Abstract
Modifying non-precious metal porphyrins at the meso-position is sufficient to further improve the ability to activate O2 and the selectivity of the corresponding redox products. In this study, a crown ether-appended Fe(III) porphyrin complex (FeTC4PCl) was formed by replacing Fe(III) porphyrin (FeTPPCl) at the meso-position. The reactions of FeTPPCl and FeTC4PCl catalysed by O2 oxidation of cyclohexene under different conditions were studied, and three main products, 2-cyclohexen-1-ol (1), 2-cyclohexen-1-one (2), and 7-oxabicyclo[4.1.0]heptane (3), were obtained. The effects of reaction temperature, reaction time, and the addition of axial coordination compounds on the reactions were investigated. The conversion of cyclohexene reached 94% at 70 °C after 12 h, and the selectivity toward product 1 was 73%. The geometrical structure optimization, molecular orbital energy level analysis, atomic charge, spin density, and density of orbital states analysis of FeTPPCl, FeTC4PCl, as well as the oxygenated complexes (Fe-O2)TCPPCl and (Fe-O2)TC4PCl formed after adsorption of O2, were carried out using the DFT method. The results of thermodynamic quantity variation with reaction temperature and Gibbs free energy variation were also analysed. Finally, based on experimental and theoretical analysis, the mechanism of the cyclohexene oxidation reaction with FeTC4PCl as a catalyst and O2 as an oxidant was deduced, and the reaction mechanism was obtained as a free radical chain reaction process.
1. Introduction
Metalloporphyrin complexes exhibit exceptional catalytic properties for various chemical transformations, including O–O bond heterolysis [1,2,3,4], H–H bond formation [5,6], O2 adsorption [4,7,8], and catalytic oxidation [9,10,11,12] due to high activity, stability, clear molecular structure, and the ability to mimic the cytochrome enzyme [13,14]. Previous studies have shown that changing the meso-position substituents of metalloporphyrins to modulate their redox properties can alter metal ions’ activities, stability, and selectivity in catalytic processes [15,16,17,18,19,20,21]. Strong electron-absorbing meso-position substituents can make the metalloporphyrins easy to reduce, while electron-donating substituents can increase the electron-cloud density on the metal ion, enhancing its binding and electron transfer ability with O2 [22,23]. Therefore, fine-tuning metal ions’ electronic structure by changing the porphyrin ring’s meso-position substituents is vital for further improving their O2-carrying ability [7,20,24]. Additionally, the planar structure of metalloporphyrins provides a good coordination environment for modifying axial ligands [2,17,18,25,26,27]. The unoccupied d orbitals of the active metal centre can receive electrons provided by the ligand, increasing the electron density of the metal ion active centre or forming an electron-deficient structure that acts as a Lewis acid centre at the axial vacant coordination site, which is conducive to O2 adsorption.
In this study, the pre-synthesized meso-5, 10, 15, and 20-tetra-(15-crown-5-benzo-3-yl-sulfonyloxophen-4-yl) Fe(III) porphyrin complex (FeTC4PCl) was used as the catalyst [28] (Figure 1a). The catalytic oxidation of cyclohexene was carried out by using O2 as an oxidant under mild conditions without any auxiliary to verify the effect of the substitution of the meso-position with the 15-crown-5-benzo-3-sulfonyl group on the catalytic ability of the Fe(III) porphyrin, and in comparison with that of Fe(III) porphyrin complex (FeTPPCl, Figure 1b) without this group, the results demonstrate that the FeTC4PCl has higher product selectivity and substrate conversion as well as exhibits the ability to shorten the reaction induction period. This change indicates that the meso-substituent can effectively influence the catalytic activity of Fe(III). In contrast, the oxidation selectivity for the product 2-cyclohexene-1-ol was significantly higher than that of the FeTPPCl complex. The changes in catalytic oxidation at different temperatures and reaction times as well as the addition of axial ligand (pyridine) were also examined.
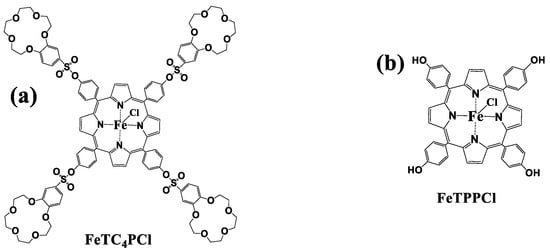
Figure 1.
Structure of the FeTC4PCl (a) and FeTPPCl (b) complexes.
To further analyse the effect of crown ether ring substituents in FeTC4PCl on the properties of Fe(III) porphyrins, the geometric configurations and corresponding orbital energy level distributions of the FeTC4PCl and FeTPPCl complexes were calculated separately using the density functional theory (DFT), and the changes of the thermodynamic quantities of FeTC4PCl at different temperatures were analysed separately. On this basis, the stable geometries of the oxygenated complexes formed after the adsorption of O2 by the FeTC4PCl and FeTPPCl complexes were also constructed. The geometric configurations and corresponding properties such as chemical bond lengths and density-of-states were calculated. Finally, the reaction mechanism of the catalytic oxidation of cyclohexene by FeTC4PCl in the presence of O2 was deduced by combining the above analysis results, and the reaction process was obtained as a free radical chain reaction-based reaction process.
2. Experimental Section
2.1. Materials and Catalytic Property Studies
All chemicals were obtained commercially and were further purified. High-purity O2 gas was purchased from a reagent supplier and purified before use. Catalysts FeTPPCl and FeTC4PCl were prepared according to the previous synthesis method of our group [28] and the corresponding structures of the complexes are shown in Figure 1.
The catalytic activity of FeTC4PCl and FeTPPCl complexes was evaluated for the oxidation of cyclohexene with O2 following the literature procedure [28,29]. FeTC4PCl (2.0 mg, 0.00096 mmol) and cyclohexene (2.0 mL, 19.76 mmol) were added to a round bottom flask. The flask was evacuated and charged with anhydrous oxygen. The reaction mixture was heated in an oil bath under stirring, and the corresponding experimental reaction apparatus is shown in Scheme S1. The consumption of oxygen was measured, and the corresponding products were analysed by gas chromatography using an Agilent 19091S-433 HP-5MS phenyl methyl siloxane column with a 30.0 m × 0.25 μm column at a temperature range of 70–230 °C (10 °C/min), Inj. 230 °C (Dec. 230 °C). All products were analysed by mass spectrometry as well as compared to the standard masses of organic compounds and their fragmentation patterns. The main oxidation products were 2-cyclohexen-1-ol (1), 2-cyclohexen-1-one (2), and 7-oxabicyclo[4.1.0]heptane (3) (Scheme 1).
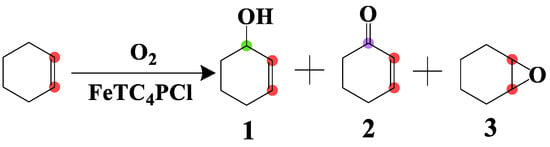
Scheme 1.
Oxidation of cyclohexene in the presence of O2.
