


Interaction of Camptothecin Anticancer Drugs with Ribosomal Proteins L15 and L11: A Molecular Docking Study

Abstract
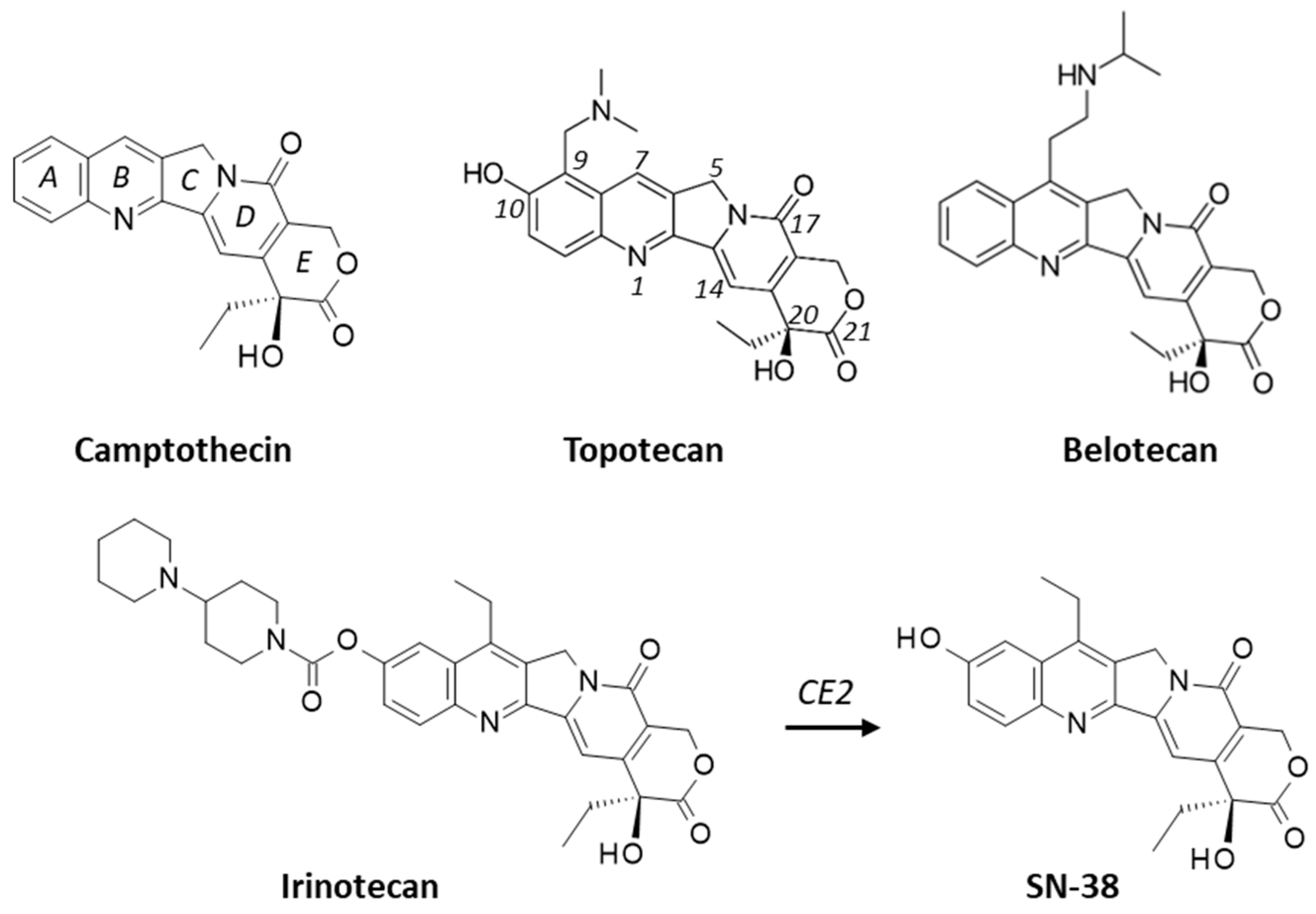
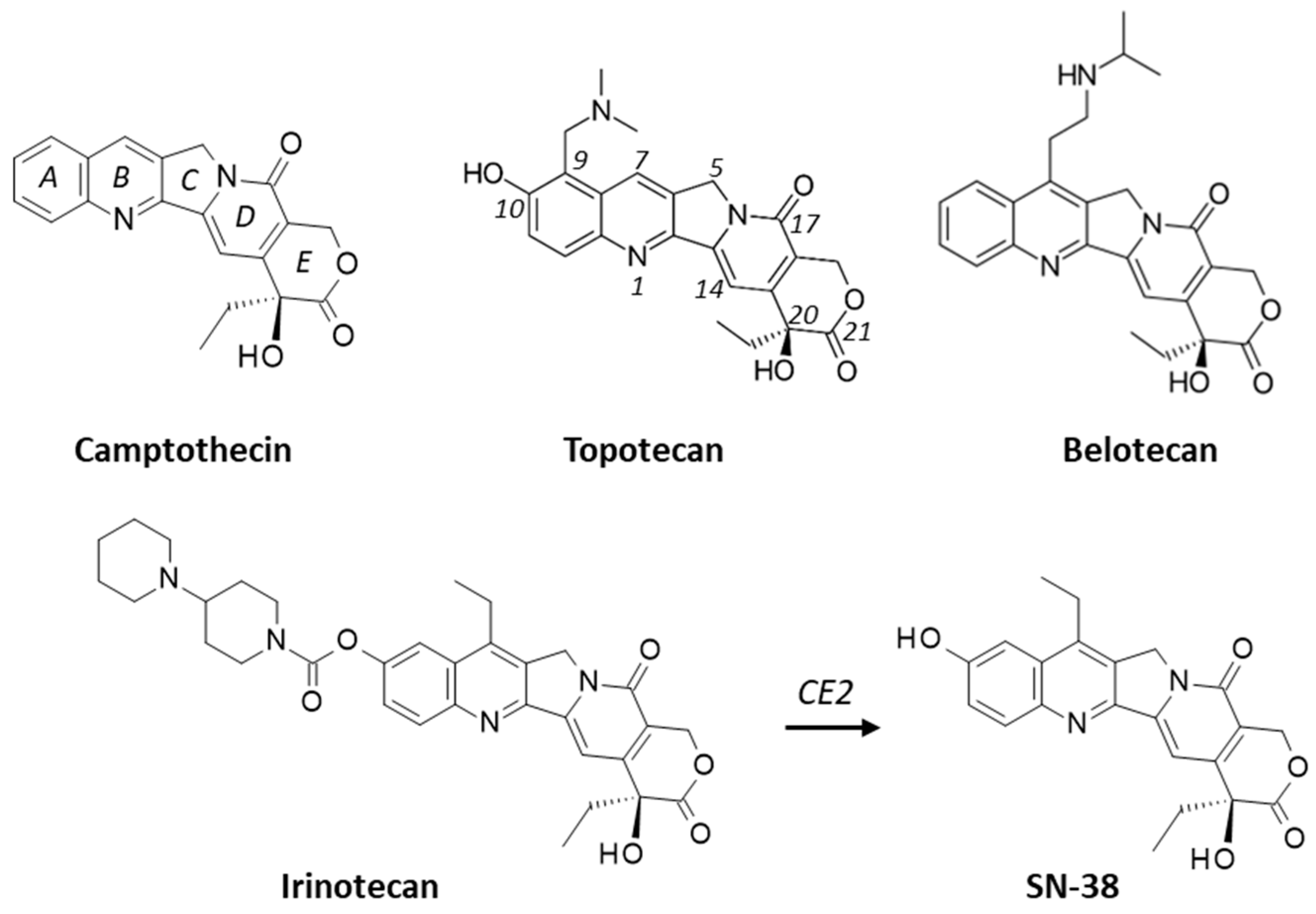
:1. Introduction
2. Results
2.1. Interaction of Camptothecins with RPL15
2.2. Drug Binding to RPL15 versus Topoisomerase I and ABCG2
2.3. Interaction of Camptothecins with RPL11
3. Discussion
4. Materials and Methods
4.1. Molecular Structures and Software
4.2. In Silico Molecular Docking Procedure
- (1)
- Monte Carlo (MC) conformational search of the ligand using the BOSS (Biochemical and Organic Simulation System) software v4.9 http://zarbi.chem.yale.edu/software.html (accessed on 20 January 2023), freely available to academic users. The structure of the ligand was optimized using a classical MC conformational search procedure, as described in BOSS [64]. A conformational analysis has been performed to define the best starting geometries for each compound. Energy minimization was carried out to identify all minimum-energy conformers, leading to the identification of a unique conformer for the free ligand. Within BOSS, MC simulations were performed in the constant-temperature and constant-pressure ensemble (NPT).
