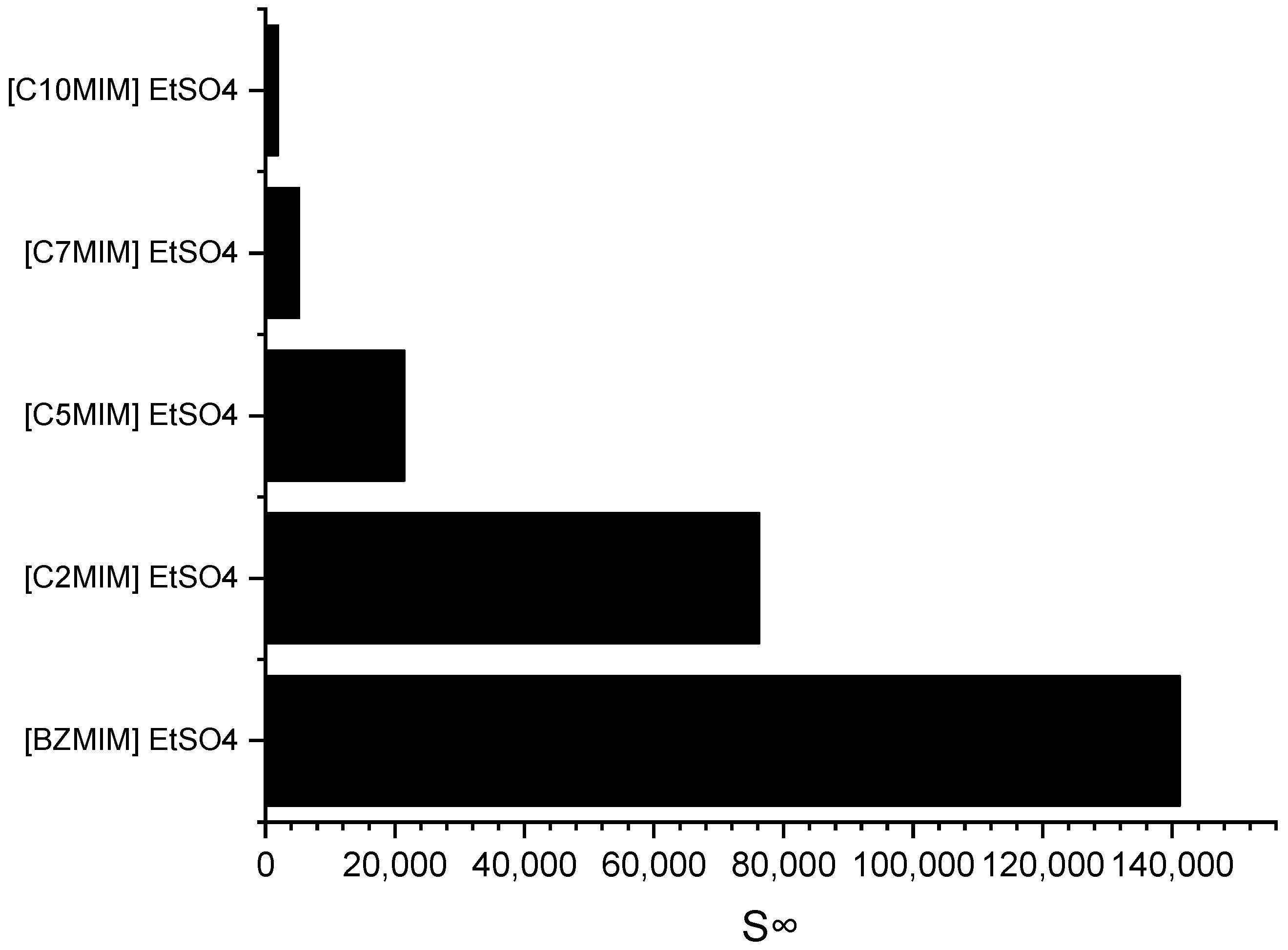
Imidazolium cations are widely used in the field of ILs application due to their ability to exhibit tuneable properties, such as miscibility, melting point, and viscosity. Consequently, an imidazolium-based cation was selected for investigating the impact of the alkyl chain length on selectivity.
Figure 4 depicts the effect of the alkyl chain length on selectivity, as determined through the COSMO-RS screening predictions.
It can be observed that as the alkyl chain length increases, the selectivity at infinite dilution decreases in the following order: C
10MIM < C
7MIM < C
5MIM < C
2MIM < BZMIM. The observed decrease in selectivity can be attributed to the reduced accommodation capacity of the pyrrole molecules, which arises from the increased steric hindrance on the imidazolium ring. However, interestingly, the results for BZMIM show a deviation from this trend. The presence of the benzyl group in BZMIM is expected to enhance selectivity due to its electron-rich nature and the potential for stronger π-π interactions between the aromatic and pyrrole compounds, facilitated by an additional π system. This stronger interaction is anticipated because the aromatic ring possesses an extended electron ring system, further contributing to the unique behavior of BZMIM in this study [
29].
The COSMO-RS prediction results are consistent with those of a prior study by Ferreira et al. (2012). This source reported a decrease in the predicted aromatic selectivity as the alkyl chain length of the cation increased, specifically for
n-hexane-benzene-[cation][TF2N] systems with imidazolium-based cations, and the order of selectivity was [EMIM] > [BMIM] > [OMIM] > [DMIM] > [C
12MIM] [
30]. In general, ILs that exhibit high selectivity have low capacity. In this case, the ILs with a BZMIM cation exhibited the lowest capacity at infinite dilution, as shown in
Table 2. This can be attributed to the presence of a benzyl group, which renders the cation more cationic, meaning a higher positive charge, thereby increasing the coulombic force between the cation and anion. This, in turn, weakens the ability of the IL to accommodate the pyrrole compound at infinite dilution [
29].