2.2. Computational Details
In this paper, all the geometry optimizations and single-point calculations were performed using the Gaussian 09 package [30]. The optimization for FeTC4PCl was carried out using three hybrid functionals, namely PBE0 [31], ωB97XD [32], and B3LYP [33], in conjunction with the def2-SVP basis set [34] for geometry and the def2-TZVPP basis set for single-point calculations in the gas phase. The DFT-D dispersion correction proposed by Grimme [35] was added to the calculation process to remedy the deficiencies in the description of the electron-correlation interaction by the two general functionals, PBE0 and B3LYP. All initial geometries were in the sextet spin state, and the optimized structures discussed were characterized as local minima without imaginary frequencies. In order to select the most appropriate functional, the IR spectra of FeTC4PCl were calculated under the def2-SVP basis set to facilitate comparison with the experimental values. The structure of FeTC4PCl is presented in Figure S1, which corresponds to the calculated bond lengths and angles shown in Table S1 along with the experimental values. The computational results using ωB97XD/def2-SVP are in agreement with the experimental values. Therefore, the optimizations in this work were performed using the ωB97XD functional. Based on the optimised structure, the single-point energy was obtained at the ωB97XD/def2-TZVPP level. To further analyse the binding ability of FeTC4PCl and FeTPPCl complexes with O2 molecules and the geometric configuration of the oxygenated complex, and to understand the influence of meso-site substituent groups on the adsorption of O2 molecules, the geometric configurations of the oxygenated complex (Fe-O2)TPPCl and (Fe-O2)TC4PCl formed by the above two complexes were simulated and constructed. Their geometric optimization and analysis were carried out using ωB97XD/def2-SVP, and single-point calculation was carried out at the TZVPP level. The entire optimized structure was a local minimum without imaginary frequencies.
The electronic structure analyses were performed using the Multiwfn 3.8 (dev) code [36], and the isosurface maps of various orbitals and real space functions were plotted using Visual Molecular Dynamics (VMD) [37] software based on the files exported from Multiwfn. In addition, Shermo [38] analysis software was used to analyse thermodynamic quantities throughout the process of forming the oxygenated complex (Fe-O2)TPPCl and (Fe-O2)TC4PCl during the combination of FeTPPCl and FeTC4PCl with O2. The variation trends of internal energy (U), enthalpy (H), and Gibbs free energy (G) with increasing temperature was calculated.
3. Results and Discussion
3.1. Catalytic Property of Complexes
Table 1 summarizes the main results obtained under different reaction conditions. The results indicate that conversion was considerably enhanced when the reaction temperature was increased from 30 °C to 70 °C (entries 1–5). However, attempts to further enhance the conversion by increasing the reaction temperature were unsuccessful, and the conversion decreased from 94% to 91% compared to entry 5 (entry 6). These results suggest that the optimal reaction temperature for the oxidation of cyclohexene is 70 °C.

Table 1.
Oxidation of cyclohexene catalysed by FeTC4PCl complex a.
The effect of product selectivity at different reaction temperatures was also discussed. As shown in Table 1, the highest product selectivity for 1 was observed at 50 °C (entry 3) rather than 70 °C. It was concluded that product 1 could be a possible intermediate product [18]. Compared to the catalytic oxidation of cyclohexene by FeCl3, FeTPPCl complexes, and meso-5,10,15,20-tetra-(15-crown-5-benzo-3-yl-sulfonyloxophen-4-yl)porphyrin compound (TC4HPP), it is evident that the FeTC4PCl complex demonstrates superior catalytic performance for the mild oxidation of cyclohexene with O2 (entries 5, 7–9). Importantly, the conversion reached up to 94% after four crown ether rings were appended to the porphyrin framework (entry 5). This phenomenon also supports the conclusion that benzo-15-crown-5 possessed a larger electron-donating environment compared to the Fe(Ⅲ) porphyrin ring, as it favours that the O2 molecule approaches the coordination centre of the Fe(Ⅲ) complex and stabilises the Fe–O2 bond. Compared to the uncrowned analogue Fe (Ⅲ) porphyrin (FeTPPCl), the reaction could not take place without the active centre (entry 10), strongly implying that O2 could not be activated without the FeTC4PCl complex or other catalysts.
3.2. Effect of meso-Substituents
FeTPPCl and FeTC4PCl were used as catalysts, and O2 was used as the oxidant to investigate the conversion of cyclohexene and product selectivity over time, as shown in Figure 2 and Figure 3. Figure 2 depicts the catalytic oxidation process of cyclohexene using FeTC4PCl as the catalyst, utilizing O2 as the oxidizing agent in the absence of any additives. FeTC4PCl, a complex featuring a 15-crown-5-benzo-3-sulfonyl group, demonstrated remarkable efficiency in enhancing the conversion rate of cyclohexene, as evidenced by the red line in Figure 2. The conversion rate increased significantly and rapidly with increasing reactivity. For instance, after 1 h of reaction time, the conversion rate was 8%, and it reached 49% after 2 h. Furthermore, after a reaction time of 12 h, the conversion rate of cyclohexene increased to 93%. In contrast, the utilization of FeTPPCl (a catalyst lacking the 15-crown-5-benzo-3-sulfonyl group) for the O2 oxidation of cyclohexene under the same reaction conditions (represented by the blank line in Figure 2) resulted in a substrate conversion rate of 7% after 1 h, which is similar to that obtained using FeTC4PCl. However, after 3 and 12 h of reaction time, the substrate conversion rates were 35% and 76%, respectively. Based on a comparison of the catalytic effects of the two catalysts, it can be concluded that FeTC4PCl exhibits superior catalytic activity. Furthermore, both catalysts significantly affected the selectivity toward the oxidation product of cyclohexene. Figure 3 shows that FeTC4PCl resulted in 70% and 53% selectivity for product 1 after 7 and 12 h, respectively, while FeTPPCl showed a lower selectivity of 56% and 40%, respectively. The effects of both catalysts on the selectivity of products 2 and 3 could have been more pronounced, which is possibly due to their low contents in the reaction mixture. Overall, the results demonstrate that FeTC4PCl with 15-crown-5-benzo-3-sulfonyl group substitution is a superior catalyst for conversion and selectivity compared to FeTPPCl.
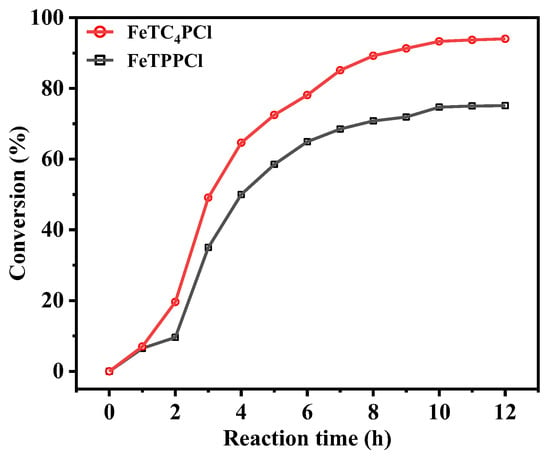
Figure 2.
Effect of reaction time on cyclohexene conversion using FeTC4PCl and FeTPPCl as the catalysts. Reaction conditions: cyclohexene (2.0 mL, 19.76 mmol), catalyst (2.0 mg, 0.00096 mmol), O2 at 1 atm, and 70 °C for 12 h.
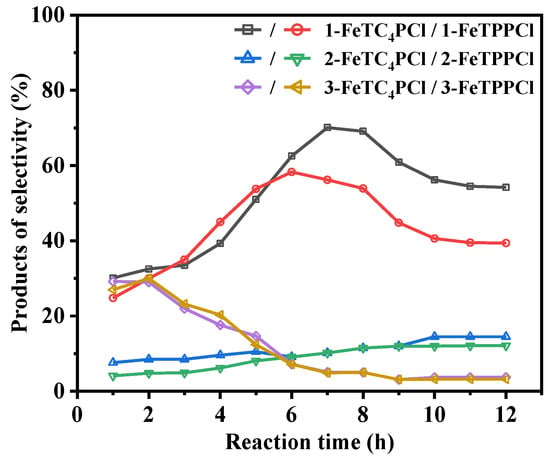
Figure 3.
Effect of reaction time on cyclohexene selectivity using FeTC4PCl and FeTPPCl as the catalysts. Reaction conditions: substrate (2.0 mL, 19.76 mmol), catalyst (2.0 mg, 0.00096 mmol), O2 at 1 atm, and 70 °C for 12 h.