- (2)
- Evaluation of the free energy of hydration for the chosen structure of the ligand. The molecular mechanics/generalized Born surface area (MM/GBSA) procedure was used to evaluate the free energies of hydration (ΔG) [65]. MC search and computation of ΔG were performed within BOSS using the xMCGB script according to procedures given in references [65,66]. The best ligand structure was then used in the docking procedure.
- (3)
- Definition of the ribosomal protein–ligand sites of interaction. Drug-binding sites were searched using CASTp 3.0, a convenient tool for active site prediction. With the 4XXB (RPL11) structure, based on shape complementarity criteria, the flexible amino acids are Asn23, Cys25, Ser51, Arg54, Ile68, His71, Ser317, His318, Asn320 and Trp323. With the 4UG0 (RPL15) structure, the flexible amino acids are (i) Lys54, Lys56, Glu57, Tyr59, Ile135, Asp136, His139, Ile142, Thr148, and Trp150 (site Ile135) and (ii) Trp11, Leu23, Arg26, Gln29, Tyr30, Leu33, His37, Thr43, Arg63, Phe129 (site Phe129). Shape complementarity and geometry considerations favor a docking grid centered in the volume defined by the central amino acid. Within the binding site, the side chains of the specific amino acids were considered fully flexible during docking.
- (4)
- Docking procedure using GOLD. In our typical docking process, 100 energetically reasonable poses (according to the ChemPLP scoring function) are retained while searching for the correct binding mode of the ligand. The decision to maintain a trial pose is based on ranked poses, using the PLP fitness scoring function (which is the default in GOLD version 5.3 used here) [67]. Six poses are kept. The empirical potential energy of the interaction ΔE for the ranked complexes was evaluated using the simple expression ΔE(interaction) = E(complex) − [E(protein) + E(ligand)]. Calculations of the final energy are performed on the basis of the SPASIBA spectroscopic force field. The corresponding parameters are derived from vibrational wavenumbers obtained in the infrared and Raman spectra of a large series of compounds including organic molecules, amino acids, saccharides, nucleic acids and lipids.