2.1.2. Effect of Anions
Increasing the COSMO volume of the anions enhances the selectivity of the ILs for nitrogen species in general [
31]. The COSMO volume quantifies the spatial extent or size of the anion in the IL. It provides information about the anion’s ability to interact with the surrounding cations and the other species within the IL. However, the observed trend differs, as the increase is dependent on the number of heteroatoms present in most anions, such as fluorine, sulfur, nitrogen, and oxygen [
32]. Another aspect of the anion that can be quantified is the presence of double bonds or triple bonds like thiocyanate, or the size of the anion or the alkyl chain length of the anion, which plays an important role in determining selectivity and capacity. Thus, in this screening, [EMIM][PF
6]
, [EMIM][SNC], [EMIM][C
7H
7O
3S] (tosylate), [EMIM][Br], and [EMIM][C
10H1
9O
2] (decanoate), were selected to study the effect of the anion on the
S∞ and
C∞.
Table 3 represents the chosen ILs, based on the COSMO-RS screening, used to study the effect of the anion.
Based on
Table 3, the
S∞ increases in the order of [EMIM][PF
6] > [EMIM][SNC] > [EMIM][C
7H
7O
3S] > [EMIM][C
10H
19O
2] > [EMIM][Br]. As stated earlier, the presence of heteroatoms influences selectivity, with [EMIM][PF
6] exhibiting the highest selectivity, followed by [SCN]. This can be explained by the fact that higher selectivity is typically associated with a number of heteroatoms. The lowest selectivity at
S∞ was observed with the halide anion [Br]
−, due to its lower number of heteroatoms, resulting in a lower charge value. Regarding the
C∞, [EMIM][Br] and [EMIM][C
10H
19O
2] (decanoate) displayed a higher
C∞ compared to the other ILs. The compactness of the cation and anion in [EMIM][Br] results in less steric hindrance, allowing for the easier accommodation of the pyrrole compound. Conversely, with [EMIM][C
10H
19O
2], as the length of the anion increases, the coulombic and inductive forces weaken, leading to a loss of stacking structure and the easier accommodation of the pyrrole compound. Further studies are required to support this assertion by calculating the interaction energy. A lower interaction energy between the cation and anion would weaken their interaction and increase the free volume effect [
33].
2.1.3. Effect of Cations