3.3. Effect of Reaction Temperature
The oxidation of cyclohexene using FeTC4PCl as the catalyst showed the best catalytic activity. Therefore, the catalytic oxidation of cyclohexene using the FeTC4PCl catalyst at various temperatures was studied in further detail.
The catalytic activity of FeTC4PCl for the oxidation of cyclohexene by activated O2 was investigated in the temperature range of 30–77 °C at 10 °C increments. The conversion, turnover numbers (TON), and product selectivity of cyclohexene oxidation at various temperatures are listed in Table 1. The conversion of cyclohexene increased from 24% to 94% with a gradual increase in temperature from 30 °C to 70 °C (Entries 1 and 5 of Table 1), indicating that temperature had a noticeable effect on increasing the catalytic activity of FeTC4PCl. With a further increase in temperature to 77 °C, the conversion of cyclohexene decreased by 4% (Entry 5 vs. 6 of Table 1), indicating that the optimal reaction temperature was 70 °C. At temperatures above 70 °C, the conversion of cyclohexene decreased because of the high volatility of cyclohexene and the maximum oxygen consumption at 70 °C. These results were consistent with the maximum conversion characteristics of cyclohexene.
Table 1 demonstrates the significant influence of reaction temperature on product selectivity, specifically regarding 1 and 2. An increase in temperature notably impacts their selectivity, as exemplified by the selectivity related to 1 reaching 53% and 2 decreasing to 12% (Entry 3 of Table 1) at 50 °C. These results suggest that raising the temperature enhances the conversion of cyclohexene, resulting in a relatively high yield of 1. Furthermore, it is observed that 1 is produced primarily among the oxidation products of cyclohexene and remains stable at 50 °C. At 77 °C, the selectivity towards 1 increased to 73%, while the selectivity towards 2 remained virtually unchanged (Entry 5 of Table 1), indicating that a higher temperature facilitated the formation of 1. The FeTC4PCl catalyst demonstrated relatively high catalytic activity at this temperature, thus promoting the selectivity regarding 1. However, a decrease in total conversion and selectivity of 1 was observed at 77 °C (Entry 6 of Table 1). This phenomenon could be attributed to the possible decomposition of the FeTC4PCl catalyst and intermediate peroxides in the reaction, which may have been accelerated due to the higher temperature, leading to the decomposition of the peroxides by the metal porphyrin and the decomposition of the metal porphyrin by the peroxides [19]. Additionally, it was observed that at 70 °C, 1 rapidly oxidized to other products, indicating that 70 °C exceeded the optimum reaction temperature.
Among the oxidized products, 3 can be considered as an intermediate in the oxidation reaction [39,40,41]. The amount of 3 increased slightly with increasing reaction time, reaching only 3% after 6 h (Entry 5 of Table 1). Upon adding a stoichiometric amount of FeCl3 to the oxidation system, a lower conversion of cyclohexene (40%) was observed (Entry 8 of Table 1) and the selectivity of products 1 and 2 were relatively lower. This suggests that the 15-crown-5-benzo-3-sulfonyl group enhances the catalytic activity by promoting the activity of the Fe(III) centre. Cyclohexene was not oxidized in the absence of the catalyst (Entry 9 of Table 1), indicating the excellent catalytic effect of metal porphyrin catalysts.
3.4. Effect of Axial Ligand
Pyridine was chosen as the axial ligand to investigate its impact on the catalytic oxidation of cyclohexene using FeTC4PCl as the catalyst. Adding 5% pyridine to the oxidation system significantly affected the conversion and the selectivity of the oxidation products. The impact of pyridine on the catalytic oxidation reaction was illustrated in Figure 4. The results showed that pyridine inhibited cyclohexene’s catalytic oxidation using FeTC4PCl as the catalyst, leading to a reduction in cyclohexene conversion from 94% to 55%. Furthermore, the selectivity of 1 and 2 were lower than those without pyridine, while the selectivity of 3 was relatively higher. The axial coordination between the transition metal ion in FeTC4PCl and pyridine influenced the redox potential of the active metal centre, making it difficult for the active intermediate to coordinate with oxygen and form a high-valence state or transfer activated oxygen to cyclohexene, thereby inhibiting the oxidation reaction [17].
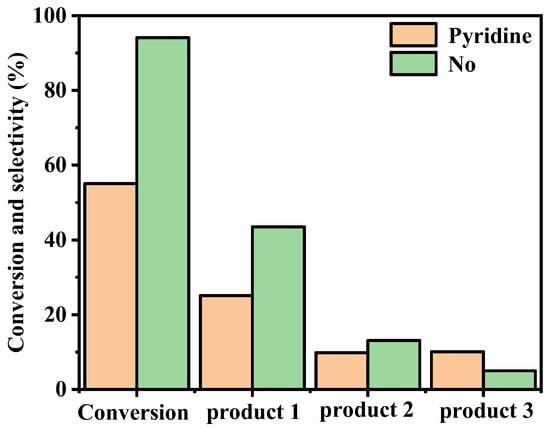
Figure 4.
Effect of pyridine on oxidation of cyclohexene catalysed by FeTC4PCl. Reaction conditions: Substrate (2.0 mL, 19.76 mmol), FeTC4PCl (2.0 mg, 0.00096 mmol), O2 at 1 atm, and 70 °C for 12 h.
3.5. Optimized Structure and Bonding Properties
The optimized geometries of FeTPPCl and FeTC4PCl in the sextet ground spin state and that of (Fe-O2)TPPCl and (Fe-O2)TC4PCl compounds in the octet ground spin state were obtained using the ωB97XD/def2-SVP geometry method. The corresponding results are presented in Figure 5 and Figure S2, and the geometry parameters are given in Table S1. All compounds were optimized using the same methodology. Previous studies have shown that in the absence of a crown ether ring attached to the meso-site of the porphyrin ring, all atoms on the porphyrin ring are arranged in a planar configuration [29,42,43,44,45]. The coordination of Fe(III) from FeCl3 with the N atom in the porphyrin ring did not significantly affect the planar configuration of the porphyrin ring. In both FeTPPCl and FeTC4PCl, the Fe(III) atom was found to be outside the porphyrin plane, and the bond lengths of N-Fe and Fe-Cl were determined to be 2.08 Å and 2.20 Å, respectively. This suggests that the structural features of the Fe(III)-porphyrin ring were not affected by the introduction of the crown ether ring. Analysis of the oxygenated complexes (Fe-O2)TPPCl and (Fe-O2)TC4PCl revealed that the O2 molecule effectively induced a change in the conformation of the Fe(III) porphyrin ring. As shown in Figure S2 and Table S1, the presence of the crown ether ring and O2 induced a non-planar “saddle-like” conformation of the Fe(III) porphyrin ring in (Fe-O2)TC4PCl. The crown ether ring and O2 had a dual-spatial-site-blocking effect, inducing a more pronounced variation of the Fe(III) porphyrin conformation seen in (Fe-O2)TC4PCl, which was confirmed by the changes in bond lengths and dihedral angles C1-N-Np-Cp1 and C2-N-Np-Cp2 in connection with Fe-O and Fe-Cl. However, the bond length of N-Fe was not significantly affected and remained at a value of 2.00 Å. In summary, the crown ether ring induced significant changes in the spatial configuration of the porphyrin ring with Fe(Ⅲ) as the coordination centre but had almost no effect on the interatomic bond length.
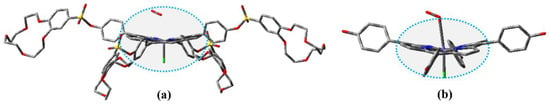
Figure 5.