- (5)
- Validation using the SPASIBA force field. This last step is considered essential to define the best protein–ligand structure. The spectroscopic SPASIBA (Spectroscopic Potential Algorithm for Simulating Biomolecular conformational Adaptability) force field has been specifically developed to provide refined empirical molecular mechanics force field parameters [68]. SPASIBA empirical energies of interaction are calculated as described [69,70]. SPASIBA (integrated into CHARMM) [71] has been shown to be excellent at reproducing crystal-phase infrared data. The same procedure was used to establish molecular models for the various drug–protein complexes.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined cytotoxic chemotherapy and immunotherapy of cancer: Modern times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Tariq, S.; Kim, S.Y.; Monteiro de Oliveira Novaes, J.; Cheng, H. Update 2021: Management of Small Cell Lung Cancer. Lung 2021, 199, 579–587. [Google Scholar] [CrossRef]
- Petrelli, F.; Ghidini, A.; Luciani, A. Topotecan or other agents as second-line therapy for relapsed small-cell lung cancer: A meta-analysis of randomized studies. Mol. Clin. Oncol. 2021, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Liposomal Irinotecan: A Review in Metastatic Pancreatic Adenocarcinoma. Drugs 2020, 80, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Milano, G.; Innocenti, F.; Minami, H. Liposomal irinotecan (Onivyde): Exemplifying the benefits of nanotherapeutic drugs. Cancer Sci. 2022, 113, 2224–2231. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. ASCENT Clinical Trial Investigators. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Fontes, M.S.; Vargas Pivato de Almeida, D.; Cavalin, C.; Tagawa, S.T. Targeted Therapy for Locally Advanced or Metastatic Urothelial Cancer (mUC): Therapeutic Potential of Sacituzumab Govitecan. OncoTargets Ther. 2022, 15, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B., Jr.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar] [CrossRef] [Green Version]
- Talukdar, A.; Kundu, B.; Sarkar, D.; Goon, S.; Mondal, M.A. Topoisomerase I inhibitors: Challenges, progress and the road ahead. Eur. J. Med. Chem. 2022, 236, 114304. [Google Scholar] [CrossRef] [PubMed]
- Martín-Encinas, E.; Selas, A.; Palacios, F.; Alonso, C. The design and discovery of topoisomerase I inhibitors as anticancer therapies. Expert. Opin. Drug Discov. 2022, 17, 581–601. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Ni, D.; Jackson, S.M.; Manolaridis, I.; Stahlberg, H.; Locher, K.P. Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, A.; Yu, Q.; Dehghani-Ghahnaviyeh, S.; Wen, P.C.; Kowal, J.; Locher, K.P.; Tajkhorshid, E. Differential dynamics and direct interaction of bound ligands with lipids in multidrug transporter ABCG2. Proc. Natl. Acad. Sci. USA 2023, 120, e2213437120. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Baig, G.A.; Alshawli, A.S.; Sait, K.H.W.; Hafeez, B.B.; Tripathi, M.K.; Alghamdi, B.S.; Mohammed Ali, H.S.H.; Rasool, M. Interaction Analysis of MRP1 with Anticancer Drugs Used in Ovarian Cancer: In Silico Approach. Life 2022, 12, 383. [Google Scholar] [CrossRef]
- Wong, D.V.T.; Holanda, R.B.F.; Cajadov, A.G.; Bandeira, A.M.; Pereira, J.F.B.; Amorim, J.O.; Torres, C.S.; Ferreira, L.M.M.; Lopes, M.H.S.; Oliveira, R.T.G.; et al. TLR4 deficiency upregulates TLR9 expression and enhances irinotecan-related intestinal mucositis and late-onset diarrhoea. Br. J. Pharmacol. 2021, 178, 4193–4209. [Google Scholar] [CrossRef]