The interaction between cations is mainly facilitated through N (heteroaromatic)-H (cation) hydrogen bonds [
34], where N and H refer to the atoms present in the nitrogen compound and the cation, respectively. The interaction is also facilitated by a CH (cation)-π (nitrogen species) interaction [
35]. In this section, six cation families, namely, imidazolium (EMIM), pyrrolidinium (EPYRO), piperidinium (EPIP), morpholinium (EMMOR), pyridinium (EPY), and pyrazolinium (TMPYZO), with the same anion [Br]
−, will be discussed in general to determine the effect of the cation family.
Table 4 presents the ionic liquids with different types of cation families to see the effect on
S∞ and
C∞.
Based on
Table 4, the
S∞ increases in the order of TMPYZO > EPY > EMIM > EPIP > EMMOR > EPYRO. This is due to the aromatic rings in the imidazolium, pyridinium, and pyrazolium cations having π-electron systems, which exhibit increased selectivity towards pyrrole [
29]. In fact, for TMPYZO, the methyl groups in TMPYZO can alter the electronic properties of the pyrazolium ring. They can induce local electronic effects that influence the electrostatic interactions between TMPYZO and pyrrole. These electronic effects can enhance the selectivity of TMPYZO towards pyrrole by promoting favorable interactions, and reducing interactions with other molecules that do not possess compatible electronic characteristics. In summary, a notable trade-off exists between capacity and selectivity in ionic liquids (ILs). Those with high capacity typically demonstrate low selectivity, while ILs with low capacity often exhibit high selectivity. This trend is commonly observed in solvent screening studies for extraction. Thus, the performance index, PI, is pivotal to obtain both criteria (capacity + selectivity).
2.1.4. Effect of Protic Ionic Liquids on Capacity and Selectivity
This section provides a brief discussion of the impact of protic ionic liquids (PILs) on the
S∞ and
C∞, considering their distinct characteristics compared to aprotic ionic liquids (APILs). PILs are formed through the transfer of protons from a Brønsted acid to a Brønsted base, enabling the formation of hydrogen bonding [
36]. While PILs have shown promising results in gas absorption studies, particularly in H
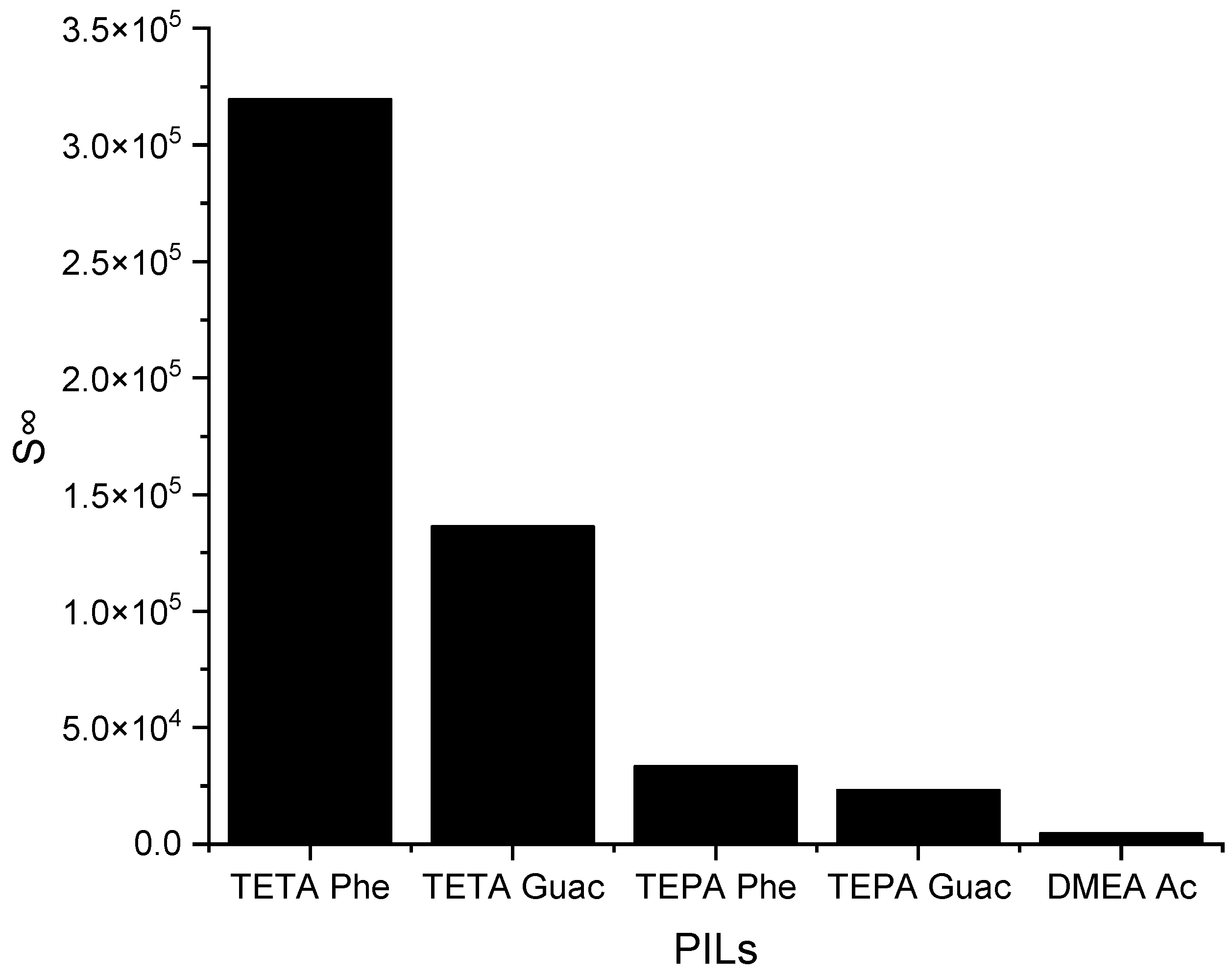