Optimized structure of FeTC4PCl (a) and FeTPPCl (b). The H atom is hidden for clarity.
3.6. Atomic Charge Analysis
An atomic charge is a fundamental and intuitive descriptor of the charge distribution in a given chemical system. However, due to the unobservable nature of the atomic charge and the lack of an objective and unique definition, numerous methods exist to calculate atomic charge [46,47]. The Atomic Dipole Corrected Hirshfeld Atomic Charge (ADCH) method is a more accurate approach for atomic charge analysis. This method is based on the Hirshfeld charge analysis, which defines atomic charge (Equation (1)) as a weighted sum of electron density contributions from neighbouring atoms:
where
where and (r) represents the electron density of all atoms in the free state; represents the deformation density, which shows the variation of the electron density during the chirality process after the atoms form molecules. is the A-atom weight function, defined as the region in the whole real space belonging to the A-atoms. However, the Hirshfeld charge data are generally small [48], and the dipole moment and electrostatic potential are poorly reproducible [49] mainly because the influence of the atomic dipole moment in the calculation process is neglected. Therefore, Lu Tian [50] proposed the ADCH method, which is a method that defines the atomic dipole moment () as:
In this method, the Hirshfeld charge of each atom and its are calculated first, and then each is expanded into the calibrated positive charge of the surrounding atoms according to Equation (3).
denotes the calibrated positive charge of the of the unfolded A-atom on the B-atom. Finally, after unfolding the of all atoms into the correctional charge and then accumulating it to the original Hirshfeld charge, the ADCH charge is obtained.
To investigate the influence of various types of meso-substituents on the active centres (mainly Fe(III), Npyrr, and Npyri) in the Fe(III) porphyrin ring, the four complexes above were analysed using the ADCH method. The results in Table 2 and Figure S3 demonstrate that the total number of atomic charges on meso-substituents in FeTPPCl and FeTC4PCl were identical, with each complex having a total atomic charge of 0.042 a.u and 0.033 a.u, respectively. This suggests that the electron-donating ability of the same type of substituents to the entire Fe(III) porphyrin ring is similar within each complex. Notably, the meso-substituents in FeTC4PCl exhibited a significantly greater electron-donating property than FeTPPCl, leading to a higher atomic charge of Fe(III) in FeTC4PCl and a slightly lower atomic charge regarding the Cl atom. It may explain the superior catalytic activity of FeTC4PCl over FeTPPCl.
Table 2.
ADCH atomic charge analysis of FeTPPCl, FeTC4PCl, (Fe-O2)TPPCl, and (Fe-O2)TC4PCl at ωB97XD/def2-SVP level of theory.
After analysing the changes in atomic charge in complexes formed by FeTPPCl and FeTC4PCl following O2 adsorption, it was found that the most significant change in atomic charge occurred in (Fe-O2)TC4PCl compared to (Fe-O2)TPPCl. The data presented in Table 2 indicate that the charge numbers of Npyrr and Npyri were averaged out, while the atomic charge of Fe(III) decreased by 0.029 atomic units (a.u.). This decrease was primarily due to the insertion of O2 on top of Fe(III), resulting in non-uniform atomic charge distribution, with the most significant effect being on meso-substituents. This effect further decreased the positive charges carried by the four substituents, as depicted in Figure S3. The primary reason for this change was the partial delocalization of electrons in the O2 molecule into the Fe(III) porphyrin ring, which was confirmed by the positive charge of 0.045 a.u. in the O2 molecule. Furthermore, in (Fe-O2)TPPCl, the charge number of the O2 molecule was 0.025 a.u., which was smaller than that of (Fe-O2)TC4PCl, indicating that the crown ether ring substituent further activated the O2 molecule and facilitated the catalytic reaction.
3.7. Molecular Orbital and Spin Density Analysis
To investigate the influence of the suspension of the crown ether ring by the porphyrin ring on the coordination centre Fe(III) and its bound O2, we analysed the electronic interactions of Fe(III)-porphyrin and its corresponding complexes using frontier orbital theory for the highest energy molecular orbital (highest occupied orbital HOMO) and the lowest energy molecular orbital (lowest unoccupied orbital LUMO), respectively. The molecular orbital energy levels of the complexes (Figure S4) showed that in the FeTPPCl and FeTC4PCl complex systems, the HOMO–LUMO band gap remained the same, indicating that the introduction of the crown ether ring at the meso-position of the porphyrin ring did not affect the electron density distribution in the orbitals, which was mainly concentrated in the porphyrin ring and its corresponding Fe(III) ion.
Upon further interaction of the FeTPPCl and FeTC4PCl complexes with molecular oxygen to form dioxygen complexes ((Fe-O2)TPPCl and (Fe-O2)TC4PCl), we observed that the HOMO–LUMO band gap corresponding to the (Fe-O2)TPPCl complex that was formed after FeTPPCl interacted with O2 did not change significantly, as seen in Figure S4a,c. However, the dioxygen complex (Fe-O2)TC4PCl formed after the interaction of FeTC4PCl with O2 exhibited a significant reduction in the HOMO–LUMO band gap, particularly in the HOMO and LUMO orbital energy of the β molecular orbital, as observed in Figure 6. This may be attributed to the electron-donating ability of the crown ether ring, which increases the electron density of the Fe(III) ion, thereby enhancing its binding and electron transfer ability in connection with O2 and ultimately improving its catalytic activity.
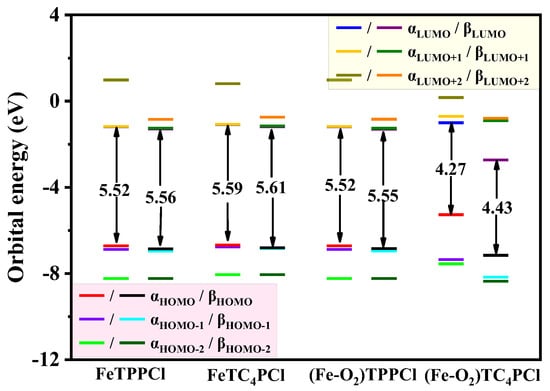
Figure 6.
Comparison of the calculated molecular orbital energy level (eV) and energy gaps between the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) of the four complexes using ωB97XD/def2-SVP level calculation.
In summary, our results demonstrate that introducing the crown ether ring at the meso-position of the porphyrin ring can significantly enhance the catalytic activity of Fe(III) complexes regarding O2, which may have important implications for the development of novel catalysts for various chemical reactions.
To further illustrate the distribution of α and β electron densities in the four open-shell aforementioned complexes, and to examine the distribution of unpaired electrons in three-dimensional space, this study employed the Multiwfn software to conduct spin density analysis. Figure 7 presents the three-dimensional spatial isosurface after full-space integration, highlighting the difference between the electron density of α orbitals (α) and β orbitals (β), which is indicated by green and blue colours, respectively. A positive value in green indicates a higher number of α orbital electrons than β, while a negative value in blue indicates the opposite.
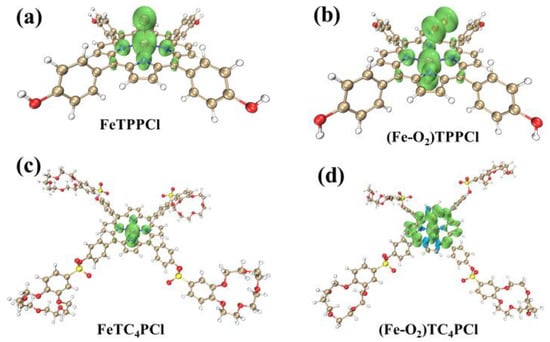
Figure 7.