- Wong, D.V.T.; Ribeiro-Filho, H.V.; Wanderley, C.W.S.; Leite, C.A.V.G.; Lima, J.B.; Assef, A.N.B.; Cajado, A.G.; Batista, G.L.P.; González, R.H.; Silva, K.O.; et al. SN-38, the active metabolite of irinotecan, inhibits the acute inflammatory response by targeting toll-like receptor 4. Cancer Chemother. Pharmacol. 2019, 84, 287–298. [Google Scholar] [CrossRef]
- Tam, J.S.Y.; Pei, J.V.; Coller, J.K.; Prestidge, C.A.; Bowen, J.M. Structural insight and analysis of TLR4 interactions with IAXO-102, TAK-242 and SN-38: An in silico approach. In Silico Pharmacol. 2022, 11, 1. [Google Scholar] [CrossRef]
- Gasimli, K.; Raab, M.; Becker, S.; Sanhaji, M.; Strebhardt, K. The Role of DAPK1 in the Cell Cycle Regulation of Cervical Cancer Cells and in Response to Topotecan. J. Cancer 2022, 13, 728–743. [Google Scholar] [CrossRef]
- Khageh Hosseini, S.; Kolterer, S.; Steiner, M.; von Manstein, V.; Gerlach, K.; Trojan, J.; Waidmann, O.; Zeuzem, S.; Schulze, J.O.; Hahn, S.; et al. Camptothecin and its analog SN-38, the active metabolite of irinotecan, inhibit binding of the transcriptional regulator and oncoprotein FUBP1 to its DNA target sequence FUSE. Biochem. Pharmacol. 2017, 146, 53–62. [Google Scholar] [CrossRef]
- Hoang, V.T.; Verma, D.; Godavarthy, P.S.; Llavona, P.; Steiner, M.; Gerlach, K.; Michels, B.E.; Bohnenberger, H.; Wachter, A.; Oellerich, T.; et al. The transcriptional regulator FUBP1 influences disease outcome in murine and human myeloid leukemia. Leukemia 2019, 33, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Min, J.A.; Nashed, A.; Lee, S.O.; Yoo, J.C.; Chi, S.W.; Yi, G.S. A novel mechanism of irinotecan targeting MDM2 and Bcl-xL. Biochem. Biophys. Res. Commun. 2019, 514, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Sharma, P.L.; Li, C.J.; Dezube, B.J.; Pardee, A.B.; Crumpacker, C.S. Topotecan inhibits human immunodeficiency virus type 1 infection through a topoisomerase-independent mechanism in a cell line with altered topoisomerase I. Antimicrob. Agents Chemother. 1997, 41, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Kitai, Y.; Tadokoro, T.; Takahashi, R.; Shoji, H.; Maemoto, T.; Ishiura, M.; Muromoto, R.; Kashiwakura, J.I.; Ishii, K.J.; et al. Identification of RPL15 60S Ribosomal Protein as a Novel Topotecan Target Protein That Correlates with DAMP Secretion and Antitumor Immune Activation. J. Immunol. 2022, 209, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Kitai, Y.; Kawasaki, T.; Sueyoshi, T.; Kobiyama, K.; Ishii, K.J.; Zou, J.; Akira, S.; Matsuda, T.; Kawai, T. DNA-Containing Exosomes Derived from Cancer Cells Treated with Topotecan Activate a STING-Dependent Pathway and Reinforce Antitumor Immunity. J. Immunol. 2017, 198, 1649–1659. [Google Scholar] [CrossRef] [Green Version]
- Kitai, Y. Elucidation of the Mechanism of Topotecan-induced Antitumor Immune Activation. Yakugaku Zasshi 2022, 142, 911–916. [Google Scholar] [CrossRef]
- Ishihara, Y.; Nakamura, K.; Nakagawa, S.; Okamoto, Y.; Yamamoto, M.; Furukawa, T.; Kawahara, K. Nucleolar Stress Response via Ribosomal Protein L11 Regulates Topoisomerase Inhibitor Sensitivity of P53-Intact Cancers. Int. J. Mol. Sci. 2022, 23, 15986. [Google Scholar] [CrossRef]