2S removal, their application to EDN remains limited. Therefore, this section investigates the effects of five different PILs, namely, triethylenetetramine phenolate ([TETA][Phe]), triethylenetetramine guaiacol ([TETA][Guac]), tetraethylenepentamine phenolate ([TEPA][Phe]), tetraethylenepentamine guaiacol ([TEPA][Guac]), and dimethylethanolamine acetate ([DMEA][Ac]), on EDN.
The selected PILs are listed based on the results obtained from the COSMO-RS screening, from largest to smallest. According to these results, all the PILs are capable of forming hydrogen bonds, but [TETA][Phe] exhibits a higher influence on the
S∞ compared to the other PILs. The reason behind this is due to the additional effect of its anion, which can contribute to selectivity through π-π interactions. Another factor is its cation structure that influences the ionization of the PIL. Primary amines, like TETA, tend to have higher proton transfer tendencies compared to DMEA, which has a tertiary amine structure, reducing the ionization of the PIL and, thus, providing two different hydrogen bond environments. Additionally, an increase in the alkyl chain length of the cation results in a reduction in ionization, as depicted in
Figure 5, regarding TETA and TEPA.
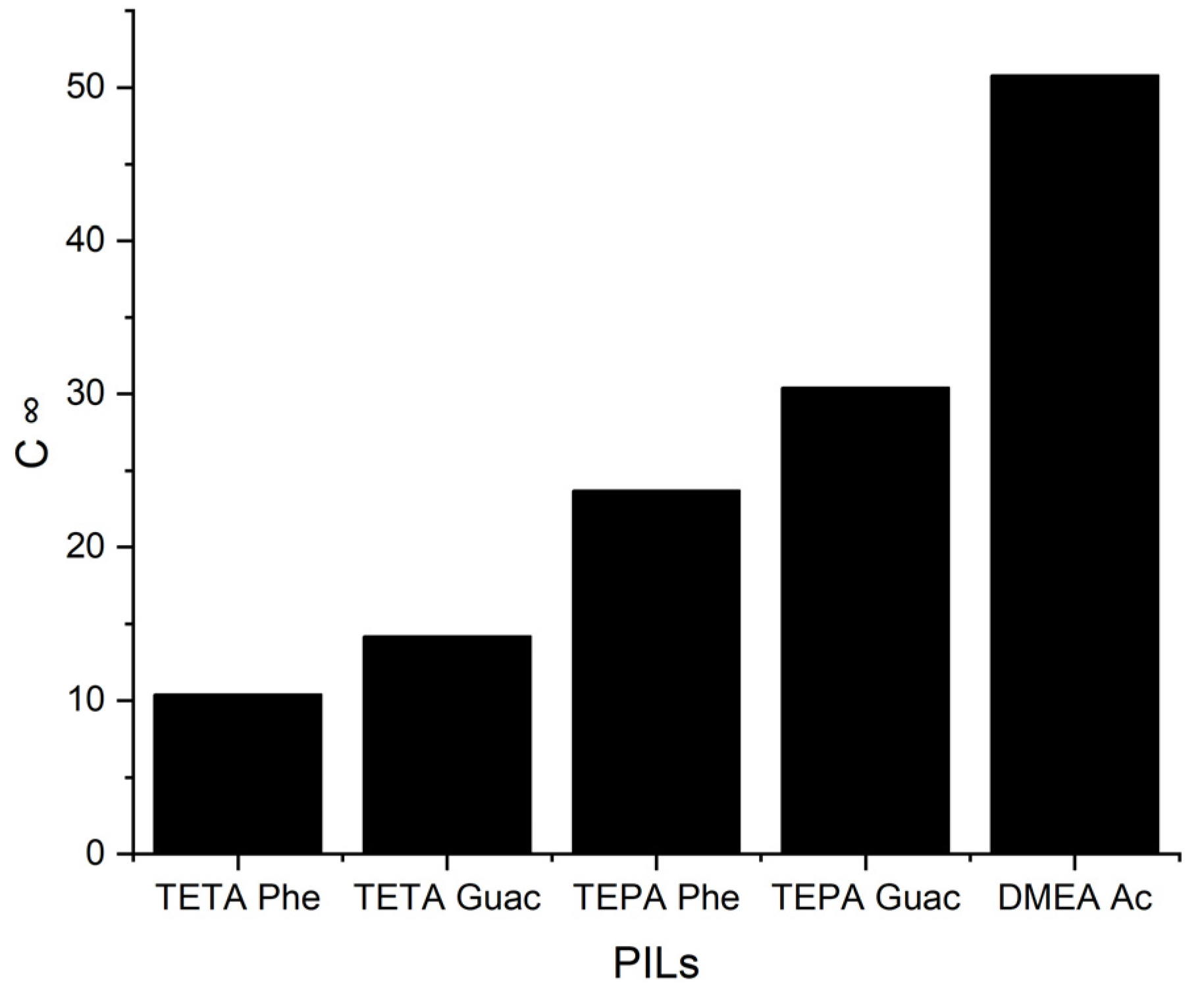
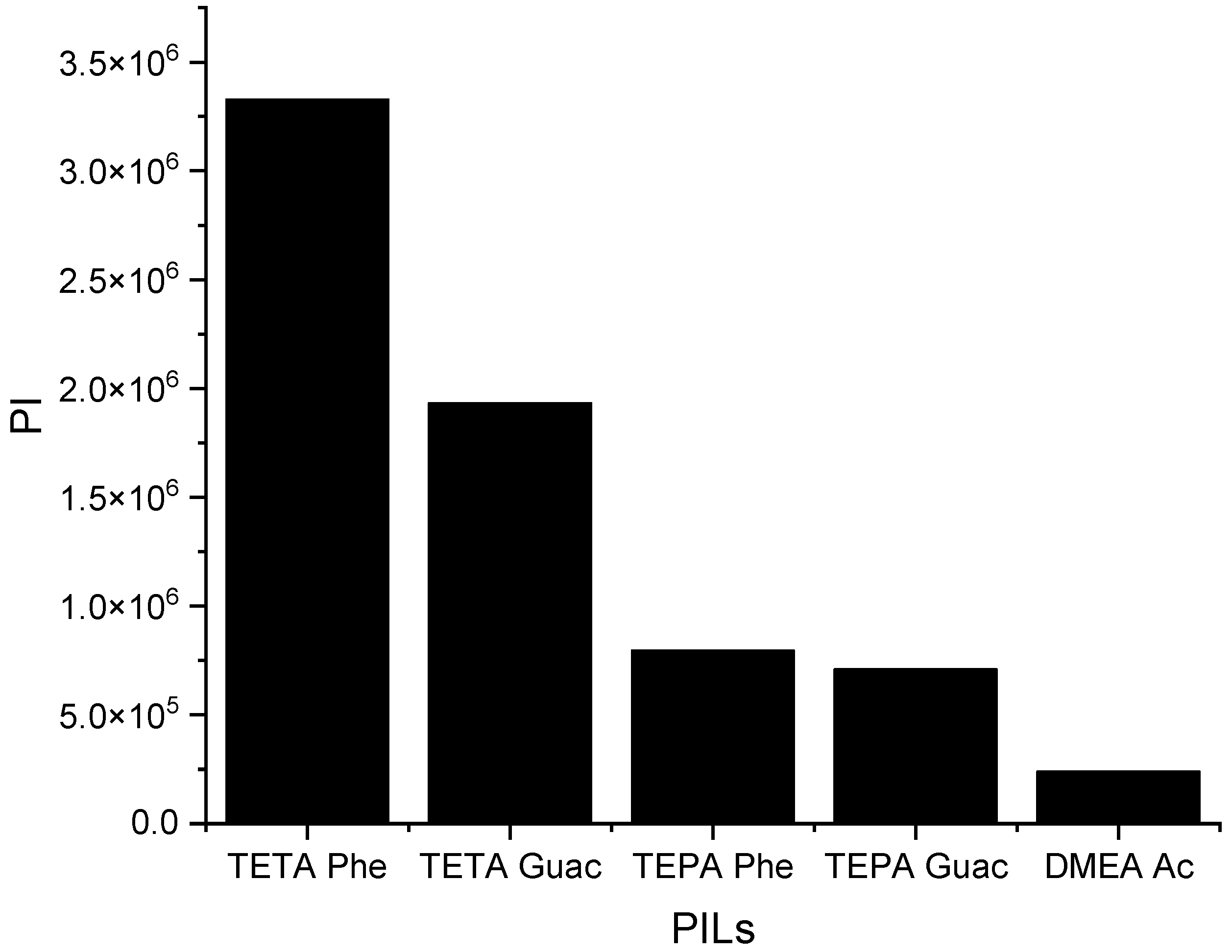
Figure 6 below illustrates the
C∞ of the five PILs.
It can be observed from
Figure 6 that when the
S∞ increases, the
C∞ decreases and vice versa. This trend is similar to that of the APILs. It shows that PILs that encompass an aromatic anion influence the capacity, since it increases the steric hindrance to accommodate pyrrole; this contrasts with DMEA that contains a normal acetate anion. In addition, the longer alkyl chain of cations like TEPA weakens the columbic force and leads to a loosening of the stacking structure, which is able to accommodate the pyrrole compound. Finally, the performance index (PI) analysis demonstrates that [TETA][Phe] is the most favorable PIL in terms of both selectivity and capacity, as depicted in
Figure 7. However, for industrial utilization, it is crucial to consider criteria such as cost-effectiveness and low toxicity. Guaiacol, a natural and nontoxic compound, serves as an exemplary candidate for the synthesis of anions in PILs. Hence, this screening confirms the capability of PILs as solvents for extracting
N-containing compounds.