Isosurface maps of the spin density of FeTPPCl (a), FeTC4PCl (b), (Fe-O2)TPPCl (c), and (Fe-O2)TC4PCl (d) (isovalue = 0.005). The green colour indicates that there are more α electrons than β electrons, and the blue colour indicates that there are more β electrons than α electrons.
Based on Figure 7, all four complexes show a higher concentration of α electrons than β electrons, which is consistent with the previous computational results. Further examination reveals that in FeTPPCl and FeTC4PCl, α electrons are mainly located on Fe(III), Cl, Npyrr, and Npyri atoms. Upon interaction with O2, α electrons transfer to the α-C atoms of pyridine and pyrrole rings. Of particular note is that the introduction of the meso-substituted crown ether ring in (Fe-O2)TC4PCl affects the electron distribution of the Fe(III) porphyrin ring, changes the HOMO orbital energy, activates molecules, and reduces the HOMO–LUMO energy difference (as shown in Figure 6). However, this modification also results in an increased instability of the entire molecular system. Therefore, further discussion is required to explore this research aspect.
3.8. Density-of-State Analysis
The density-of-state (DOS) diagram depicts the distribution of energy levels for molecular orbitals (MOs) in a chemical system. The DOS curve reflects the number of MOs within a given energy level in a unit energy interval [36,51,52,53]. Figure 8 and Figure S5 exhibit the TDOS and PDOS diagrams and maps for FeTPPCl, FeTC4PCl, (Fe-O2)TPPCl, and (Fe-O2)TC4PCl, along with the contributions from different MO groups. Since the four complex systems have an open-shell configuration, α and β MOs were plotted separately in the DOS and PDOS curves. The upper half of the graph frame displays the curves of α MOs as solid lines, while the lower half shows the curves of β MOs as dotted lines. Symmetric α and β MOs signify that electrons in both orbitals have no spin hybridization in the same energy range, although spin polarization arises otherwise.
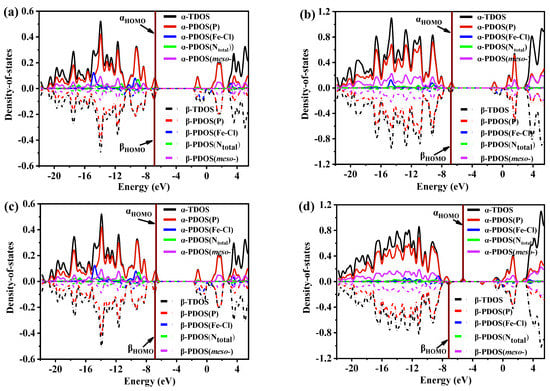
Figure 8.
Density-of-state (DOS) map and Mos degeneracy of the (a) FeTPPCl, (b) FeTC4PCl, (c) (Fe-O2)TPPCl, and (d) (Fe-O2)TC4PCl complexes. The location of the HOMO is the wine vertical line.
The changes observed in Figure 8 and Figure S5 demonstrate that the TDOS curves of α and β MOs for each complex are nearly symmetrical, indicating almost equal spin polarization of electronic states in α and β orbitals as well as a uniform contribution of molecular orbitals to TDOS. However, the contribution of each group to TDOS varies. To further analyse the impact of different groups on TDOS in various complexes, PDOS was used to examine the Fe–Cl bond, pyridine N (Npyri) on the porphyrin ring, substituted groups on the meso-position, and the angular momentum of S, P, and D in the complex. As shown in Figure 8, P orbitals contribute significantly to the entire orbital energy level in all complexes, particularly regarding the molecular frontier orbitals of HOMO and LUMO. On the other hand, S orbitals only contribute to TDOS in the lower energy level.
Additionally, the spherically symmetric distribution of S orbitals in Figure S5 suggests the symmetric spin MOs of α and β. By analysing the contribution of d orbitals to α and β MOs, it was found that the contribution of d orbitals to TDOS is minimal, but that the evident spin polarization occurring in α and β MOs is primarily due to Fe(III) being the only provider of d orbitals occupied by electrons in all systems. Among all the contributions of angular momentum to TDOS, the proportion of d orbitals is the smallest. The spin multiplicity of all studied systems was set to sextet states for FeTPPCl and FeTC4PCl complexes and octet for (Fe-O2)TPPCl and (Fe-O2)TC4PCl, resulting in different electron degeneracies on α and β MOs.
The contribution of porphyrin Ntotal (the total of the Npyri and Npyrr) and meso-substituent (meso-) to the total density of states (TDOS) in Fe–Cl and porphyrin rings shown in Figure 8 was analysed, which revealed that while these groups contribute to the orbital energy levels throughout the entire interval, their overall contribution is not significant. A further comparison revealed that the meso-crown ether ring-substituted group contributed most significantly to TDOS (Figure 8 and Figure S5). The (Fe-O2)TC4PCl complex was especially analysed, and the difference in highest occupied molecular orbital (HOMO) between α and β MOs was found to be 1.89 eV, which can be determined by analysing the HOMO difference of α and β MOs in FeTPPCl (0.13 eV), which indicates that the introduction of the crown ether ring primarily caused this change. Combined with the analysis in Section 3.6, it can be seen that the crown ether ring helps to influence the coordination field environment of the FeTC4PCl complex binding O2, alter the electron spin polarization of Fe(III), and improve its catalytic activity.
3.9. Thermodynamic Quantities Analysis
The study of thermodynamic quantities involves electron energy, vibration energy, and geometry concerning the structure and other information in quantum calculations for the electron energy obtained under a specific geometry. The study of practical problems needs to be more accurate. Gibbs free energy (G), internal energy (U), entropy (S), enthalpy (H), and other thermodynamic quantities such as those that are a function of temperature, and the temperature setting affects their values [3,38]; therefore, in this paper, the thermodynamic changes of the complexes during the reaction as well as their differences before and after the reaction were studied in depth using Shermo software. At the same time, the changes in thermodynamic quantities at different temperatures were calculated.
Table 3 summarizes the electronic energy [Eele], internal energy [U], enthalpy [H], and free energy [G] of each complex at a steady state, and Figure 9 plots the corresponding thermodynamic amounts with temperature regarding the thermal corrections to internal energy (Ucorr), enthalpy (Hcorr), and Gibbs free energy (Gcorr). It can be seen from Table 3 that when the FeTPPCl and FeTC4PCl complexes combine with O2 molecules to form oxygenated complexes ((Fe-O2)TPPCl and (Fe-O2)TC4PCl), their corresponding thermodynamic quantities significantly decrease, especially ΔG < 0, indicating that the two complexes can find adsorption with O2 and form lower-energy oxygenated complexes. However, it was found from the previous experimental results that an increase in reaction temperature has a more significant enhancement on the reaction rate and product selectivity. To explore the reason for this, the process of G, U, and H was analysed using Shermo software. It can be seen from Figure 9 that when the temperature was further increased, the change of thermodynamic quantities (Eele, U, H, and G) was found to be nonlinear with increasing temperature. The trend of the G curve was more extensive than that of the U and H curves (see Figure 9a), and the trend of the curve of Gcorr was also more extensive than that of Ucorr and Hcorr, which shows that the effect of temperature on this reaction cannot be neglected.
Table 3.
The thermodynamic data of FeTPPCl, FeTC4PCl, (Fe-O2)TPPCl, and (Fe-O2)TC4PCl complexes calculated at ωB97XD/def2-TZVPP level of theory under 298.15 K.
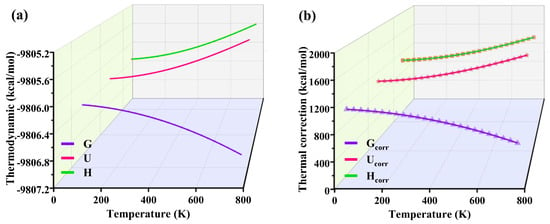
Figure 9.
