

Exploring the Therapeutic Potential of Petiveria alliacea L. Phytochemicals: A Computational Study on Inhibiting SARS-CoV-2’s Main Protease (Mpro)

 ,
,  , and
, and
Abstract
1. Introduction
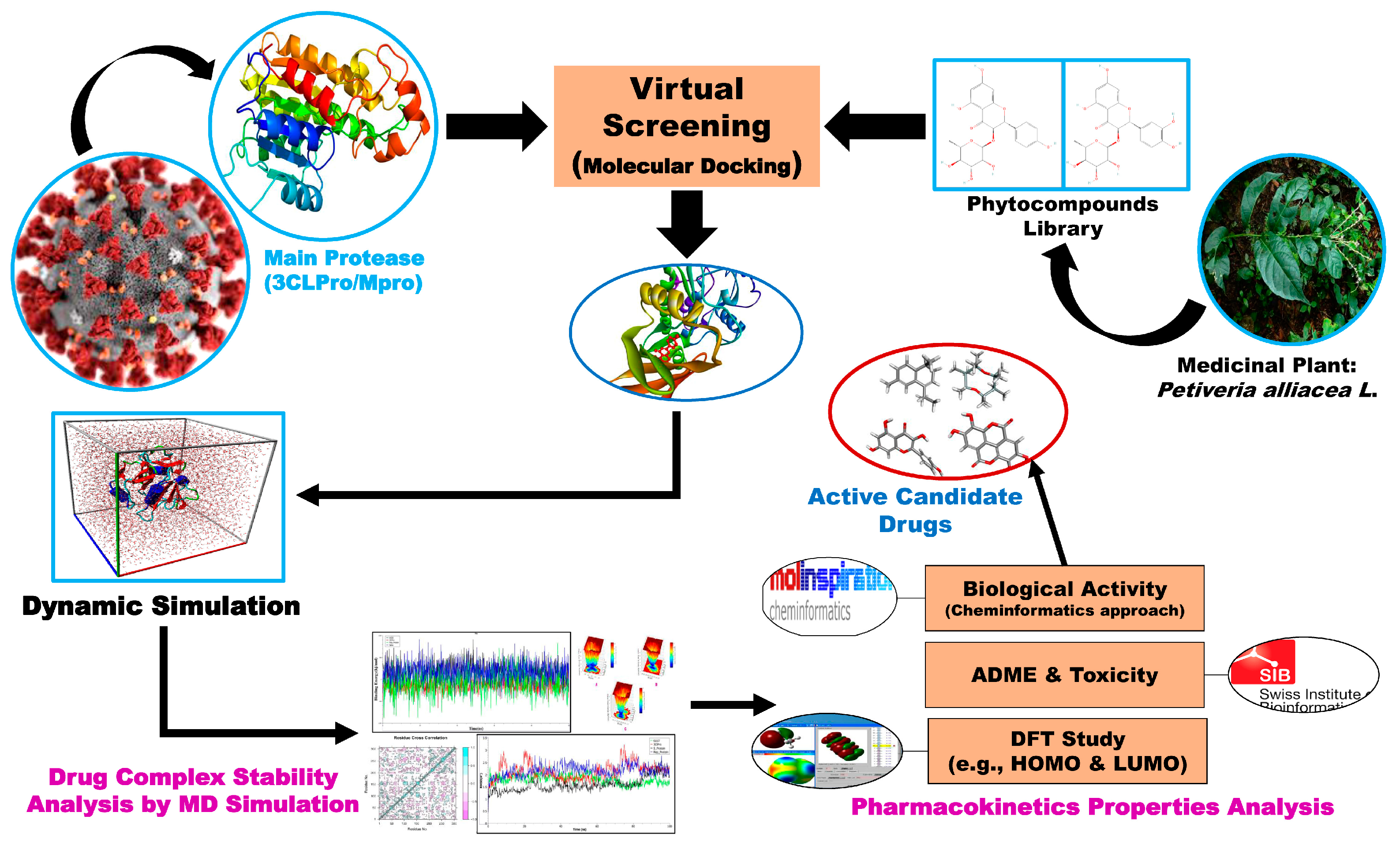
2. Materials and Methods
2.1. Target Protein Retrieval and Preparation
2.2. Phytocompound Collection and Preparation
2.3. Molecular Docking
2.4. Molecular Dynamic Simulation
MM-PBSA-Based Binding Energy Analysis
2.5. Principal Component Analysis (PCA)
Gibbs Free Energy Landscape (FEL) Analyses
2.6. ADMET and Druglikeness Properties Analysis
2.7. Prediction of Biological Activity
2.8. Density Functional Theory Analysis
3. Results and Discussion
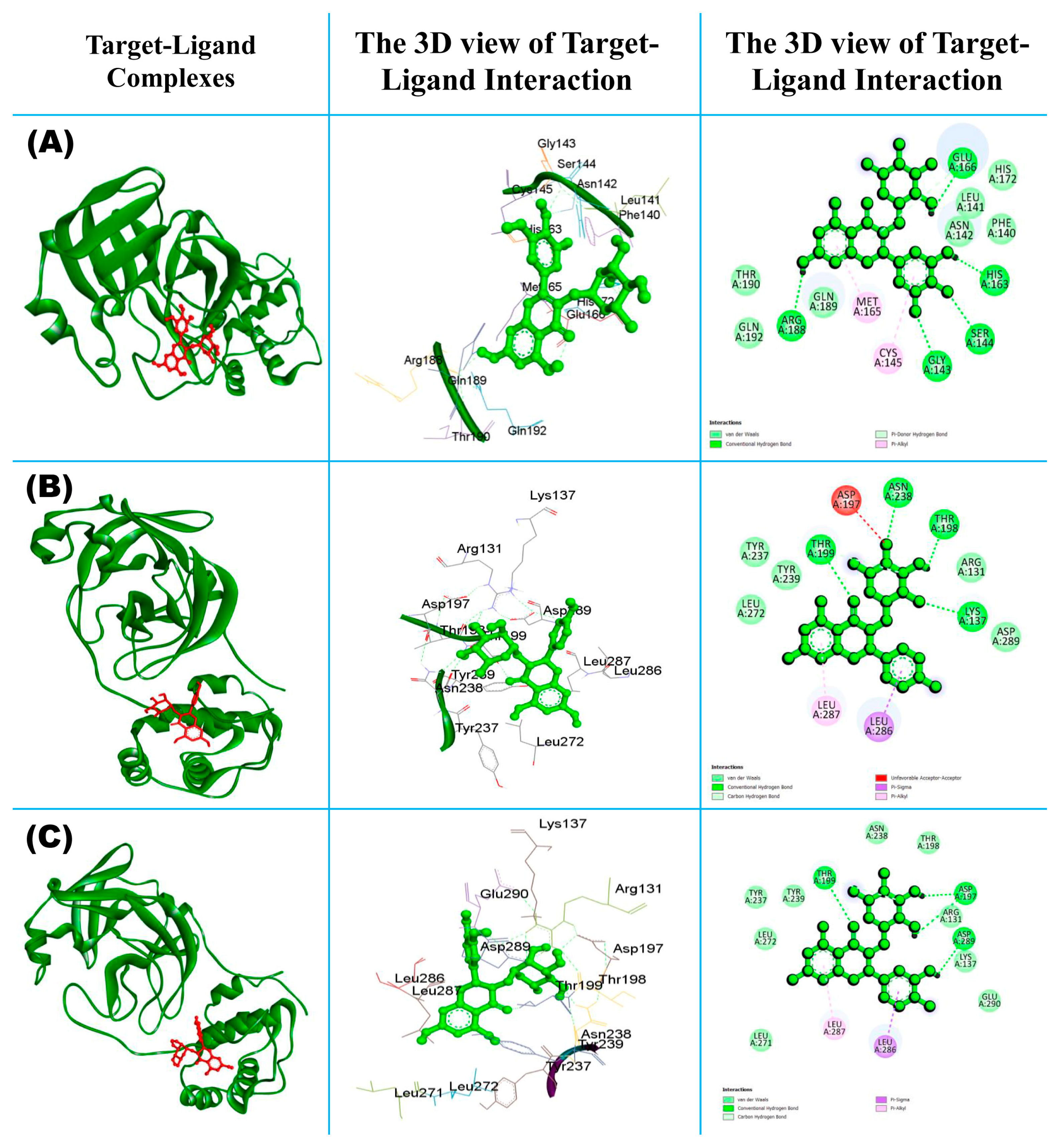
3.1. Molecular Docking between Mpro and Phytocompounds
3.1.1. Molecular Interaction of Top-Ranked Protein–Ligand Complexes
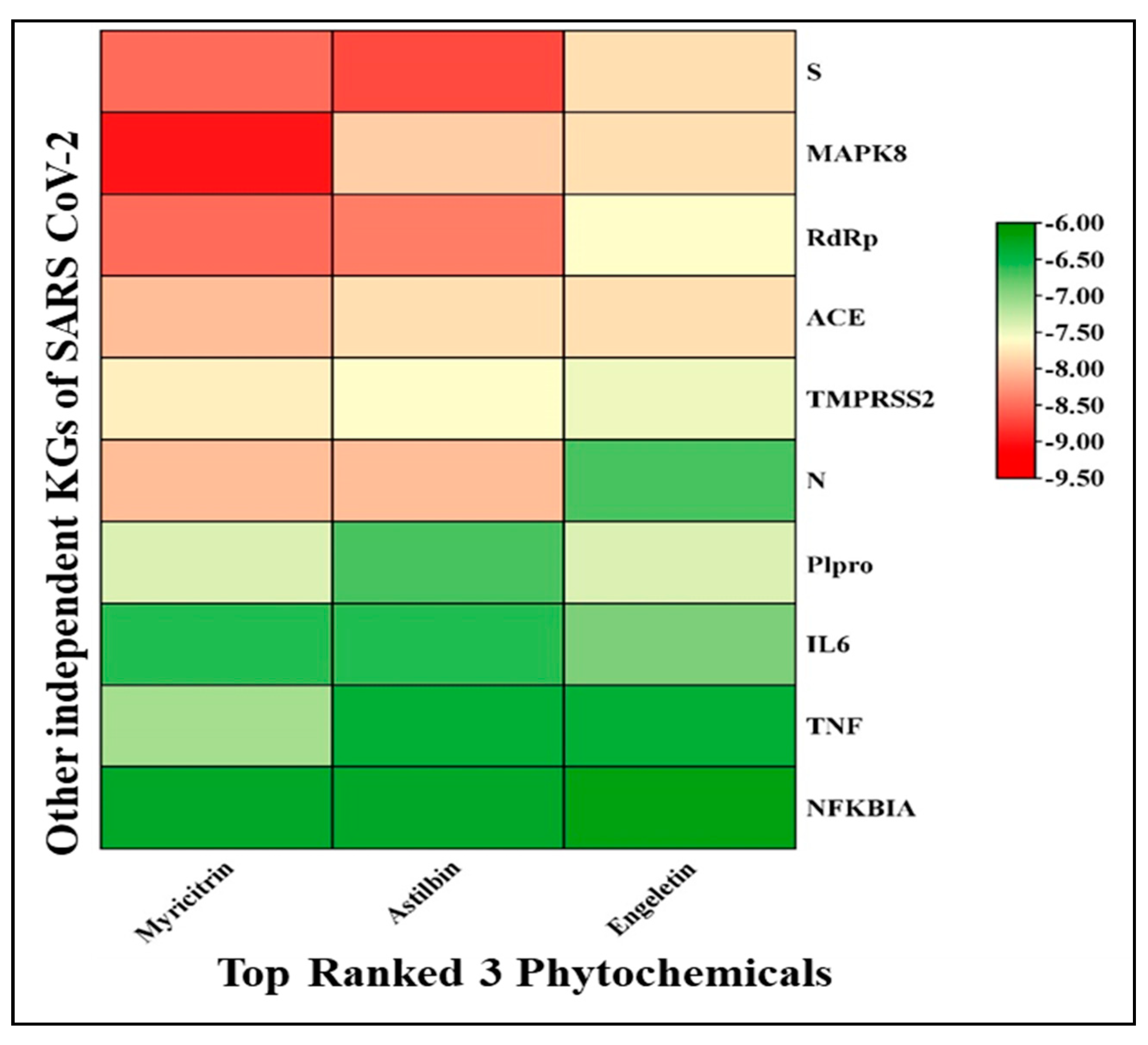
3.1.2. Performance against Some Other SARS-CoV-2 Infection-Causing Genes
3.2. Molecular Dynamics Simulation
3.2.1. Root Mean Square Deviation (RMSD)
3.2.2. The Root Mean Square Fluctuation (RMSF)
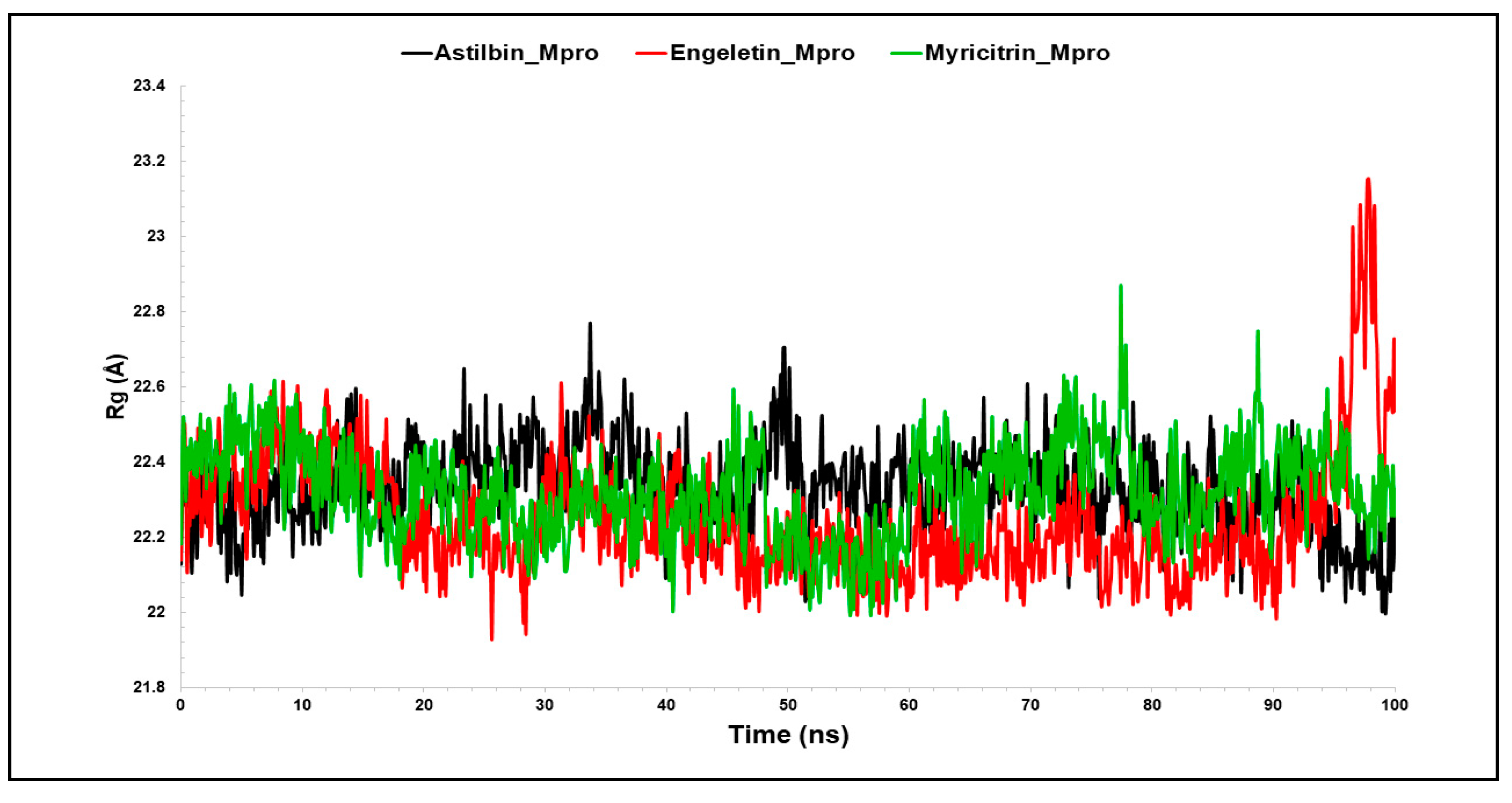
3.2.3. The Radius of Gyration (Rg)
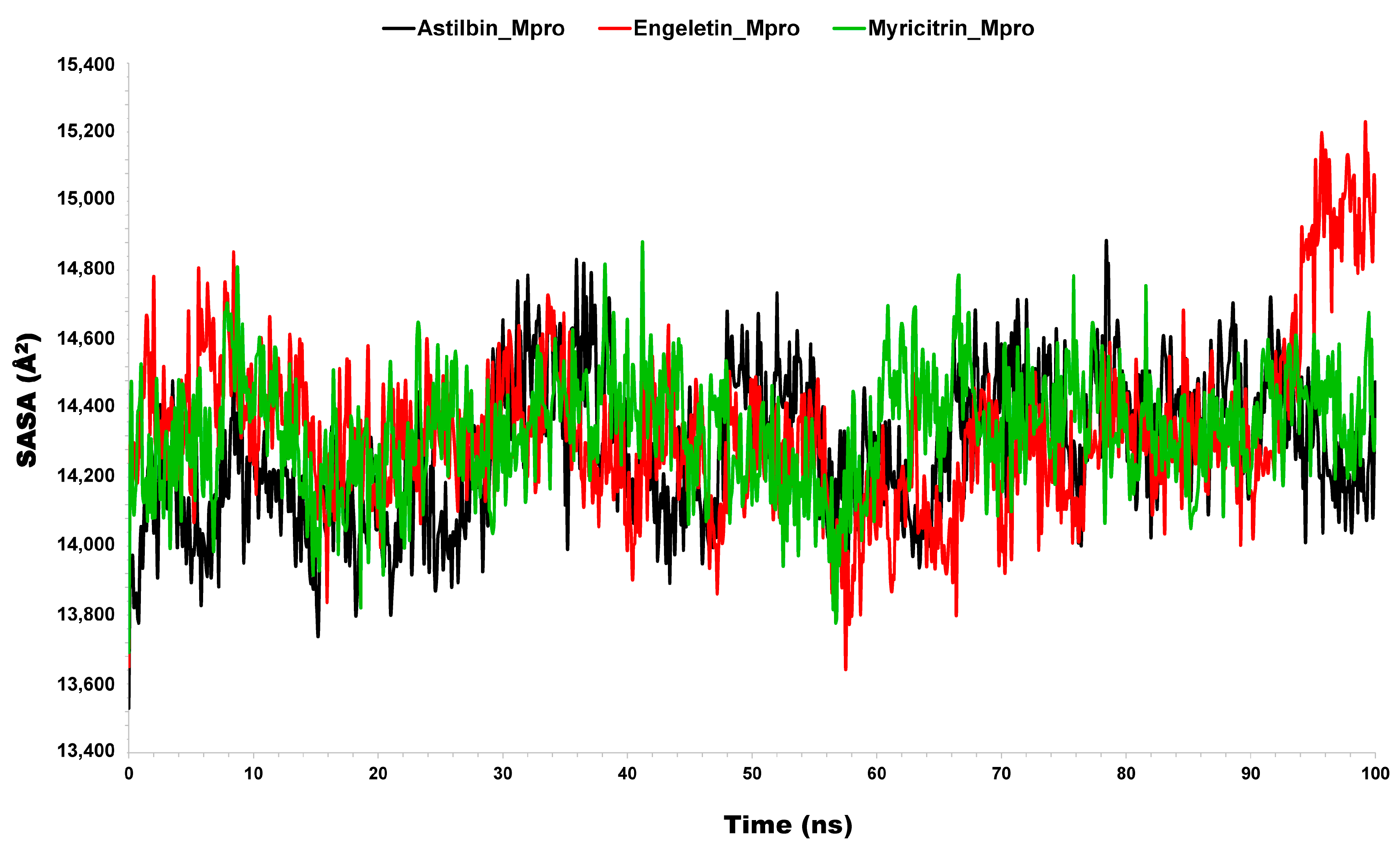
3.2.4. Solvent-Accessible Surface Area (SASA)
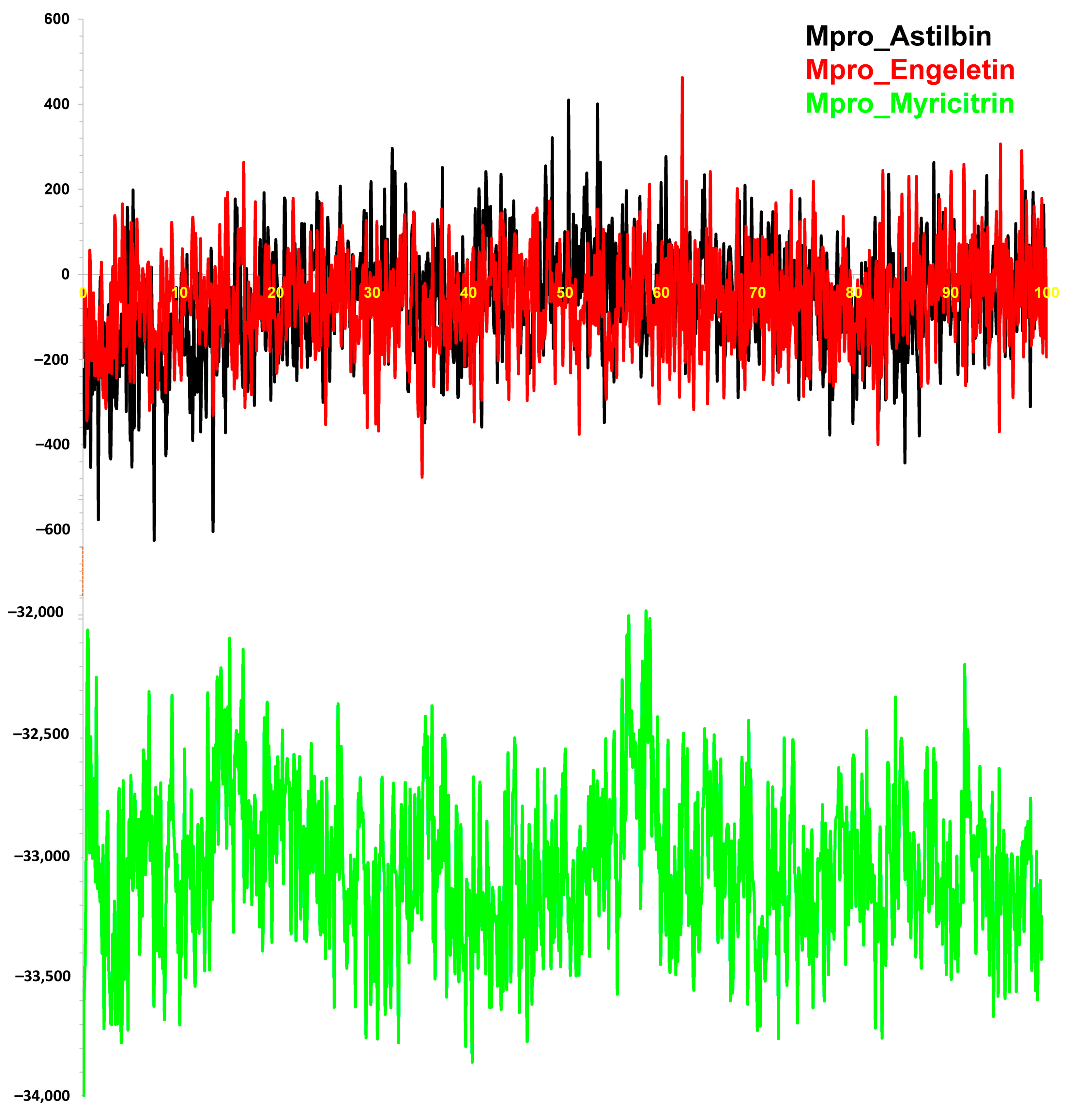
3.2.5. Binding Free Energy (MM-PBSA) Calculation
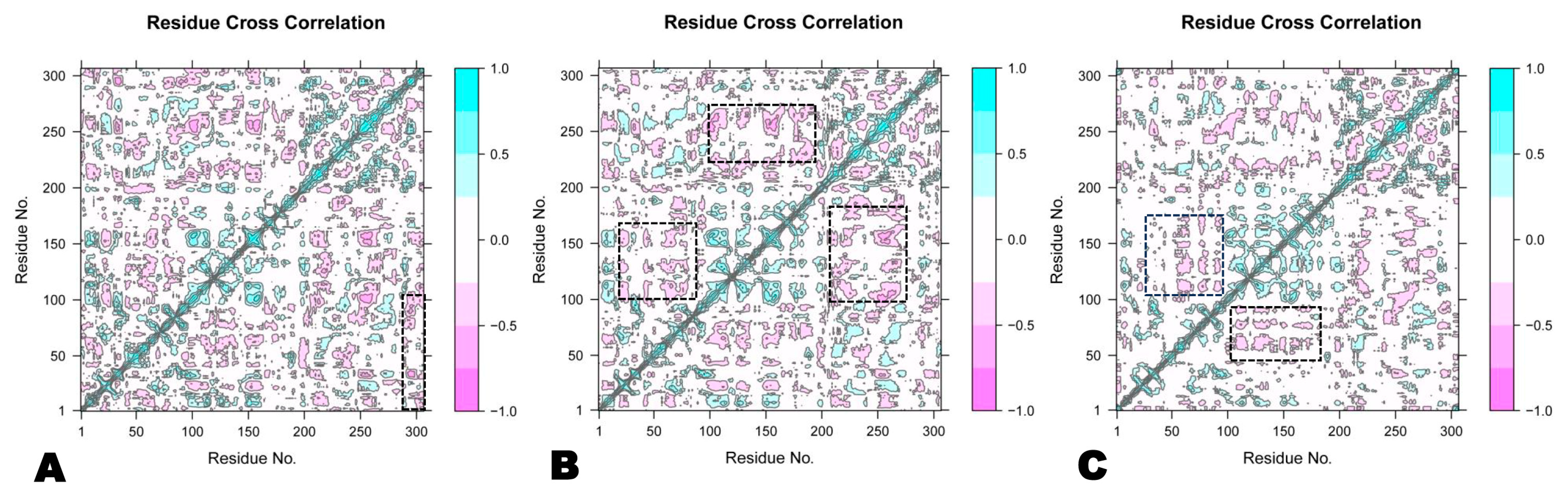
3.2.6. Dynamic Cross-Correlation Matrix
3.2.7. Principal Component Analysis (PCA)
3.2.8. Gibbs Free Energy Landscape (FEL) Analysis
3.3. Pharmacokinetics Properties Analysis
3.3.1. Drug-Likeness Profile (ADMET)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Absorption | Desorption | Excretion | Metabolism CYP | Toxicity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Caco2 Log cm/s | HIA | BBB | VDss | CNS | TC | Substrate | Inhibitors | AMES | Skin Sensitization | ||||||
| 2D6 | 3A4 | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 | |||||||||
| Astilbin | 0.34 | 49.00 | −1.19 | 1.59 | −4.17 | −0.28 | No | No | No | No | No | No | No | No | No |
| Myricitrin | −0.98 | 43.33 | −1.81 | 1.55 | −4.37 | 0.30 | No | No | No | No | No | No | No | No | No |
| Engeletin | 0.41 | 58.66 | −0.99 | 1.12 | −4.01 | 0.05 | No | No | No | No | No | No | No | No | No |
3.3.2. Biological Activity Analysis
3.4. Density Functional Theory (DFT)
3.4.1. Frontier Molecular Orbital Study
3.4.2. Quantum Chemical Descriptors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verma, D.; Mitra, D.; Paul, M.; Chaudhary, P.; Kamboj, A.; Thatoi, H.; Janmeda, P.; Jain, D.; Panneerselvam, P.; Shrivastav, R.; et al. Potential inhibitors of SARS-CoV-2 (COVID-19) proteases PLpro and Mpro/3CLpro: Molecular docking and simulation studies of three pertinent medicinal plant natural components. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100038. [Google Scholar] [CrossRef] [PubMed]