- Khatter, H.; Myasnikov, A.G.; Natchiar, S.K.; Klaholz, B.P. Structure of the human 80S ribosome. Nature 2015, 520, 640–645. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, M.L. Solvent-accessible surfaces of proteins and nucleic acids. Science 1983, 221, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Lang, Y.; Zhang, Q.; Cui, D.; Sun, H.; Jiang, L.; Chen, Z.; Zhang, R.; Gao, Y.; Tian, W.; et al. Structure of human MDM2 complexed with RPL11 reveals the molecular basis of p53 activation. Genes Dev. 2015, 29, 1524–1534. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.S.; Kumar, A.; Warner, J.R. Ribosome formation is blocked by camptothecin, a reversible inhibitor of RNA synthesis. Proc. Natl. Acad. Sci. USA 1971, 68, 3009–3014. [Google Scholar] [CrossRef] [Green Version]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [CrossRef]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Tong, Q.; Liu, G.; Sang, X.; Zhu, X.; Fu, X.; Dou, C.; Jian, Y.; Zhang, J.; Zou, S.; Zhang, G.; et al. Targeting RNA G-quadruplex with repurposed drugs blocks SARS-CoV-2 entry. PLoS Pathog. 2023, 19, e1011131. [Google Scholar] [CrossRef]
- Lieberman, K.R.; Noller, H.F. Ribosomal protein L15 as a probe of 50 S ribosomal subunit structure. J. Mol. Biol. 1998, 284, 1367–1378. [Google Scholar] [CrossRef]
- Dong, Z.; Jiang, H.; Liang, S.; Wang, Y.; Jiang, W.; Zhu, C. Ribosomal Protein L15 is involved in Colon Carcinogenesis. Int. J. Med. Sci. 2019, 16, 1132–1141. [Google Scholar] [CrossRef]
- Yan, T.T.; Fu, X.L.; Li, J.; Bian, Y.N.; Liu, D.J.; Hua, R.; Ren, L.L.; Li, C.T.; Sun, Y.W.; Chen, H.Y.; et al. Downregulation of RPL15 may predict poor survival and associate with tumor progression in pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 37028–37042. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Liu, Z. RPL15 promotes hepatocellular carcinoma progression via regulation of RPs-MDM2-p53 signaling pathway. Cancer Cell Int. 2022, 22, 150. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, L.N.; Li, K.Z.; Ling, R.; Li, X.J.; Wang, L. Overexpression of ribosomal protein L15 is associated with cell proliferation in gastric cancer. BMC Cancer 2006, 6, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.A.; Lin, H.J.; Sheu, J.J.; Shieh, F.K.; Chen, S.Y.; Lai, C.H.; Tsai, F.J.; Wan, L.; Chen, B.H. A novel interaction between interferon-inducible protein p56 and ribosomal protein L15 in gastric cancer cells. DNA Cell Biol. 2011, 30, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Li, X.; Nian, W.; Wang, J.; Wang, X.; Sun, L.; Zhu, Y.; Tong, Z. Ribosome Proteins Represented by RPL27A Mark the Development and Metastasis of Triple-Negative Breast Cancer in Mouse and Human. Front. Cell Dev. Biol. 2021, 9, 716730. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Shi, Y.; Xu, L.; Zeng, Y.; Cui, X.; Wang, Y.; Yang, N.; Zhou, F.; Zhou, Y. Prognostic Value and Related Regulatory Networks of MRPL15 in Non-Small-Cell Lung Cancer. Front. Oncol. 2021, 11, 656172. [Google Scholar] [CrossRef]
- Xu, H.; Zou, R.; Li, F.; Liu, J.; Luan, N.; Wang, S.; Zhu, L. MRPL15 is a novel prognostic biomarker and therapeutic target for epithelial ovarian cancer. Cancer Med. 2021, 10, 3655–3673. [Google Scholar] [CrossRef]
- He, S.J.; Shu, L.P.; Zhou, Z.W.; Yang, T.; Duan, W.; Zhang, X.; He, Z.X.; Zhou, S.F. Inhibition of Aurora kinases induces apoptosis and autophagy via AURKB/p70S6K/RPL15 axis in human leukemia cells. Cancer Lett. 2016, 382, 215–230. [Google Scholar] [CrossRef]