The curve of thermodynamics with temperature (a) and thermal with temperature (b).
3.10. Discussion on the Mechanism of the Oxidation of Cyclohexene
A proposed reaction mechanism is presented in this study to investigate the oxidation process of cyclohexene with a carbon–carbon double bond using FeTC4PCl and O2 as oxidants at standard atmospheric pressure without any reducing agent and additives. By combining experimental data, theoretical analysis, and product analysis, it is concluded that the oxidation of cyclohexene by O2 occurs mainly in the allylic position. Previous research on this reaction mechanism (particularly the study by Mohebi et al. [54]) was considered in proposing the mechanism presented in Scheme 2. The proposed mechanism includes multiple steps [14,55,56,57], with the primary oxidation process being a free radical chain reaction. Initially, the catalyst FeTC4PCl was activated at a specific temperature, as observed in Figure 2 and Figure 3. The low conversion rate of the substrate within 1 h of the reaction verifies this activation process. Fe(III) in FeTC4PCl and O2 undergo surface adsorption at a specific temperature to form a compound containing O2 molecules ((Fe-O2)TC4PCl). This compound can combine with protons in the system to form an unstable product ((FeIV-OOH)TC4PCl), which then loses H2O molecules to form a catalytically active high-valence iron intermediate [(FeIV = O)TC4PCl]+•. This high-valence complex can interact with the carbon–carbon double bond with cyclohexene, leading to the removal of a proton from the allylic carbon and the generation of a relatively stable allyl radical and a high-valence intermediate ((FeIV-O-H)TC4PCl (Step 1)). This step completes the initiation process of the cyclohexene radical, which is considered the chain initiation of free radicals. After the formation of the allyl radical, it rapidly reacts with O2 molecules in the system to generate an allyl peroxygen radical (step 2). It then undergoes a radical reaction with the substrate cyclohexene molecule (step 3). These reactions produce an allyl radical and product 3 or generate the transition-state product of 2-cyclohexyl-1-yl-hydrogen peroxide. The allyl free radical generated can either continue the process triggered by the free radical chain or undergo subsequent reactions. The 2-cyclohexyl-1-yl-hydrogen peroxide produced can react with FeTC4PCl to generate (FeIV-O-H)TC4PCl and an alkoxy free radical (step 4). The alkoxy radical can react quickly with cyclohexene to form product 1 (Step 5), which is then further oxidized at higher temperatures to form product 2 (Step 10). During step 5, the substrate cyclohexene loses the protons on the allylic carbon to form an allyl radical, part of which comes from the previous step. These allyl radicals can combine hydrogen and oxygen to oxidize the active 2-cyclohexen-1-yl-hydroperoxide (step 6) in the oxidation system. In the presence of the active species (FeIV-O-H)TC4PCl, a homolytic reaction occurs by forming the alkoxy radical and the hydroxide radical, which are the main products (Step 7). Subsequently, the alkoxy radical reacts with the previously generated 2-cyclohexen-1-yl-hydroperoxide, producing product 2 and a new alkoxy radical (Step 8). The newly formed alkoxy radical undergoes a propagation reaction similar to the reactions described in Steps 5 and 8. Some may combine it with the hydroxyl radical to yield products 1 and 2 (Step 9). The concentration of the alkoxy radical decreases due to the formation of products 1 and 2, resulting in a decrease in the oxidation reaction rate. This mechanism demonstrates the crucial role played by FeTC4PCl in initiating and propagating radicals. Additionally, the catalytically active Fe(III) centre may promote the change in its valency [42], facilitating the formation of the active intermediate in the catalytic oxidation reaction.
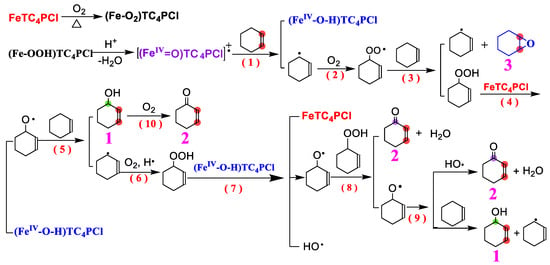
Scheme 2.
The possible mechanism for cyclohexene oxidation catalysed by FeTC4PCl.
4. Conclusions
This paper investigates the impact of meso-position substituents on the catalytic activity of Fe(III) porphyrin complexes as well as examines the catalytic oxidation of cyclohexene under mild and without reductant conditions. The results show that meso-substituents effectively influence the catalytic activity of Fe(III) and shorten the reaction induction period. FeTC4PCl with meso-5,10,15,20-tetra-(15-crown-5-benzo-3-yl-sulfonyloxophen-4-yl) substituents exhibit higher conversion rates and selectivity compared to FeTPPCl with meso-phenylhydroxy substituents. Moreover, introducing pyridine ligands inhibits the oxidation reaction by affecting the redox potential of the active metal centre.
Furthermore, the paper employs the DFT method to optimize the geometric structure, molecular orbital energy level analysis, atomic charge, spin density, and density of orbital states of FeTPPCl, FeTC4PCl, and the oxygenated complexes formed after the adsorption of O2. The (Fe-O2)TCPPCl and (Fe-O2)TC4PCl oxygenated complexes exhibit distinct characteristics, with the planar structure of Fe(III)-porphyrin changing to a nonplanar “saddle-like” conformation in (Fe-O2)TC4PCl. The crown ether ring substituent impacts the electron distribution of the Fe(III) porphyrin ring, modifies the HOMO orbital energy, activates the molecule, and further reduces the energy level difference of HOMO–LUMO, indicating its auxiliary activation effect on the catalytic system composed of the Fe(III) porphyrin ring. However, additional research is necessary to fully comprehend the intricacies of these changes.
In summary, this study provides new insights into the design of meso-substituted Fe(III) porphyrin catalysts for catalytic oxidation reactions. Based on experimental and theoretical analyses, this paper deduced the mechanism of the cyclohexene oxidation reaction using FeTC4PCl as the catalyst and O2 as the oxidant. The reaction proceeds by generating the high-valent iron intermediate [(FeIV = O)TC4PCl]+ initially, followed by the formation of (FeIV-O-H)TC4PCl and an alkoxy radical transition state. Radical transfer and transfer subsequently generate the main three products. The study also reveals that the reaction mechanism follows a free radical chain reaction process, and the crown ether ring affects the activity of the catalytic centre of Fe(III) and demonstrates the property of shortening the reaction induction period. These findings provide new theoretical insights into modifying metalloporphyrins with meso-substituent groups.
Supplementary Materials
Supplementary material associated with this article can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28083452/s1. Scheme S1 Experimental schematic diagram of the catalytic oxidation reaction; Figure S1. Simulated infrared spectrum of FeTC4PCl. The frequencies have been scaled by the fundamental scaling factor; Table S1. The selected bond length of FeTPPCl, FeTC4PCl and (Fe-O2)TC4PCl complexes calculated at ωB97XD/def2-SVP level of theory; Figure S2. Optimized geometry of (a) FeTC4PCl, (b) (Fe-O2)TC4PCl, (c) FeTPPCl and FeTC4PCl using ωB97XD/def2-SVP level calculation; Figure S3. ADCH atomic charge analysis of (a) FeTPPCl, (b) FeTC4PCl, (c) (Fe-O2)TPPCl, and (d) (Fe-O2)TC4PCl at ωB97XD/def2-SVP level of theory; Figure S4. The frontier molecular orbital distributions of (a)FeTPPCl, (b) FeTC4PCl, (c) Fe-O2TPPCl, and (d) Fe-O2TC4PCl; Figure S5. Density-of-state (DOS) map and MOs degeneracy of the (a,b) FeTPPCl, FeTC4PCl (c,d), (e,f) (Fe-O2)TPPCl, and (g,h) (Fe-O2)TC4PCl complexes.