- Astuti, I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, B.; Ma, S.; Wang, H.; Shang, L.; Zhu, C.; Ye, S. Discovery of SARS-CoV-2 3CLPro Peptidomimetic Inhibitors through the Catalytic Dyad Histidine-Specific Protein–Ligand Interactions. Int. J. Mol. Sci. 2022, 23, 2392. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Joshi, T.; Mathpal, S.; Joshi, T.; Pundir, H.; Chandra, S.; Tamta, S. Identification of natural inhibitors against Mpro of SARS-CoV-2 by molecular docking, molecular dynamics simulation, and MM/PBSA methods. J. Biomol. Struct. Dyn. 2022, 40, 2757–2768. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Hassam, M.; Bashir, M.A.; Shafi, S.; Zahra, N.-U.; Khan, K.; Jalal, K.; Siddiqui, H.; Uddin, R. Identification of potent compounds against SARS-CoV-2: An in-silico based drug searching against Mpro. Comput. Biol. Med. 2022, 151, 106284. [Google Scholar] [CrossRef] [PubMed]
- Worldmeter. COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 10 February 2024).
- Muralidharan, N.; Sakthivel, R.; Velmurugan, D.; Gromiha, M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 2673–2678. [Google Scholar] [CrossRef] [PubMed]
- Hamad, M.N.M. Minocycline Superior to Chloroquine Phosphate as COVID-19 Treatment. Saudi J. Biomed. Res. 2020, 5, 46–47. [Google Scholar] [CrossRef]
- Karimi, A.; Tabatabaei, S.R.; Rajabnejad, M.; Pourmoghaddas, Z.; Rahimi, H.; Armin, S.; Ghanaie, R.M.; Kadivar, M.R.; Fahimzad, S.A.; Sedighi, I.; et al. An algorithmic approach to diagnosis and treatment of coronavirus disease 2019 (COVID-19) in children: Iranian expert’s consensus statement. Arch. Pediatr. Infect. Dis. 2020, 8, e102400. [Google Scholar] [CrossRef]
- Alam, M.N. Recent Progress in the Treatment of Coronavirus Disease. OSF Prepr. 2020. [Google Scholar] [CrossRef]
- Esposito, S.; Noviello, S.; Pagliano, P. Update on treatment of COVID-19: Ongoing studies between promising and disappointing results. Infez Med. 2020, 28, 198–211. [Google Scholar]
- Noordin, S.S.; Yusoff, N.M.; Karim, F.A.; Chong, S.E. Blood transfusion services amidst the COVID-19 pandemic. J. Glob. Health 2021, 11, 03053. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Kawase, M.; Nao, N.; Shirato, K.; Ujike, M.; Kamitani, W.; Shimojima, M.; Fukushi, S. The inhaled corticosteroid ciclesonide blocks coronavirus RNA replication by targeting viral NSP15. BioRxiv 2020, 95, e01648-20. [Google Scholar] [CrossRef]
- Avchaciov, K.; Burmistrova, O.; Fedichev, P.O. AI for the repurposing of approved or investigational drugs against COVID-19. Res. Gate 2020, 10. [Google Scholar] [CrossRef]
- Farooq, S.; Ngaini, Z. Natural and Synthetic Drugs as Potential Treatment for Coronavirus Disease 2019 (COVID-2019). Chem. Afr. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Behzadi, A.; Imani, S.; Deravi, N.; Taheri, Z.M.; Mohammadian, F.; Moraveji, Z.; Shavysi, S.; Mostafaloo, M.; Hadidi, F.S.; Nanbakhsh, S.; et al. Antiviral Potential of Melissa officinalis L.: A Literature Review. Nutr. Metab. Insights 2023, 16, 11786388221146683. [Google Scholar] [CrossRef]
