
Trichostatin A Promotes Cytotoxicity of Cisplatin, as Evidenced by Enhanced Apoptosis/Cell Death Markers

 ,
,
Abstract

1. Introduction
2. Results
2.1. TSA Promoted Cisplatin-Induced Apoptosis and Arrest at the G2/M Phase of A549 Cells
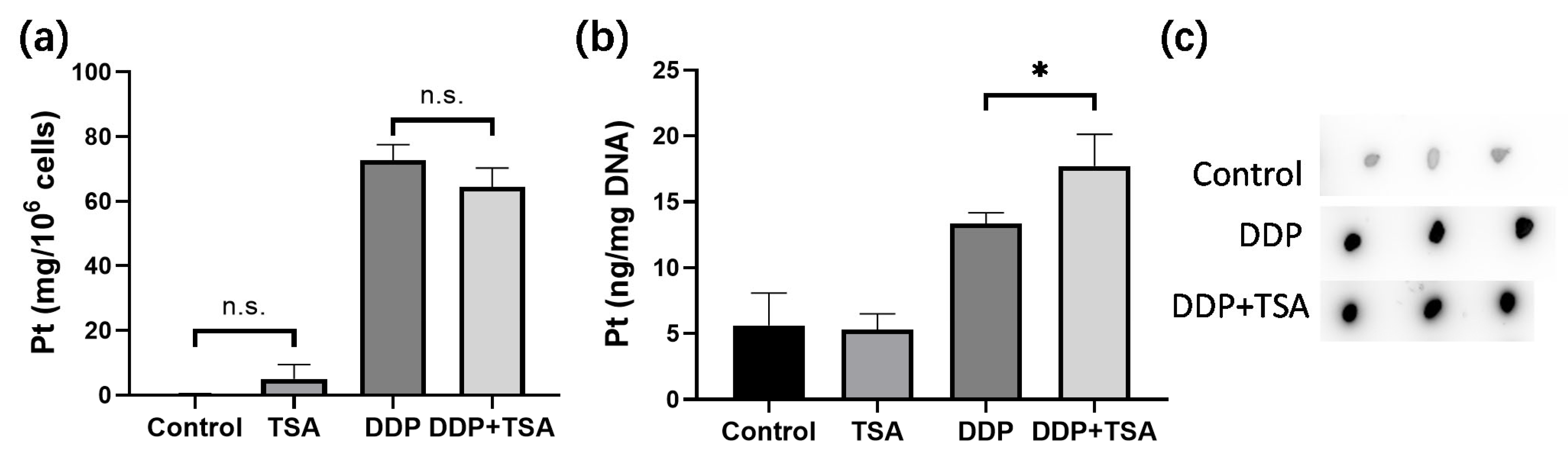