Author Contributions
Conceptualization, Y.Z.; Funding acquisition, A.F.; Methodology, F.H.; Supervision, A.F.; and Validation, P.L.; Writing—review & editing, X.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 51801001), China Postdoctoral Science Foundation (No. 2016M601878), Provincial Key Research and Development Program of Shaanxi (No. 2019GY-197), Key Project of Baoji University of Arts and Sciences (No. 209040127, No. ZK2018051), Special Scientific Research Project of Shaanxi Education Department (No. 21JK0478, 202310561), and the Natural Science Basic Research Plan in Shanxi Province of China (No. 2015JM5215). Feng AL is supported by The Thousand Talents Plan for Young Professionals of Shannxi Province and High-level Leading Talents of Scientific and Technological Innovation of Baoji, and the Special Project of Philosophy and Social science of Baoji (BJSKZX-202263). We would like to thank the Key Laboratory of Materials Physics and Functional Devices of Baoji.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable requests.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, X.; Lei, H.; Xie, L.; Wang, N.; Zhang, W.; Cao, R. Metalloporphyrins as Catalytic Models for Studying Hydrogen and Oxygen Evolution and Oxygen Reduction Reactions. Acc. Chem. Res. 2022, 55, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, P.; Li, X.; Xie, L.; Wang, N.; Lei, H.; Zhang, C.; Zhang, W.; Lee, Y.M.; Zhang, W.; et al. Crucial Roles of a Pendant Imidazole Ligand of a Cobalt Porphyrin Complex in the Stoichiometric and Catalytic Reduction of Dioxygen. Angew. Chem. Int. Ed. Engl. 2022, 61, e202208143. [Google Scholar] [PubMed]
- Singh, P.; Denler, M.C.; Mayfield, J.R.; Jackson, T.A. Differences in chemoselectivity in olefin oxidation by a series of non-porphyrin manganese(IV)-oxo complexes. Dalton Trans. 2022, 51, 5938–5949. [Google Scholar] [CrossRef]
- Lu, X.; Wang, S.; Qin, J.H. Isolating Fe-O2 Intermediates in Dioxygen Activation by Iron Porphyrin Complexes. Molecules 2022, 27, 4690. [Google Scholar] [CrossRef]
- Guo, X.; Wang, N.; Li, X.; Zhang, Z.; Zhao, J.; Ren, W.; Ding, S.; Xu, G.; Li, J.; Apfel, U.P.; et al. Homolytic versus Heterolytic Hydrogen Evolution Reaction Steered by a Steric Effect. Angew. Chem. Int. Ed. 2020, 59, 8941–8946. [Google Scholar] [CrossRef]
- Marinescu, S.C.; Winkler, J.R.; Gray, H.B. Molecular mechanisms of cobalt-catalyzed hydrogen evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 15127–15131. [Google Scholar] [CrossRef]
- Guo, H.B.; Wang, Y.N.; Guo, K.; Lei, H.T.; Liang, Z.Z.; Zhang, X.P.; Cao, R. A Co Porphyrin with Electron-Withdrawing and Hydrophilic Substituents for Improved Electrocatalytic Oxygen Reduction. J. Electrochem. 2022, 28, 2214002–2214012. [Google Scholar]
- Sinha, S.; Ghosh, M.; Warren, J.J. Changing the Selectivity of O2 Reduction Catalysis with One Ligand Heteroatom. ACS Catal. 2019, 9, 2685–2691. [Google Scholar] [CrossRef]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants for iron(iv)-oxo porphyrin cation radical complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef]
- Feng, L.; Wang, K.-Y.; Joseph, E.; Zhou, H.-C. Catalytic Porphyrin Framework Compounds. Trends Chem. 2020, 2, 555–568. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, Z.; Zhou, X.; Ji, H. Progress in the application of metalloporphyrins compounds in catalytic oxidation reactions. Sci. Sin. Chim. 2022, 52, 1224–1238. [Google Scholar] [CrossRef]
- Berijani, K.; Hosseini-Monfared, H. Aerobic enantioselective epoxidation of olefins mediated by an easy-to-prepare recyclable manganese-porphyrin. Mol. Catal. 2017, 433, 136–144. [Google Scholar] [CrossRef]
- Song, S.; Zhou, M.; Rao, Y.; Xu, L.; Song, J. Development of the design, synthesis and property of porphyrin arrays and porphyrinoids. Sci. Sin. Chim. 2022, 52, 1189–1204. [Google Scholar] [CrossRef]
- Zhou, X.-T.; Tang, Q.-H.; Ji, H.-B. Remarkable enhancement of aerobic epoxidation reactivity for olefins catalyzed by μ-oxo-bisiron(III) porphyrins under ambient conditions. Tetrahedron Lett. 2009, 50, 6601–6605. [Google Scholar] [CrossRef]
- Yuasa, M.; Nishihara, R.; Shi, C.; Anson, F.C. A Comparison of Several Meso-Tetraalkyl Cobalt Porphyrins as Catalysts for the Electroreduction of Dioxygen. Polym. Adv. Technol 2001, 12, 266–270. [Google Scholar] [CrossRef]
- Sinha, S.; Aaron, M.S.; Blagojevic, J.; Warren, J.J. Electrocatalytic Dioxygen Reduction by Carbon Electrodes Noncovalently Modified with Iron Porphyrin Complexes: Enhancements from a Single Proton Relay. Chem. A Eur. J. 2015, 21, 18072–18075. [Google Scholar] [CrossRef]
- Yang, X.; Sun, S.; Meng, L.; Li, K.; Mukherjee, S.; Chen, X.; Lv, J.; Liang, S.; Zang, H.-Y.; Yan, L.-K.; et al. Molecular single iron site catalysts for electrochemical nitrogen fixation under ambient conditions. Appl. Catal. B Environ. 2021, 285, 119794. [Google Scholar] [CrossRef]
- Yuan, R.; George, S.L.; Chen, J.; Wu, Q.; Qiu, X.; Zhao, L. Meso-substituted Metalloporphyrin-based Composites for Electrocatalytic Oxygen Reduction Reactions. ChemNanoMat 2023, e202300027. [Google Scholar] [CrossRef]
- Soury, R.; Chaabene, M.; Jabli, M.; Saleh, T.A.; Ben Chaabane, R.; Saint-Aman, E.; Loiseau, F.; Philouze, C.; Allouche, A.-R.; Nasri, H. Meso-tetrakis(3,4,5-trimethoxyphenyl)porphyrin derivatives: Synthesis, spectroscopic characterizations and adsorption of NO2. Chem. Eng. J. 2019, 375, 122005. [Google Scholar] [CrossRef]
- Ardakani, M.M.; Rahimi, P.; Dehghani, H.; Karami, P.E.; Zare, H.R.; Karami, S. Electrocatalytic Reduction of Dioxygen on the Surface of Glassy Carbon Electrodes Modified with Cobalt Porphyrin Complexes. Electroanalysis 2007, 19, 2258–2263. [Google Scholar] [CrossRef]
- Lei, H.; Li, X.; Meng, J.; Zheng, H.; Zhang, W.; Cao, R. Structure Effects of Metal Corroles on Energy-Related Small Molecule Activation Reactions. ACS Catal. 2019, 9, 4320–4344. [Google Scholar] [CrossRef]
- Wannakao, S.; Maihom, T.; Kongpatpanich, K.; Limtrakul, J.; Promarak, V. Halogen substitutions leading to enhanced oxygen evolution and oxygen reduction reactions in metalloporphyrin frameworks. Phys. Chem. Chem. Phys. 2017, 19, 29540–29548. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Wang, Y.; Wang, B.; Duan, X.; Lei, H.; Zhang, X.; Zheng, H.; Zhang, W.; Cao, R. Cobalt porphyrins supported on carbon nanotubes as model catalysts of metal-N4/C sites for oxygen electrocatalysis. J. Energy Chem. 2021, 53, 77–81. [Google Scholar] [CrossRef]