- De Sousa, J.R.; Demuner, A.J.; Pinheiro, J.A.; Breitmaier, E.; Cassels, B.K. Dibenzyl trisulphide and trans-N-methyl-4-methoxyproline from Petiveria alliacea. Phytochemistry 1990, 29, 3653–3655. [Google Scholar] [CrossRef]
- Wauchope, S.; Roy, M.A.; Irvine, W.; Morrison, I.; Brantley, E.; Gossell-Williams, M.; Timme-Laragy, A.R.; Delgoda, R. Dibenzyl trisulfide binds to and competitively inhibits the cytochrome P450 1A1 active site without impacting the expression of the aryl hydrocarbon receptor. Toxicol. Appl. Pharmacol. 2021, 419, 115502. [Google Scholar] [CrossRef] [PubMed]
- Hoyos, L.S.; Au, W.W.; Heo, M.Y.; Morris, D.L.; Legator, M.S. Evaluation of the genotoxic effects of a folk medicine, Petiveria alliacea (Anamu). Mutat. Res. Toxicol. 1992, 280, 29–34. [Google Scholar] [CrossRef]
- Alves, T.C.; Rodrigues, E.; Lago, J.H.; Prado, C.M.; Girardi, C.E.N.; Hipólide, D.C. Petiveria alliacea, a plant used in Afro-Brazilian smoke rituals, triggers pulmonary inflammation in rats. Rev. Bras. Farm. 2019, 29, 656–664. [Google Scholar] [CrossRef]
- Kubec, R.; Kim, S.; Musah, R.A. S-Substituted cysteine derivatives and thiosulfinate formation in Petiveria alliacea—Part II. Phytochemistry 2002, 61, 675–680. [Google Scholar] [CrossRef]
- Lopes-Martins, R.; Pegoraro, D.; Woisky, R.; Penna, S.; Sertié, J. The anti-Inflammatory and analgesic effects of a crude extract of Petiveria alliacea L. (Phytolaccaceae). Phytomedicine 2002, 9, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Salomón, K.d.C.; Cruz-Rodríguez, R.I.; Espinosa-Juárez, J.V.; Cruz-Salomón, A.; Briones-Aranda, A.; Ruiz-Lau, N.; Ruíz-Valdiviezo, V.M. In Vivo and In Silico Study of the Antinociceptive and Toxicological Effect of the Extracts of Petiveria alliacea L. Leaves. Pharmaceuticals 2022, 15, 943. [Google Scholar] [CrossRef] [PubMed]
- Zavala-Ocampo, L.M.; Aguirre-Hernández, E.; Érez-Hernández, N.P.; Rivera, G.; Marchat, L.A.; Ramírez-Moreno, E. Antiamoebic Activity of Petiveria alliacea Leaves and Their Main Component, Isoarborinol. J. Microbiol. Biotechnol. 2017, 27, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002; Version 2. [Google Scholar]
- Studio, D. Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5; Accelrys Software Inc.: San Diego, CA, USA, 2015. [Google Scholar]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Murillo, N.; Lasso, P.; Urueña, C.; Pardo-Rodriguez, D.; Ballesteros-Ramírez, R.; Betancourt, G.; Rojas, L.; Cala, M.P.; Fiorentino, S. Petiveria alliacea Reduces Tumor Burden and Metastasis and Regulates the Peripheral Immune Response in a Murine Myeloid Leukemia Model. Int. J. Mol. Sci. 2023, 24, 12972. [Google Scholar] [CrossRef]