- Wang, H.; Feng, J.; Zhou, T.; Wei, L.; Zhou, J. Involvement of RPL11 in the enhancement of P53 stability by a podophyllum derivative, a topoisomerase II inhibitor. Cell Biol. Int. 2018, 42, 121–129. [Google Scholar] [CrossRef]
- Rao, Z.; Shen, J.; Wang, J.; Zhang, Z.; Zhou, J.; Zhu, J.; Chen, J.; Chen, W.; Wang, H. The role of PICT1 in RPL11/Mdm2/p53 pathway-regulated inhibition of cell growth induced by topoisomerase IIα inhibitor against cervical cancer cell line. Biochem. Pharmacol. 2022, 201, 115098. [Google Scholar] [CrossRef]
- Franklin, D.A.; Liu, S.; Jin, A.; Cui, P.; Guo, Z.; Arend, K.C.; Moorman, N.J.; He, S.; Wang, G.G.; Wan, Y.Y.; et al. Ribosomal protein RPL11 haploinsufficiency causes anemia in mice via activation of the RP-MDM2-p53 pathway. J. Biol. Chem. 2022, 299, 102739. [Google Scholar] [CrossRef]
- Bailly, A.; Perrin, A.; Bou Malhab, L.J.; Pion, E.; Larance, M.; Nagala, M.; Smith, P.; O’Donohue, M.F.; Gleizes, P.E.; Zomerdijk, J.; et al. The NEDD8 inhibitor MLN4924 increases the size of the nucleolus and activates p53 through the ribosomal-Mdm2 pathway. Oncogene 2016, 35, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Goudarzi, K.M.; Nistér, M.; Lindström, M.S. mTOR inhibitors blunt the p53 response to nucleolar stress by regulating RPL11 and MDM2 levels. Cancer Biol. Ther. 2014, 15, 1499–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Tong, J.; Wang, M.; Gan, Y.; Gao, B.; Chen, J.; Liu, Y.; Hao, Q.; Zhou, X. Olaparib Induces RPL5/RPL11-Dependent p53 Activation via Nucleolar Stress. Front. Oncol. 2022, 12, 821366. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Palacin, L.; Llanos, S.; Urbano-Cuadrado, M.; Blanco-Aparicio, C.; Megias, D.; Pastor, J.; Serrano, M. Non-genotoxic activation of p53 through the RPL11-dependent ribosomal stress pathway. Carcinogenesis 2014, 35, 2822–2830. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Gao, J.; Zhao, Z.; Zhong, X.; Cui, H.; Hou, H.; Zhang, Y.; Zheng, J.; Di, J.; Liu, Y. Identification of a small-molecule RPL11 mimetic that inhibits tumor growth by targeting MDM2-p53 pathway. Mol. Med. 2022, 28, 109. [Google Scholar] [CrossRef]
- Bursać, S.; Prodan, Y.; Pullen, N.; Bartek, J.; Volarević, S. Dysregulated Ribosome Biogenesis Reveals Therapeutic Liabilities in Cancer. Trends Cancer 2021, 7, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Vergoten, G. Binding of Vialinin A and p-Terphenyl Derivatives to Ubiquitin-Specific Protease 4 (USP4): A Molecular Docking Study. Molecules 2022, 27, 5909. [Google Scholar] [CrossRef] [PubMed]
- Vergoten, G.; Bailly, C. Molecular docking study of britannin binding to PD-L1 and related anticancer pseudoguaianolide sesquiterpene lactones. J. Recept. Signal Transduct. Res. 2022, 42, 454–461. [Google Scholar] [CrossRef]
- Vergoten, G.; Bailly, C. Molecular docking study of GSK-3β interaction with nomilin, kihadanin B, and related limonoids and triterpenes with a furyl-δ-lactone core. J. Biochem. Mol. Toxicol. 2022, 36, e23130. [Google Scholar] [CrossRef]