- Venegas, R.; Recio, F.J.; Riquelme, J.; Neira, K.; Marco, J.F.; Ponce, I.; Zagal, J.H.; Tasca, F. Biomimetic reduction of O2 in an acid medium on iron phthalocyanines axially coordinated to pyridine anchored on carbon nanotubes. J. Mater. Chem. A 2017, 5, 12054–12059. [Google Scholar] [CrossRef]
- Aarabi, M.; Omidyan, R.; Soorkia, S.; Grégoire, G.; Broquier, M.; Crestoni, M.-E.; de la Lande, A.; Soep, B.; Shafizadeh, N. The dramatic effect of N-methylimidazole on trans axial ligand binding to ferric heme: Experiment and theory. Phys. Chem. Chem. Phys. 2019, 21, 1750–1760. [Google Scholar] [CrossRef]
- Ma, Z.; Nakatani, N.; Fujii, H.; Hada, M. DFT insight into axial ligand effects on electronic structure and mechanistic reactivity of oxoiron(iv) porphyrin. Phys. Chem. Chem. Phys. 2020, 22, 12173–12179. [Google Scholar] [CrossRef]
- Meng, J.; Lei, H.; Li, X.; Zhang, W.; Cao, R. The Trans Axial Ligand Effect on Oxygen Reduction. Immobilization Method May Weaken Catalyst Design for Electrocatalytic Performance. J. Phys. Chem. C 2020, 124, 16324–16331. [Google Scholar] [CrossRef]
- Li, X.D.; Zhu, Y.C.; Yang, L.J. Crown ether-appended Fe (III) porphyrin: Synthesis, characterization and catalytic oxidation of cyclohexene with molecular oxygen. Chin. Chem. Lett. 2012, 23, 375–378. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Kuo, M.-C.; Lee, C.-W.; Liang, Y.-R.; Lee, G.-H.; Peng, S.-M.; Yeh, C.-Y. Synthesis, structure, and cation complexation of a novel crown ether porphyrin. Tetrahedron Lett. 2008, 49, 7223–7226. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Versions D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, T.; Grimme, S. Double-hybrid density functionals with long-range dispersion corrections: Higher accuracy and extended applicability. Phys. Chem. Chem. Phys 2007, 9, 3397–3406. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Shermo: A general code for calculating molecular thermochemistry properties. Comput. Theor. Chem. 2021, 1200, 113249. [Google Scholar] [CrossRef]
- Wang, R.M.; Li, S.B.; Wang, Y.P.; He, Y.F.; Lei, Z.Q. Sheet polymer and its complexes. II. Preparation and catalytic activity of polymeric tetrakisphenylporphyrin films crosslinked by 4,4′-biphenylene-bisulfoate. J. Appl. Polym. Sci. 1998, 67, 2027–2034. [Google Scholar] [CrossRef]
- Baricelli, P.J.; Sánchez, V.J.; Pardey, A.J.; Moya, S.A. Catalytic oxidation of 1-hexene and cyclohexene with molecular oxygen by iridium nitro complexes. J. Mol. Catal. A Chem. 2000, 164, 77–84. [Google Scholar] [CrossRef]
- Wang, R.M.; Hao, C.J.; Wang, Y.P.; Li, S.B. Amino acid Schiff base complex catalyst for effective oxidation of olefins with molecular oxygen. J. Mol. Catal. A Chem. 1999, 147, 173–178. [Google Scholar] [CrossRef]
- Cai, Y.; Castro, P.P.; Gutierrez-Tunstad, L.M. ‘Cloverleaf’ crown ether resorcin[4]arenes. Tetrahedron Lett. 2008, 49, 2146–2149. [Google Scholar] [CrossRef]
- Liu, H.; Shao, X.-B.; Jia, M.-X.; Jiang, X.-K.; Li, Z.-T.; Chen, G.-J. Selective recognition of sodium cyanide and potassium cyanide by diaza-crown ether-capped Zn-porphyrin receptors in polar solvents. Tetrahedron 2005, 61, 8095–8100. [Google Scholar] [CrossRef]
- Fathalla, M. Synthesis and characterization of a porphyrin-crown ether conjugate as a potential intermediate for drug delivery application. J. Porphyr. Phthalocyanines 2020, 25, 95–101. [Google Scholar] [CrossRef]
- Li, H.; Zheng, X.; Jia, Z.; Wang, X. Theoretical study on noncovalent interaction of molecular tweezers by Zn(II) salphen-azo-crown ether triads receptor. J. Mol. Model 2020, 26, 39. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Atomic Dipole Moment Corrected Hirshfeld Population Method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- Xia-Yu, Z.; Chun-Ying, R.; Tian, L.U.; Shu-Bin, L.I.U. Hirshfeld Charge as a Quantitative Measure of Electrophilicity and Nucleophilicity: Nitrogen-Containing Systems. Acta Physico Chim. Sin. 2014, 30, 2055–2062. [Google Scholar] [CrossRef]
- Davidson, E.R.; Chakravorty, S. A test of the Hirshfeld definition of atomic charges and moments. Theor. Chim. Acta 1992, 83, 319–330. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Rablen, P.R. Comparison of Atomic Charges Derived via Different Procedures. J. Comput. Chem. 1993, 14, 1504–1518. [Google Scholar] [CrossRef]
- Tian, L.U.; Fei-Wu, C. Comparison of Computational Methods for Atomic Charges. Acta Phys. Chim. Sin. 2012, 28, 1–18. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Bonding character, electron delocalization, and aromaticity. Carbon 2020, 165, 468–475. [Google Scholar] [CrossRef]
- Manzetti, S.; Lu, T.; Behzadi, H.; Estrafili, M.D.; Thi Le, H.-L.; Vach, H. Intriguing properties of unusual silicon nanocrystals. RSC Adv. 2015, 5, 78192–78208. [Google Scholar] [CrossRef]
- Lin, Z.; Lu, T.; Ding, X.-L. A theoretical investigation on doping superalkali for triggering considerable nonlinear optical properties of Si12C12 nanostructure. J. Comput. Chem. 2017, 38, 1574–1582. [Google Scholar] [CrossRef]
- Mohebbi, S.; Boghaei, D.M.; Sarvestani, A.H.; Salimi, A. Oxovanadium(IV) complexes as homogeneous catalyst—Aerobic epoxidation of olefins. Appl. Catal. A Gen. 2005, 278, 263–267. [Google Scholar] [CrossRef]
- Heiko Weiner, A.T.; Finke, R.G. Expanded product, plus kinetic and mechanistic, studies of polyoxoanion-based cyclohexene oxidation catalysis: The detection of ~70 products at higher conversion leading to a simple, product-based test for the presence of olefin autoxidation. J. Mol. Catal. A Chem. 2003, 191, 217–252. [Google Scholar] [CrossRef]
- Birnbaum, E.R.; Grinstaff, M.W.; Labinger, J.A.; Bercaw, J.E.; Gray, H.B. On the mechanism of catalytic alkene oxidation by molecular oxygen and halogenated iron porphyrins. J. Mol. Catal. A Chem. 1995, 104, L119–L122. [Google Scholar] [CrossRef]
- Labinger, J.A. A simplified model for catalyzed isobutane autoxidation. Catal. Lett. 1994, 26, 95–99. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).