2.1.5. Sigma Profile Analysis
This study delves into the influence of cation and anion structures on the extractability and interactions between ionic liquids (ILs) and solutes, employing the σ-profile approach. This approach provides insights into how the molecular structures of ILs affect their interactions with solutes. According to the COSMO-RS theory, the charge density of a molecule can be divided into three regions: the hydrogen bond donor region (HBD) (σ < −0.0084 e.Å
−2), the nonpolar region (−0.0084 e.Å
−2 < σ < 0.0084 e.Å
−2), and the hydrogen bond acceptor region (HBA) (σ > 0.0084 e.Å
−2). These regions serve as the fingerprint features of a molecule [
37].
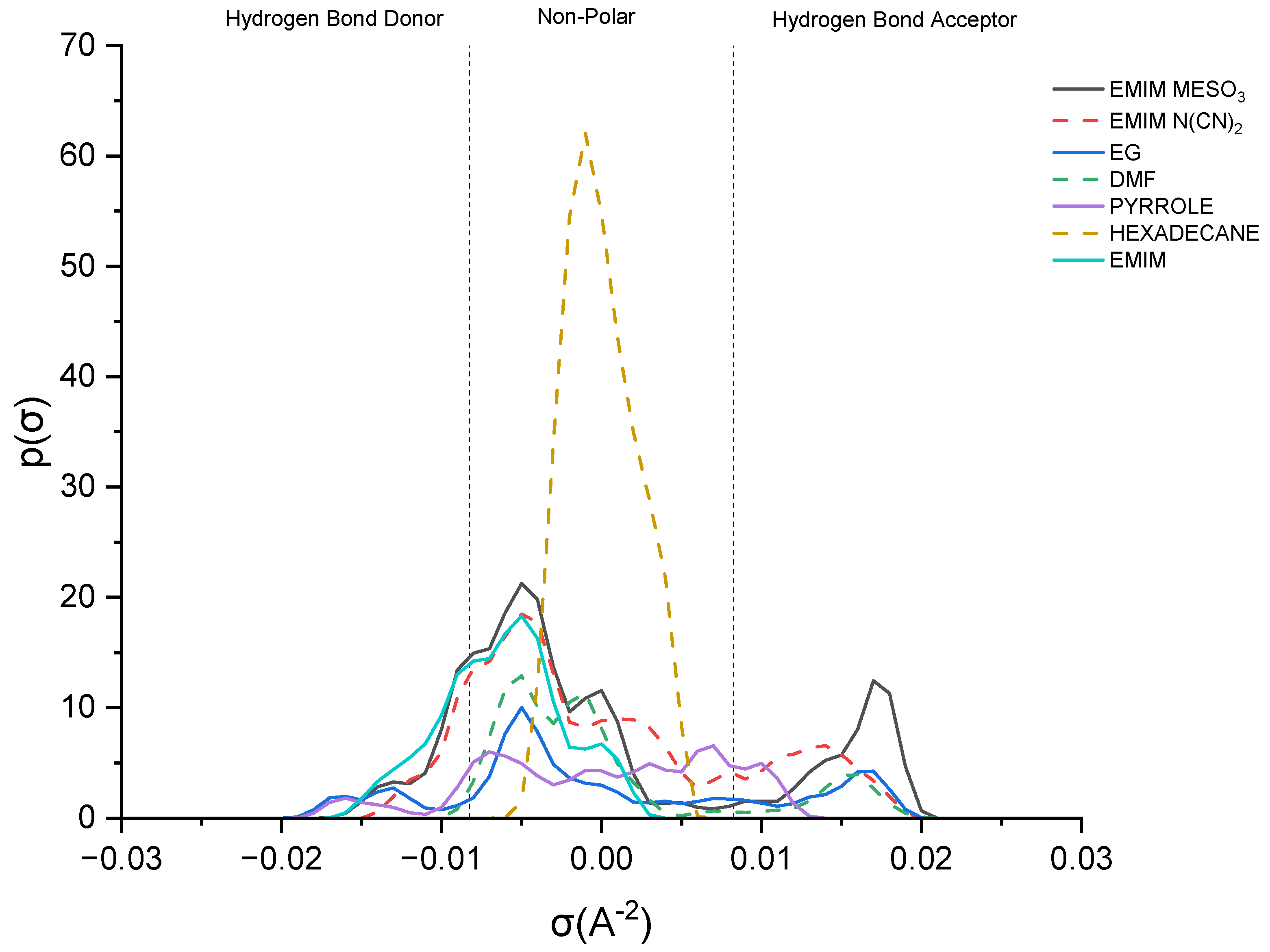
Figure 8 illustrates the sigma profile of both the industrial solvents and ILs.
As depicted in
Figure 8, pyrrole demonstrates a small distribution curve in both the hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA) regions. Therefore, an effective extracting solvent should possess both HBA and HBD characteristics in order to establish favorable interactions with pyrrole. [EMIM]
+ exhibits a peak in the negative direction, signifying the positive charge on the nitrogen atom [
38]. The extent of the interaction between the cations and pyrrole depends on the presence of accessible hydrogen bond donors or acceptors. Although the HBA region for [EMIM]
+ is non-existent, a weak hydrogen bonding interaction with pyrrole can still occur, due to a portion of the [EMIM]
+ profile being to the left of the cut-off zone.