- Urueña, C.; Cifuentes, C.; Castañeda, D.; Arango, A.; Kaur, P.; Asea, A.; Fiorentino, S. Petiveria alliacea extracts uses multiple mechanisms to inhibit growth of human and mouse tumoral cells. BMC Complement. Altern. Med. 2008, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Luz, D.A.; Pinheiro, A.M.; Silva, M.L.; Monteiro, M.C.; Prediger, R.D.; Maia, C.S.F.; Fontes-Júnior, E.A. Ethnobotany, phytochemistry and neuropharmacological effects of Petiveria alliacea L. (Phytolaccaceae): A review. J. Ethnopharmacol. 2016, 185, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.A.; Rosner, H.; Levy, H.G.; Barton, E.N. A critical review of the therapeutic potential of dibenzyl trisulphide isolated from Petiveria alliacea L. (guinea hen weed, anamu). West Indian Med. J. 2007, 56, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10039. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Allouche, A. Software News and Updates Gabedit—A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2012, 32, 174–182. [Google Scholar] [CrossRef]
- Oleg, T.; Arthur, J.O. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Hosen, S.M.Z.; Rubayed, M.; Dash, R.; Junaid, M.; Mitra, S.; Alam, M.S.; Dey, R. Prospecting and Structural Insight into the Binding of Novel Plant-Derived Molecules of Leea indica as Inhibitors of BACE1. Curr. Pharm. Des. 2018, 24, 3972–3979. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A., III; Duke, R.E.; Gohlke, H.; Goetz, A.W.; et al. The Amber Molecular Dynamics Package. Amber 2014, 14. [Google Scholar] [CrossRef]
- Gupta, S.; Biswas, A.; Akhter, M.S.; Krettler, C.; Reinhart, C.; Dodt, J.; Reuter, A.; Philippou, H.; Ivaskevicius, V.; Oldenburg, J. Revisiting the mechanism of coagulation factor XIII activation and regulation from a structure/functional perspective. Sci. Rep. 2016, 6, 30105. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Abbasi, H.W.; Shahid, S.; Gul, S.; Abbasi, S.W. Molecular docking, simulation and MM-PBSA studies of Nigella sativa compounds: A computational quest to identify potential natural antiviral for COVID-19 treatment. J. Biomol. Struct. Dyn. 2021, 39, 4225–4233. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. Methods Mol. Biol. 2018, 1685, 2018. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Tavernelli, I.; Cotesta, S.; Di Iorio, E.E. Protein dynamics, thermal stability, and free-energy landscapes: A molecular dynamics investigation. Biophys. J. 2003, 85, 2641–2649. [Google Scholar] [CrossRef]
- R Core Team. Award Number: W81XWH-12-2-0022 TITLE: Prehospital Use of Plasma for Traumatic Hemorrhage PRINCIPAL INVESTIGATOR: Bruce D; Spiess Contracting Organization—Virginia Commonwealth University: Richmond, VA, USA, 2014. [Google Scholar]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- Kagami, L.P.; das Neves, G.M.; Timmers, L.F.S.M.; Caceres, R.A.; Eifler-Lima, V.L. Geo-Measures: A PyMOL plugin for protein structure ensembles analysis. Comput. Biol. Chem. 2020, 87, 107322. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Cheminformatics, M. Molinspiration cheminformatics. Choice Rev. Online 2006, 43, 6538. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Raghavachari, K. Perspective on “Density functional thermochemistry. III. The role of exact exchange”. Theor. Chem. Acc. 2000, 103, 361–363. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision B.01. Gaussian 09, Revis B01; Gaussian, Inc.: Wallingford, UK, 2009; pp. 1–20. [Google Scholar]
- Machado, S.; Camiletti, G.; Neto, A.C.; Jorge, F.; Jorge, R.S. Gaussian basis set of triple zeta valence quality for the atoms from K to Kr: Application in DFT and CCSD(T) calculations of molecular properties. Mol. Phys. 2009, 107, 1713–1727. [Google Scholar] [CrossRef]
- Rahman, M.A.; Chakma, U.; Kumer, A.; Rahman, M.R.; Matin, M.M. Uridine-Derived 4-Aminophenyl 1-Thioglucosides: DFT Optimized FMO, ADME, and Antiviral Activities Study. Biointerface Res. Appl. Chem. 2023, 13, 52. [Google Scholar] [CrossRef]
- Tariq, A.; Nazir, S.; Arshad, A.W.; Nawaz, F.; Ayub, K.; Iqbal, J. DFT study of the therapeutic potential of phosphorene as a new drug-delivery system to treat cancer. RSC Adv. 2019, 9, 24325–24332. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. Gauss View; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- El-Shamy, N.T.; Alkaoud, A.M.; Hussein, R.K.; Ibrahim, M.A.; Alhamzani, A.G.; Abou-Krisha, M.M. DFT, ADMET and Molecular Docking Investigations for the Antimicrobial Activity of 6, 6′-Diamino-1, 1′, 3, 3′-tetramethyl-5, 5′-(4-chlorobenzylidene) bis [pyrimidine-2, 4 (1H, 3H)-dione]. Molecules 2022, 27, 620. [Google Scholar] [CrossRef]
- Odhar, H.A. Molecular docking analysis and dynamics simulation of salbutamol with the monoamine oxidase B (MAO-B) enzyme. Bioinformation 2022, 18, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Dhorajiwala, T.M.; Halder, S.T.; Samant, L. Comparative in silico molecular docking analysis of l-threonine-3-dehydrogenase, a protein target against African trypanosomiasis using selected phytochemicals. J. Appl. Biotechnol. Rep. 2019, 6, 101–108. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef]
- Tran, Q.-H.; Nguyen, Q.-T.; Vo, N.-Q.; Mai, T.T.; Tran, T.-T.; Tran, T.-D.; Le, M.-T.; Trinh, D.-T.T.; Thai, K.-M. Structure-based 3D-Pharmacophore modeling to discover novel interleukin 6 inhibitors: An in silico screening, molecular dynamics simulations and binding free energy calculations. PLoS ONE 2022, 17, e0266632. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.Z.; Al-gawati, M.; Poirier, R.A. Investigating the Potential of 6-Substituted 3-Formyl Chromone Derivatives as Anti-Diabetic Agents Using DFT, Molecular Docking and Molecular Dynamics Methods n.d.:1–32. Preprint (Version 1) Available at Research Square. Available online: https://www.researchsquare.com/article/rs-3257298/v1 (accessed on 10 April 2024).