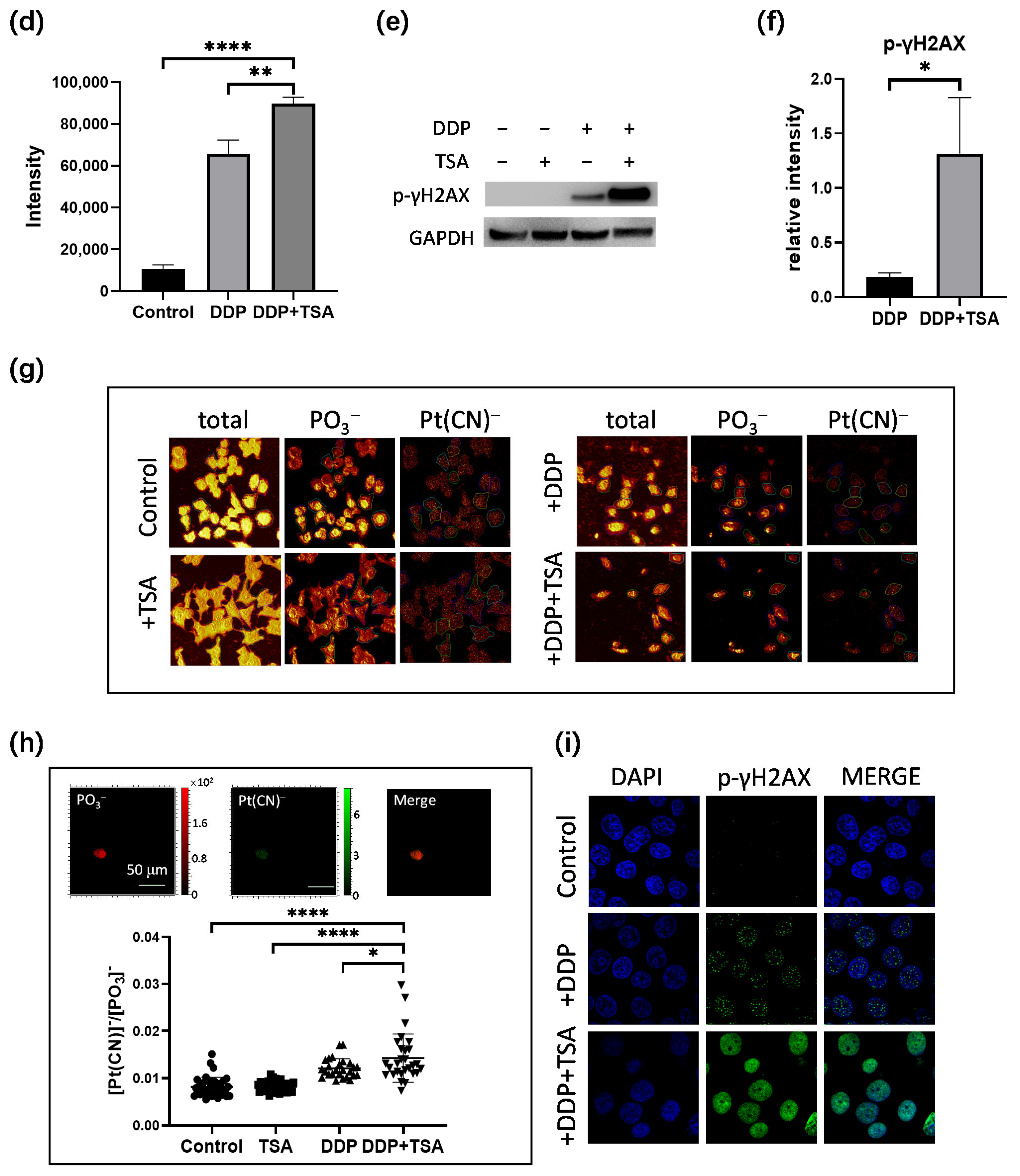
2.2. TSA Enhanced Cisplatin Binding to DNA
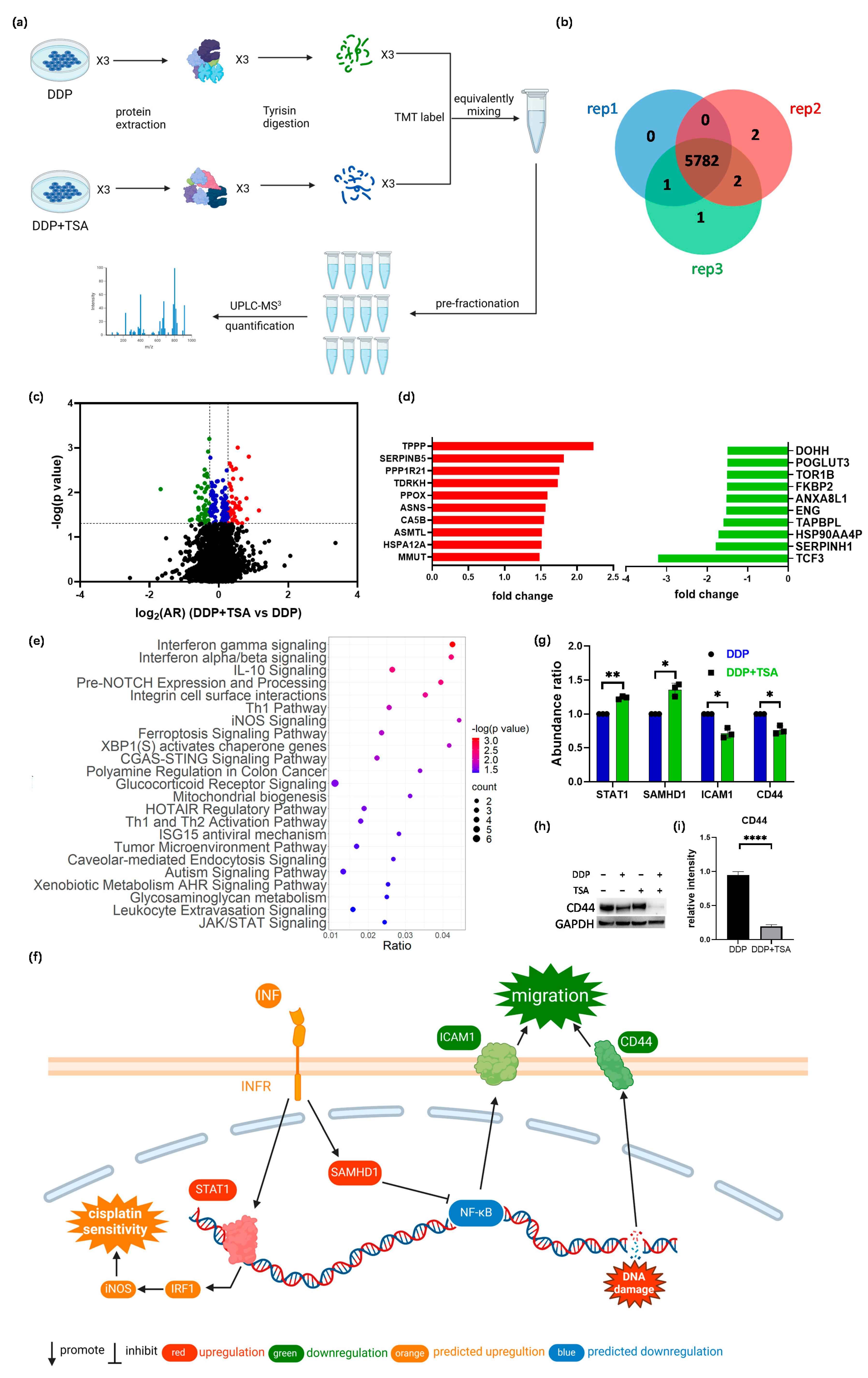
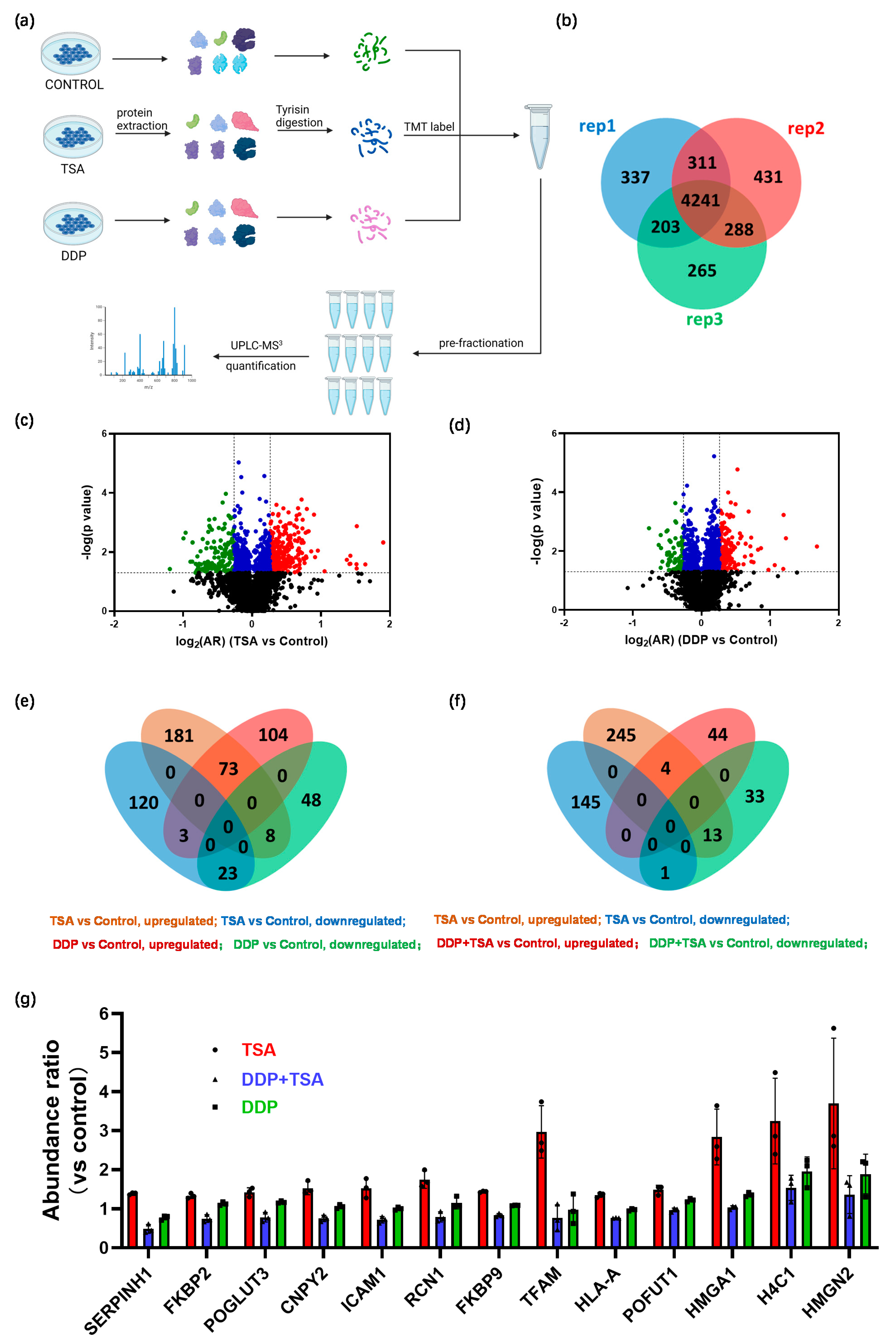
2.3. Quantitative Proteomics Deciphered the Mechanism of Action of Cisplatin in the Absence and Presence of TSA