The sigma profile of hexadecane falls within the non-polar region, since it consists solely of carbon and hydrogen atoms. Hydrocarbons, being composed of elements with similar electronegativities, exhibit a relatively uniform distribution of electron density, resulting in weak or non-existent interactions with pyrrole. Moving on to the ion pairs, [EMIM][N(CN)
2] and [EMIM][MeSO
3], both demonstrate a distribution curve in both the HBD and HBA regions. However, there is a slight dominance in the HBA region, primarily due to the inherent negative charge of the anions [
22].
[EMIM][MeSO3] exhibits a higher polarization, with a charge density ranging up to 0.021 σ (e/Å2), compared to that of [EMIM][N(CN)2], with 0.019 σ (e/Å2). Additionally, [EMIM][MeSO3] provides a larger area for interaction with pyrrole, resulting in a higher capacity. Despite pyrrole exhibiting weak polarization, it can still form weak hydrogen bonding interactions with both ion pairs.
Lastly, focusing on the industrial solvents, EG (ethylene glycol) demonstrates an almost symmetrical sigma profile in both the HBA and HBD regions, whereas DMF (dimethylformamide) exhibits an asymmetrical sigma profile. Nevertheless, both solvents complement pyrrole in forming mutual interactions.
2.1.6. Sigma Potential Analysis
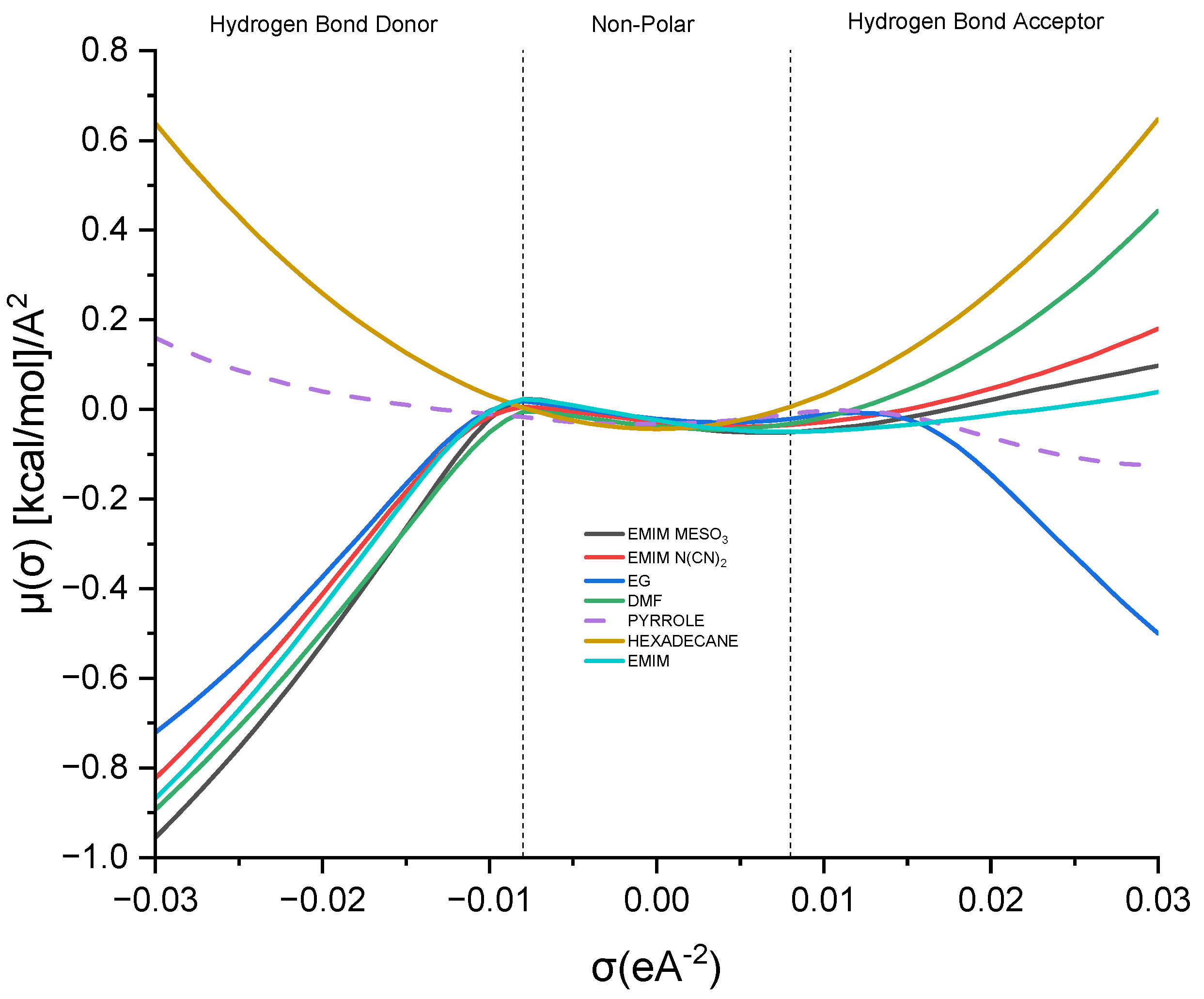
The sigma potential (σ) provides insights into the affinity between solvents in a mixture. A greater negative value of μ (σ) signifies a stronger attraction between molecules, whereas a higher positive value suggests repulsive behavior [
39]. Examining
Figure 9, the σ-potential curve for hexadecane resembles a parabola, while that for pyrrole demonstrates almost symmetrical characteristics, with a slight affinity towards the hydrogen bond acceptor region.
Analyzing the σ-potential of the [EMIM]+ cation reveals a similar pattern to that of the σ-profile, where [EMIM]+ exhibits a high affinity as a hydrogen bond donor. Moving on to the other ionic liquids, such as [EMIM][MeSO3] and [EMIM][N(CN2)], they predominantly occupy the hydrogen bond donor region. This differs slightly from their σ-profile analysis, where both solvents display a dominant presence in the hydrogen bond acceptor region. Nonetheless, these solvents possess high negative values, indicating a complementary interaction with the solute.