- Joshi, T.; Sharma, P.; Joshi, T.; Pundir, H.; Mathpal, S.; Chandra, S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2021, 25, 1665–1677. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Bahadur, R.P.; Chakrabarti, P.; Janin, J. Hydration of protein–protein interfaces. Proteins Struct. Funct. Bioinform. 2005, 60, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Charles, S.; Edgar, M.P. Geometric Deep Learning Prioritization and Validation of Cannabis Phytochemicals as Anti-HCV Non-Nucleoside Direct-Acting Inhibitors 2024. Preprint (Version 1) Available at Research Square. Available online: https://assets.researchsquare.com/files/rs-3961716/v1/1b19e533-615e-4f50-8be5-768fe8652848.pdf?c=1712713502 (accessed on 10 April 2024).
- Gerlt, J.A.; Kreevoy, M.M.; Cleland, W.; Frey, P.A. Understanding enzymic catalysis: The importance of short, strong hydrogen bonds. Chem. Biol. 1997, 4, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Moradi, S.; Shahlaei, M. A molecular dynamics simulation study on the mechanism of loading of gemcitabine and camptothecin in poly lactic-co-glycolic acid as a nano drug delivery system. J. Mol. Liq. 2018, 269, 110–118. [Google Scholar] [CrossRef]
- Akash, S.; Hosen, E.; Mahmood, S.; Supti, S.J.; Kumer, A.; Sultana, S.; Jannat, S.; Bayıl, I.; Nafidi, H.-A.; Bin Jardan, Y.A.; et al. Anti-parasitic drug discovery against Babesia microti by natural compounds: An extensive computational drug design approach. Front. Cell. Infect. Microbiol. 2023, 13, 1222913. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Ghosh, A.; Yan, H. Correlated Motions and Dynamics in Different Domains of Epidermal Growth Factor Receptor with L858R and T790M Mutations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Akash, S.; Bayıl, I.; Hossain, S.; Islam, R.; Hosen, E.; Mekonnen, A.B.; Nafidi, H.-A.; Bin Jardan, Y.A.; Bourhia, M.; Bin Emran, T. Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and DNA polymerase theta receptor by Apigenin derivatives. Sci. Rep. 2023, 13, 16565. [Google Scholar] [CrossRef] [PubMed]
- Ngidi, N.T.P.; Machaba, K.E.; Mhlongo, N.N. In Silico Drug Repurposing Approach: Investigation of Mycobacterium tuberculosis FadD32 Targeted by FDA-Approved Drugs. Molecules 2022, 27, 668. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, K.K.; Ahmad, I.; Pati, S.; Ghosh, A.; Sarkar, T.; Rabha, B.; Patel, H.; Baishya, D.; Edinur, H.A.; Kari, Z.A.; et al. Potent Bioactive Compounds From Seaweed Waste to Combat Cancer Through Bioinformatics Investigation. Front. Nutr. 2022, 9, 889276. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.K.; Das, P.; Purohit, R. A computational approach for rational discovery of inhibitors for non-structural protein 1 of SARS-CoV-2. Comput. Biol. Med. 2021, 135, 104555. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, D.; Canipa, S.; Wolber, G. HuskinDB, a database for skin permeation of xenobiotics. Sci. Data 2020, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Alvi, S.S.; Azaz, T.; Khan, A.R.; Tiwari, B.; Bin Hafeez, B.; Nasibullah, M. Novel Functionalized Spiro [Indoline-3,5′-pyrroline]-2,2′dione Derivatives: Synthesis, Characterization, Drug-Likeness, ADME, and Anticancer Potential. Int. J. Mol. Sci. 2023, 24, 7336. [Google Scholar] [CrossRef] [PubMed]
- Netzeva, T.I.; Worth, A.P.; Aldenberg, T.; Benigni, R.; Cronin, M.T.; Gramatica, P.; Jaworska, J.S.; Kahn, S.; Klopman, G.; Marchant, C.A.; et al. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52. Altern. Lab. Anim. 2005, 33, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Boulenc, X.; Nicolas, O.; Hermabessière, S.; Zobouyan, I.; Martin, V.; Donazzolo, Y.; Ollier, C. CYP3A4-based drug–drug interaction: CYP3A4 substrates’ pharmacokinetic properties and ketoconazole dose regimen effect. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Yergaliyeva, E.M.; Bazhykova, K.B.; Abeuova, S.B.; Vazhev, V.V.; Langer, P. In silico drug-likeness, biological activity and toxicity prediction of new 3,5-bis(hydroxymethyl)tetrahydro-4H-pyran-4-one derivatives. Chem. Bull. Kazakh Natl. Univ. 2022, 107, 14–20. [Google Scholar] [CrossRef]
- Velloso, J.P.L.; Ascher, D.B.; Pires, D.E.V. pdCSM-GPCR: Predicting potent GPCR ligands with graph-based signatures. Bioinform. Adv. 2021, 1, vbab031. [Google Scholar] [CrossRef]
- Mahmud, S.; Afrose, S.; Biswas, S.; Nagata, A.; Paul, G.K.; Mita, M.A.; Hasan, R.; Shimu, M.S.S.; Zaman, S.; Uddin, S.; et al. Plant-derived compounds effectively inhibit the main protease of SARS-CoV-2: An in silico approach. PLoS ONE 2022, 17, e0273341. [Google Scholar] [CrossRef]
- Jordaan, M.A.; Ebenezer, O.; Damoyi, N.; Shapi, M. Virtual screening, molecular docking studies and DFT calculations of FDA approved compounds similar to the non-nucleoside reverse transcriptase inhibitor (NNRTI) efavirenz. Heliyon 2020, 6, e04642. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T.; Song, J.-W.; Suzuki, S.; Hirao, K. On Koopmans’ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.M.; Farag, A.M. Synthesis, antimicrobial evaluation, molecular docking and theoretical calculations of novel pyrazolo [1,5-a]pyrimidine derivatives. J. Mol. Struct. 2020, 1199, 127025. [Google Scholar] [CrossRef]
- Karton, A.; Spackman, P.R. Evaluation of density functional theory for a large and diverse set of organic and inorganic equilibrium structures. J. Comput. Chem. 2021, 42, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Szymański, S.; Majerz, I. Theoretical Studies on the Structure and Intramolecular Interactions of Fagopyrins—Natural Photosensitizers of Fagopyrum. Molecules 2022, 27, 3689. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R. Theoretical Studies on the Molecular Properties, Toxicity, and Biological Efficacy of 21 New Chemical Entities. ACS Omega 2021, 6, 24891–24901. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-J.; Chao, T.-L.; Kao, H.-C.; Tsai, Y.-M.; Liu, Y.-K.; Wang, L.H.-C.; Hsieh, M.-C.; Chang, S.-Y.; Liang, P.-H. Kinetic characterization and inhibitor screening for the proteases leading to identification of drugs against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 18, e02577-20. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Di Filippo, J.I. In silico Drug Repurposing for COVID-19: Targeting SARS-CoV-2 Proteins through Docking and Consensus Ranking. Mol. Inform. 2021, 40, 2000115. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- Guedes, I.A.; Costa, L.S.C.; dos Santos, K.B.; Karl, A.L.M.; Rocha, G.K.; Teixeira, I.M.; Galheigo, M.M.; Medeiros, V.; Krempser, E.; Custodio, F.L.; et al. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci. Rep. 2021, 11, 1–20. [Google Scholar] [CrossRef]
- Liang, H.; Zhao, L.; Gong, X.; Hu, M.; Wang, H. Virtual screening FDA approved drugs against multiple targets of SARS-CoV-2. Clin. Transl. Sci. 2021, 14, 1123–1132. [Google Scholar] [CrossRef]