- Sharma, P.P.; Bansal, M.; Sethi, A.; Poonam Pena, L.; Goel, V.K.; Grishina, M.; Chaturvedi, S.; Kumar, D.; Rathi, B. Computational methods directed towards drug repurposing for COVID-19: Advantages and limitations. RSC Adv. 2021, 11, 36181–36198. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, Y.; Higo, J.; Kasahara, K. Computer simulation of molecular recognition in biomolecular system: From in silico screening to generalized ensembles. Biophys. Rev. 2022, 14, 1423–1447. [Google Scholar] [CrossRef]
- Gentile, F.; Oprea, T.I.; Tropsha, A.; Cherkasov, A. Surely you are joking, Mr Docking! Chem. Soc. Rev. 2023, 52, 872–878. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Monte Carlo versus Molecular Dynamics for conformational sampling. J. Phys. Chem. 1996, 100, 14508–14513. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem. 2005, 26, 1689–1700. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Ulmschneider, J.P.; Tirado-Rives, J. Free energies of hydration from a generalized Born model and an ALL-atom force field. J. Phys. Chem. B 2004, 108, 16264–16270. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Meziane-Tani, M.; Lagant, P.; Semmoud, A.; Vergoten, G. The SPASIBA force field for chondroitin sulfate: Vibrational analysis of D-glucuronic and N-acetyl-D-galactosamine 4-sulfate sodium salts. J. Phys. Chem. A 2006, 110, 11359–11370. [Google Scholar] [CrossRef]
- Vergoten, G.; Mazur, I.; Lagant, P.; Michalski, J.C.; Zanetta, J.P. The SPASIBA force field as an essential tool for studying the structure and dynamics of saccharides. Biochimie 2003, 85, 65–73. [Google Scholar] [CrossRef]
- Lagant, P.; Nolde, D.; Stote, R.; Vergoten, G.; Karplus, M. Increasing Normal Modes Analysis Accuracy: The SPASIBA Spectroscopic Force Field Introduced into the CHARMM Program. J. Phys. Chem. A 2004, 108, 4019–4029. [Google Scholar] [CrossRef]
- Homans, S.W. A molecular mechanical force field for the conformational analysis of oligosaccharides: Comparison of theoretical and crystal structures of Man alpha 1-3Man beta 1-4GlcNAc. Biochemistry 1990, 29, 9110–9118. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ΔE (kcal/mol) | ΔG (kcal/mol) | ΔE (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|---|---|
| Site | Site Phe129 | Site Ile135 | ||
| Belotecan | −63.30 | −15.70 | −80.40 | −11.90 |
| Camptothecin | −51.90 | −15.50 | −65.10 | −19.20 |
| SN38 | −83.70 | −16.00 | −79.30 | −14.50 |
| Topotecan | −65.90 | −16.70 | −66.30 | −11.30 |
| Compounds | Target | PDB | ΔE (kcal/mol) |
|---|---|---|---|
| SN38 | RPL15 | 4UGO | −83.7 * |
| Camptothecin | TopoI-DNA complex | 1TI8 | −114.20 |
| Topotecan | TopoI-DNA complex | 1K4T | −80.10 |
| Topotecan | ABCG2 transporter | 7NEZ | −67.55 |
| Compounds | ΔE (kcal/mol) | ΔG (kcal/mol) |
|---|---|---|
| SN38 | −112.70 | −20.30 |
| Belotecan | −93.50 | −30.40 |
| Topotecan | −80.80 | −21.20 |
| Camptothecin | −67.55 | −23.20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailly, C.; Vergoten, G. Interaction of Camptothecin Anticancer Drugs with Ribosomal Proteins L15 and L11: A Molecular Docking Study. Molecules 2023, 28, 1828. https://doi.org/10.3390/molecules28041828
Bailly C, Vergoten G. Interaction of Camptothecin Anticancer Drugs with Ribosomal Proteins L15 and L11: A Molecular Docking Study. Molecules. 2023; 28(4):1828. https://doi.org/10.3390/molecules28041828
Chicago/Turabian StyleBailly, Christian, and Gérard Vergoten. 2023. "Interaction of Camptothecin Anticancer Drugs with Ribosomal Proteins L15 and L11: A Molecular Docking Study" Molecules 28, no. 4: 1828. https://doi.org/10.3390/molecules28041828