Considering the conventional industrial solvents, EG and DMF, it can be observed that EG exhibits almost symmetrical curves in both regions, with a slight dominance in the hydrogen bond donor area. On the other hand, DMF deviates slightly from the σ-profile analysis, tending towards the hydrogen bond donor region. This suggests that DMF has the potential to donate a hydrogen bond to the pyrrole nitrogen atom, facilitating a hydrogen bond interaction between the two molecules.
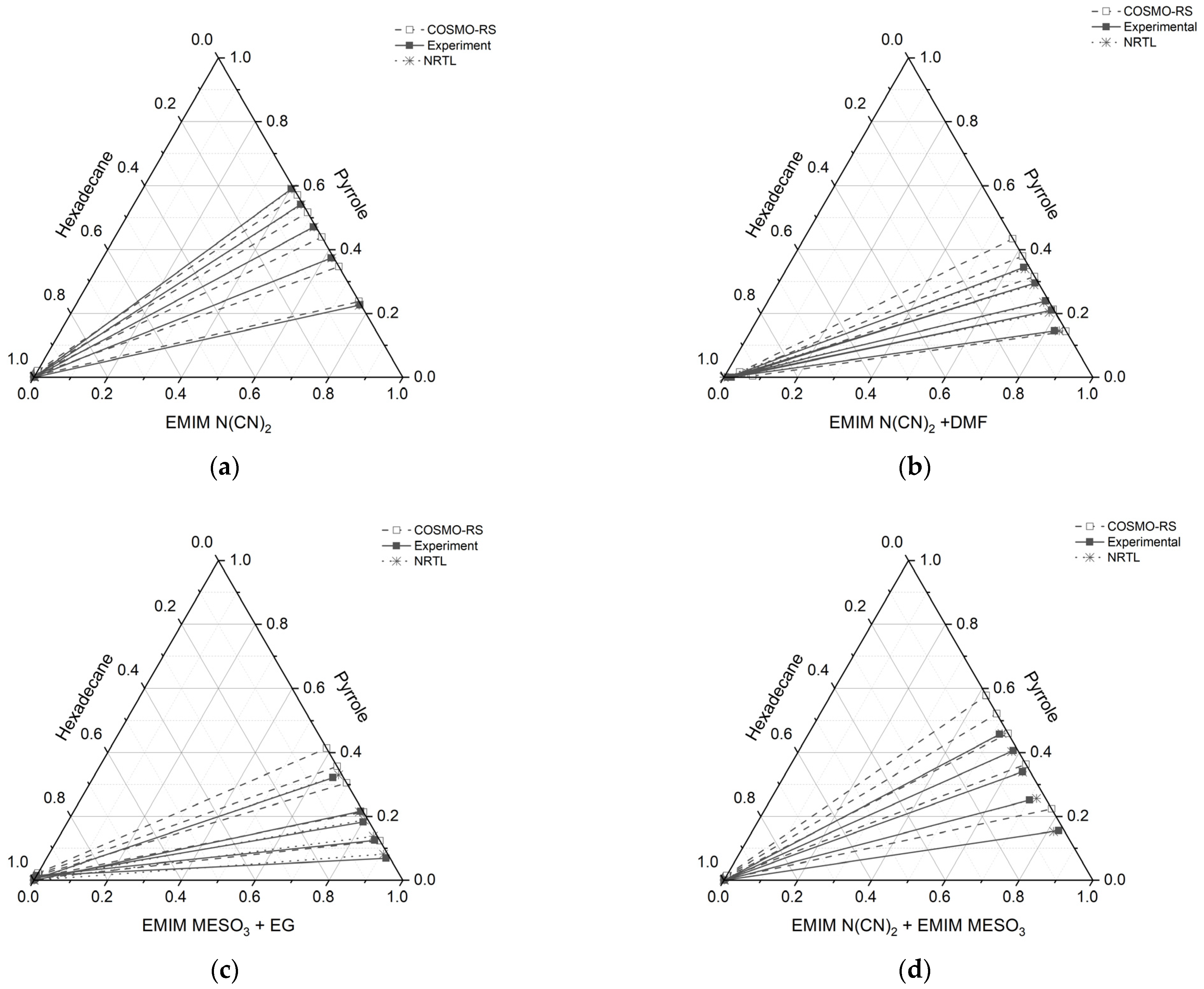
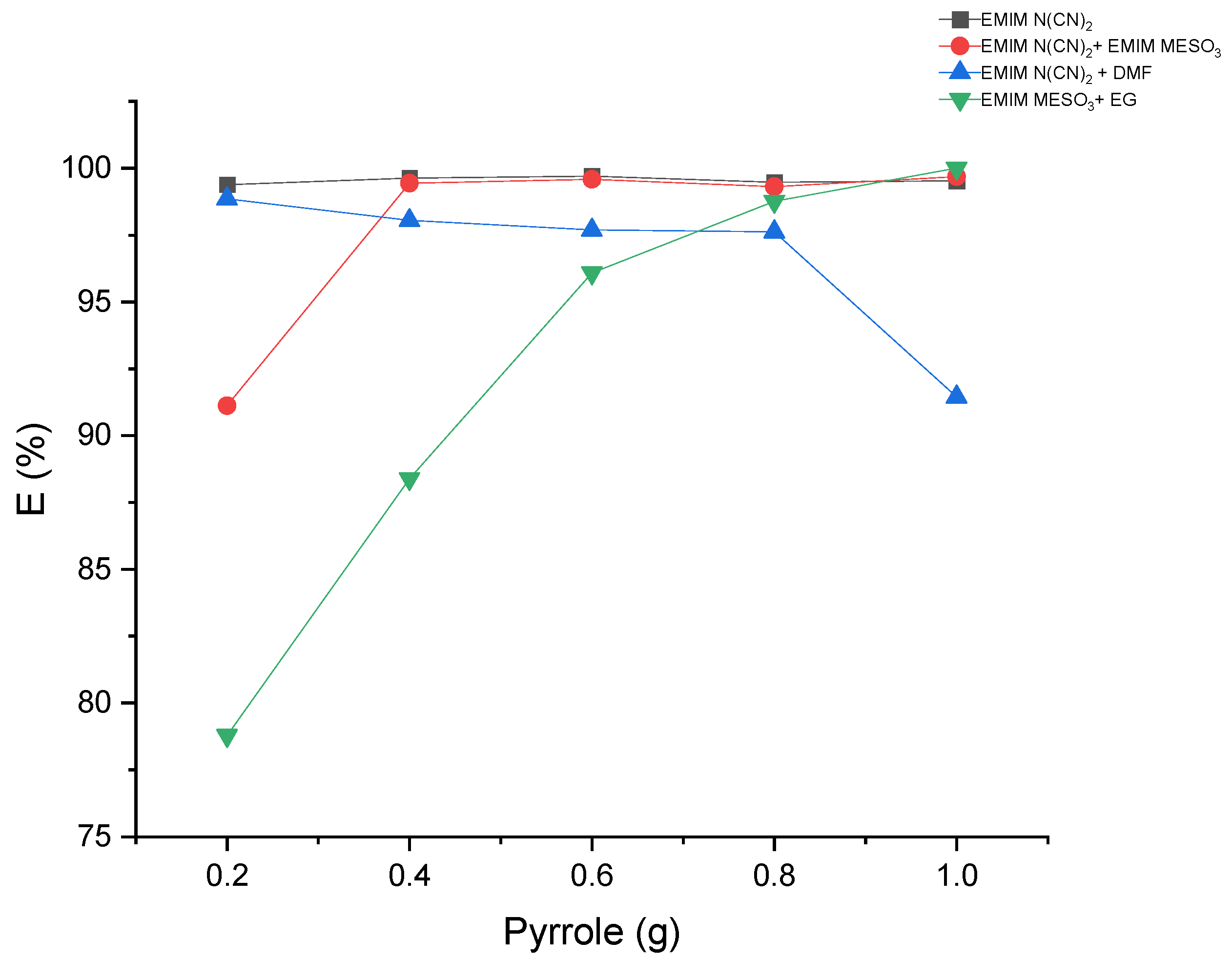
2.3. COSMO-RS Prediction vs. Experimental Data
The results obtained from
Figure 10 reveal a satisfactory agreement between the COSMO-RS predictions and the experimental data for the system [EMIM][N(CN)
2], particularly for the raffinate and extract phases, which are rich in hexadecane and IL solvent, respectively. However, for three systems ([EMIM][N(CN)
2] + DMF, [EMIM][MeSO
3] + EG, and [EMIM][N(CN)
2] + [EMIM][MeSO
3]), slight discrepancies were observed. In these systems, the experimental tie lines exhibited a small amount of hexadecane, which was not predicted by the COSMO-RS model. Conversely, in the raffinate phase of the [EMIM][N(CN)
2] + DMF system, the COSMO-RS predicted a small amount of pyrrole that was not observed experimentally. It should be noted that the quantities of hexadecane in all the systems were less than 0.01 mole, as shown in
Table 5.
In the case of the [EMIM][MeSO3] + EG system, at the lowest feed concentration of pyrrole, the experimental tie line displayed a lower slope and gradient compared to the COSMO-RS predictions. Additionally, this system exhibited a relatively high discrepancy between the COSMO-RS and experimental values. However, the positive slope observed in the tie lines indicates that [EMIM][MeSO3] + EG has an affinity for pyrrole.
The disparities observed between the COSMO-RS predictions and the experimental data imply the existence of additional factors or complexities that could potentially influence the extraction behavior within these systems. Further investigation and refinement of the model may be necessary to improve the accuracy of the predictions and to better capture the experimental observations.
2.4. NRTL Modeling
The LLE phase compositions were determined by solving an isothermal liquid–liquid flash at a specified temperature and pressure, involving the following system of equations:
In the given equations, ω represents the liquid–liquid splitting ratio, xi is the quantity of component i in the mixture, denotes the quantity of component i in the liquid phase L1; represents the quantity of component i in the liquid phase L2, and NC is the number of constituents in the liquid phases. The parameters and signify the activity coefficients of component i in L1 and L2, respectively.
In this study, the activity coefficients were computed using the non-random two-liquid (NRTL) model. For a system with multiple components, the activity coefficient of component
i is determined using the following general expression:
Here,
τij and
τji represent the binary interaction parameters, and α
ij corresponds to the non-randomness parameter. The level of non-randomness in the mixture is characterized by the parameter α
ij. In numerous prior studies involving ILs, a value of 0.2 for the non-randomness parameter has been proven to yield precise fittings for ternary LLE systems. The same value is retained in this study. The model was constructed in the Simulis
® environment [[
40] In this environment, the binary interaction parameters
τij and
τji are determined by minimizing the root-mean-square deviation (RMSD) between the calculated and experimental solubilities of each constituent in each phase, as described by the equation:
Here, x represents the concentration of a species in mole fraction. The subscripts i, j, and k denote the component, phase, and tie line, respectively. m is the number of tie lines, c is the number of components, and j is the number of phases, which is two in this case.
The respective RMSD values that correlate with each system are presented in
Table 6. The RMSD values for the NRTL correlation consistently fell below 1%, suggesting that the NRTL correlation effectively represents the experimental data. This is further evident in the ternary diagrams, where the tie lines calculated using the NRTL model closely coincide with the experimental tie lines, indicating a high level of agreement between the calculated and experimental data. The NRTL binary interaction parameters regressed for each ternary system are provided in
Table 7. To ensure coherence and consistency with our prior findings, the binary interaction parameters between the pyrrole and
n-hexadecane were adopted from our previous study [
27] and remained constant across all the ternary systems, regardless of the IL or IL mixture utilized.



 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}