- Rahman, F.; Tabrez, S.; Ali, R.; Alqahtani, A.S.; Ahmed, M.Z.; Rub, A. Molecular docking analysis of rutin reveals possible inhibition of SARS-CoV-2 vital proteins. J. Tradit. Complement. Med. 2021, 11, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Kumar, S.; Jeyakanthan, J.; Srivastava, V. Searching for target-specific and multi-targeting organics for COVID-19 in the Drugbank database with a double scoring approach. Sci. Rep. 2020, 10, 19125. [Google Scholar] [CrossRef]
- Manikyam, H.K.; Joshi, S.K. Whole Genome Analysis and Targeted Drug Discovery Using Computational Methods and High Throughput Screening Tools for Emerged Novel Coronavirus (2019-nCoV). J. Pharm. Drug Res. 2020, 3, 341–361. [Google Scholar]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Basset, M.; Hawash, H.; Elhoseny, M.; Chakrabortty, R.K.; Ryan, M. Deeph-DTA: Deep learning for predicting drug-target interactions: A case study of COVID-19 drug repurposing. IEEE Access 2020, 8, 170433–170451. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, K.; Maskey, A.R.; Huang, W.; Toutov, A.A.; Yang, N.; Srivastava, K.; Geliebter, J.; Tiwari, R.; Miao, M.; et al. A small molecule compound berberine as an orally active therapeutic candidate against COVID-19 and SARS: A computational and mechanistic study. FASEB J. 2021, 35, e21360. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Pandey, P.; Sharma, R.D.; Malik, Z.; Mongre, R.K.; Lynn, A.M.; Prasad, R.; Jeon, R.; Prakash, A. Identifying the natural polyphenol catechin as a multi-targeted agent against SARS-CoV-2 for the plausible therapy of COVID-19: An integrated computational approach. Briefings Bioinform. 2021, 22, 1346–1360. [Google Scholar] [CrossRef] [PubMed]
- Nelakuditi, B.; Shrivastava, A. Drug Repurposing 57 well-known drugs for three COVID-19 targets: Mpro, Spike, RdRp. Biol. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Mhatre, S.; Naik, S.; Patravale, V. A molecular docking study of EGCG and theaflavin digallate with the druggable targets of SARS-CoV-2. Comput. Biol. Med. 2021, 129, 104137. [Google Scholar] [CrossRef]
- Joshi, R.S.; Jagdale, S.S.; Bansode, S.B.; Shankar, S.S.; Tellis, M.B.; Pandya, V.K.; Chugh, A.; Giri, A.P.; Kulkarni, M.J. Discovery of potential multi-target-directed ligands by targeting host-specific SARS-CoV-2 structurally conserved main protease. J. Biomol. Struct. Dyn. 2020, 39, 3099–3114. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.-M.; Guo, R.; Wang, Q.; Zhou, J.-R. Screening and molecular modeling evaluation of food peptides to inhibit key targets of COVID-19 virus. Biomolecules 2021, 11, 330. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.-G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef] [PubMed]
- Jena, S.; Munusami, P.; Mm, B.; Chanda, K. Computationally approached inhibition potential of Tinospora cordifolia towards COVID-19 targets. VirusDisease 2021, 32, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Du, J.; Li, X.; Zeng, J.; Tan, B.; Xu, J.; Lin, W.; Chen, X.-L. Network pharmacology and molecular docking analysis on molecular targets and mechanisms of Huashi Baidu formula in the treatment of COVID-19. Drug Dev. Ind. Pharm. 2020, 46, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Duverger, E.; Herlem, G.; Picaud, F. A potential solution to avoid overdose of mixed drugs in the event of COVID-19: Nanomedicine at the heart of the COVID-19 pandemic. J. Mol. Graph. Model. 2021, 104, 107834. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.; Zhang, L.; Ran, Q.; Xiong, A.; Liu, S.; Wu, D.; Niu, B.; Xiong, Y.; Li, G. Virtual Screening of Potential AEC2 Inhibitors for COVID-19 from Traditional Chinese Medicine. 2021. Available online: https://www.researchsquare.com/article/rs-145338/v1 (accessed on 10 April 2024).
- Li, Z.; Yang, L. Underlying Mechanisms and Candidate Drugs for COVID-19 Based on the Connectivity Map Database. Front. Genet. 2020, 11, 558557. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wei, X.-X.; Zheng, Y.-J.; Zhang, L.-L.; Wang, X.-M.; Yang, H.-Y.; Ma, X.; Zhao, L.-H.; Tong, X.-L. Potential mechanism prediction of Cold-Damp Plague Formula against COVID-19 via network pharmacology analysis and molecular docking. Chin. Med. 2020, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, O.V.; Rocha, G.B.; Paluch, A.S.; Costa, L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020, 39, 3924–3933. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Hajjo, R.; Sabbah, D.A. Sitagliptin: A potential drug for the treatment of COVID-19? Acta Pharm. 2021, 71, 175–184. [Google Scholar] [CrossRef]
- Kabir, E.R.; Mustafa, N.; Nausheen, N.; Siam, M.K.S.; Syed, E.U. Exploring existing drugs: Proposing potential compounds in the treatment of COVID-19. Heliyon 2021, 7, e06284. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Aftab, S.O.; Ghouri, M.Z.; Masood, M.U.; Haider, Z.; Khan, Z.; Ahmad, A.; Munawar, N. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. 2020, 18, 275. [Google Scholar] [CrossRef]
- Elfiky, A. A SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2021, 39, 3204–3212. [Google Scholar] [CrossRef]
- Pirzada, R.H.; Haseeb, M.; Batool, M.; Kim, M.; Choi, S. Remdesivir and Ledipasvir among the FDA-Approved Antiviral Drugs Have Potential to Inhibit SARS-CoV-2 Replication. Cells 2021, 10, 1052. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, L.; Poullikkas, T.; Eisenhower, S.; Monsanto, C.; Bakku, R.K.; Chen, M.-H.; Kalra, R.S. Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for Potential Therapeutic Targets. Antibodies 2021, 10, 3. [Google Scholar] [CrossRef]
- Sun, Y.J.; Velez, G.; Parsons, D.E.; Li, K.; Ortiz, M.E.; Sharma, S.; McCray, P.B.; Bassuk, A.G.; Mahajan, V.B. Structure-based phylogeny identifies avoralstat as a TMPRSS2 inhibitor that prevents SARS-CoV-2 infection in mice. J. Clin. Investig. 2021, 131, e147973. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.; Han, H.-S.; Jeong, J.; Park, E.-M.; Shim, K.-S. A novel computational approach for the discovery of drug delivery system candidates for COVID-19. Int. J. Mol. Sci. 2021, 22, 2815. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2020, 11, 237–245. [Google Scholar] [CrossRef]
- Zhao, Y.; Du, X.; Duan, Y.; Pan, X.; Sun, Y.; You, T.; Han, L.; Jin, Z.; Shang, W.; Yu, J.; et al. High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell 2021, 12, 877–888. [Google Scholar] [CrossRef]
- Weglarz-Tomczak, E.; Tomczak, J.M.; Talma, M.; Burda-Grabowska, M.; Giurg, M.; Brul, S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021, 11, 3640. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pei, R.-J.; Li, H.; Ma, X.-N.; Zhou, Y.; Zhu, F.-H.; He, P.-L.; Tang, W.; Zhang, Y.-C.; Xiong, J.; et al. Identification of SARS-CoV-2 entry inhibitors among already approved drugs. Acta Pharmacol. Sin. 2020, 42, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, Y.; Yang, Y.; Li, M.; Du, Y.; Zhang, Y.; Wang, J.; Shi, Y. Study on mechanism of matrine in treatment of COVID-19 combined with liver injury by network pharmacology and molecular docking technology. Drug Deliv. 2021, 28, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ruan, S.; Zhao, X.; Liu, Q.; Dou, Y.; Mao, F. Transcriptomic signatures and repurposing drugs for COVID-19 patients: Findings of bioinformatics analyses. Comput. Struct. Biotechnol. J. 2020, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, K.M.; Farah, M.A.; Hor, Y.-Y. Multi-targeted approaches and drug repurposing reveal possible SARS-CoV-2 inhibitors. Vaccines 2021, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Jose, S.; Gupta, M.; Sharma, U.; Quintero-Saumeth, J.; Dwivedi, M. Potential of phytocompounds from Brassica oleracea targeting S2-domain of SARS-CoV-2 spike glycoproteins: Structural and molecular insights. J. Mol. Struct. 2022, 1254, 132369. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Banerjee, A.; Nazmeen, A.; Kanwar, M.; Das, S. Active-site Molecular docking of Nigellidine with nucleocapsid- NSP2-MPro of COVID-19 and to human IL1R-IL6R and strong antioxidant role of Nigella-sativa in experimental rats. J. Drug Target. 2022, 30, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, B.; Xiao, Z.; Zhou, M.; Ge, L.; Jia, F.; Liu, Y.; Jin, H.; Zhu, X.; Gao, J.; et al. Computational and Experimental Studies Reveal That Thymoquinone Blocks the Entry of Coronaviruses into In Vitro Cells. Infect. Dis. Ther. 2021, 10, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.J.; Sehgal, N.; Dogra, N.; Saxena, S.; Katare, D.P. Deciphering underlying mechanism of SARS-CoV-2 infection in humans and revealing the therapeutic potential of bioactive constituents from Nigella sativa to combat COVID-19: In-silico study. J. Biomol. Struct. Dyn. 2020, 40, 2417–2429. [Google Scholar] [CrossRef]
- Mir, S.A.; Firoz, A.; Alaidarous, M.; Alshehri, B.; Bin Dukhyil, A.A.; Banawas, S.; A Alsagaby, S.; Alturaiki, W.; Bhat, G.A.; Kashoo, F.; et al. Identification of SARS-CoV-2 RNA-dependent RNA polymerase inhibitors from the major phytochemicals of Nigella sativa: An in silico approach. Saudi J. Biol. Sci. 2021, 29, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Banerjee, A.; Kanwar, M. In silico Nigellidine (N. sativa) bind to viral spike/active-sites of ACE1/2, AT1/2 to prevent COVID-19 induced vaso-tumult/vascular-damage/comorbidity. Vascul. Pharmacol. 2020, 138, 106856. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Ali, A.; Wei, X.; Nadeem, T.; Muhammad, S.; Al-Sehemi, A.G.; Wei, D. Inhibitory effect of thymoquinone from Nigella sativa against SARS-CoV-2 main protease. An in-silico study. Braz. J. Biol. 2024, 84, e250667. [Google Scholar] [CrossRef] [PubMed]
- Ullah, S.; Munir, B.; Al-Sehemi, A.G.; Muhammad, S.; Haq, I.-U.; Aziz, A.; Ahmed, B.; Ghaffar, A. Identification of phytochemical inhibitors of SARS-CoV-2 protease 3CLpro from selected medicinal plants as per molecular docking, bond energies and amino acid binding energies. Saudi J. Biol. Sci. 2022, 29, 103274. [Google Scholar] [CrossRef]
- Siddiqui, S.; Upadhyay, S.; Ahmad, R.; Gupta, A.; Srivastava, A.; Trivedi, A.; Husain, I.; Ahmad, B.; Ahamed, M.; Khan, M.A. Virtual screening of phytoconstituents from miracle herb nigella sativa targeting nucleocapsid protein and papain-like protease of SARS-CoV-2 for COVID-19 treatment. J. Biomol. Struct. Dyn. 2020, 40, 3928–3948. [Google Scholar] [CrossRef]





| Name of Complex | MD Simulation Study | |||||
|---|---|---|---|---|---|---|
| Average RMSD (SD) | Average RMSF (SD) | Average Rg (SD) | Average SASA (SD) | Average H-Bonds (SD) | Average Binding Energy (SD) | |
| Mpro vs. Astilbin | 1.81 (0.30) | 1.40 (1.04) | 22.34 (0.16) | 14,283.55 (213.76) | 12.82 (1.91) | −63.08 (133.93) |
| Mpro vs. Engeletin | 2.40 (0.35) | 1.32 (0.93) | 22.24 (0.16) | 14,329.47 (234.89) | 12.19 (1.71) | −60.88 (117.51) |
| Mpro vs. Myricitrin | 2.16 (0.25) | 1.30 (0.82) | 22.32 (0.12) | 14,335.72 (163.21) | 12.34 (1.93) | −33,042.16 (556.96) |
| Compound | Molecular Weight | LogPo/w | NHBA | NHBD | Log Kp (Cm/S) | Lipinski’s Rule | Synthetic Accessibility | |
|---|---|---|---|---|---|---|---|---|
| Follow | Violation | |||||||
| Astilbin | 450.396 | 0.038 | 10 | 7 | −2.735 | 4 | 1 | 5.27 |
| Myricitrin | 464.379 | 0.194 | 12 | 8 | −2.735 | 3 | 2 | 5.32 |
| Engeletin | 434.397 | 0.333 | 10 | 5 | −2.735 | 5 | 0 | 5.20 |
| Compounds | GPCR Ligand | Ion Channel Inhibitor | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|
| Astilbin | 0.11 | 0.05 | 0.03 | 0.12 | 0.15 | 0.33 |
| Myricitrin | −0.02 | −0.08 | 0.08 | 0.14 | −0.06 | 0.38 |
| Engeletin | 0.10 | 0.03 | 0.04 | 0.11 | 0.17 | 0.34 |
| Parameter | Astilbin | Engeletin | Myricitrin |
|---|---|---|---|
| Optimized energy (a.u.) | −1640.846 | −1565.628 | −1714.364 |
| Dipole moment, D | 3.1739 | 2.8213 | 5.0897 |
| HOMO energy (EHOMO) | −0.236 | −0.245 | −0.216 |
| LUMO energy (ELUMO) | −0.0846 | −0.0857 | −0.0695 |
| Energy gap (∆E = ELUMO − EHOMO) | 0.151 | 0.159 | 0.147 |
| Ionization potential (I) | 0.236 | 0.245 | 0.216 |
| Electron affinity (A) | 0.0846 | 0.0857 | 0.0695 |
| Chemical hardness (η) | 0.0757 | 0.0797 | 0.0733 |
| Softness (σ) | 13.21 | 12.55 | 13.64 |
| Electro-negativity (χ) | 0.160 | 0.165 | 0.142 |
| Chemical potential (μ) | −0.160 | −0.165 | −0.142 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.A.; Sheikh, H.; Yaseen, M.; Faruqe, M.O.; Ullah, I.; Kumar, N.; Bhat, M.A.; Mollah, M.N.H. Exploring the Therapeutic Potential of Petiveria alliacea L. Phytochemicals: A Computational Study on Inhibiting SARS-CoV-2’s Main Protease (Mpro). Molecules 2024, 29, 2524. https://doi.org/10.3390/molecules29112524
Ali MA, Sheikh H, Yaseen M, Faruqe MO, Ullah I, Kumar N, Bhat MA, Mollah MNH. Exploring the Therapeutic Potential of Petiveria alliacea L. Phytochemicals: A Computational Study on Inhibiting SARS-CoV-2’s Main Protease (Mpro). Molecules. 2024; 29(11):2524. https://doi.org/10.3390/molecules29112524
Chicago/Turabian StyleAli, Md. Ahad, Humaira Sheikh, Muhammad Yaseen, Md Omar Faruqe, Ihsan Ullah, Neeraj Kumar, Mashooq Ahmad Bhat, and Md. Nurul Haque Mollah. 2024. "Exploring the Therapeutic Potential of Petiveria alliacea L. Phytochemicals: A Computational Study on Inhibiting SARS-CoV-2’s Main Protease (Mpro)" Molecules 29, no. 11: 2524. https://doi.org/10.3390/molecules29112524
APA StyleAli, M. A., Sheikh, H., Yaseen, M., Faruqe, M. O., Ullah, I., Kumar, N., Bhat, M. A., & Mollah, M. N. H. (2024). Exploring the Therapeutic Potential of Petiveria alliacea L. Phytochemicals: A Computational Study on Inhibiting SARS-CoV-2’s Main Protease (Mpro). Molecules, 29(11), 2524. https://doi.org/10.3390/molecules29112524