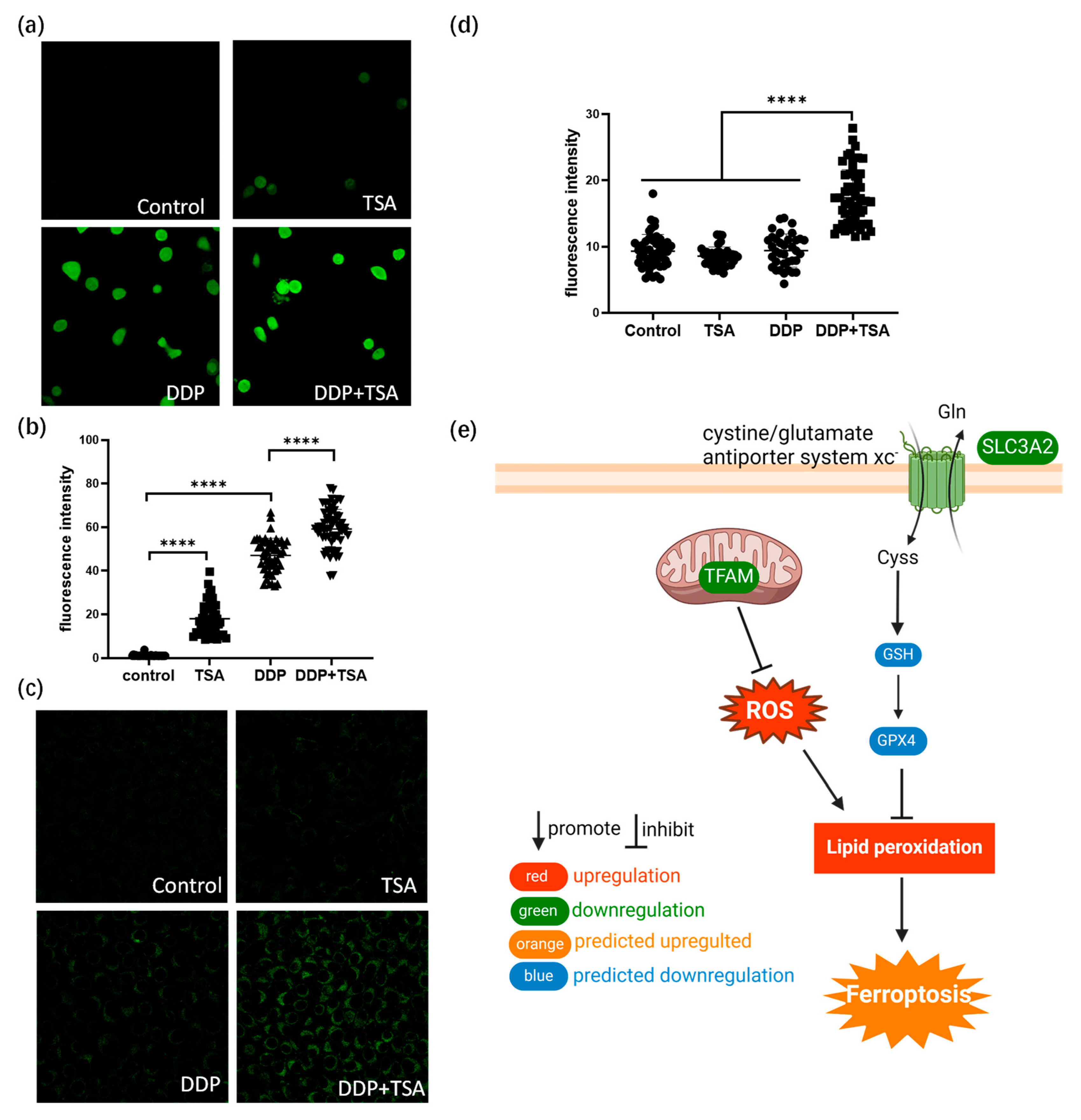
2.4. TSA Elevated the Acetylation of H4K8 to Promote Cisplatin Activity
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Cell Cultures
3.3. CCK-8 Assay and Determination of IC50
3.4. Cell Apoptosis and Cell Cycle Assay
3.5. Caspase Activity Assay
3.6. Western Blotting
3.7. Dot Blot Assay
3.8. Immunofluorescence Imaging
3.9. Wound Healing Assay
3.10. ROS and Lipid Peroxidation Detection
3.11. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
3.12. Time of Flight Secondary Ion Mass Spectrometry (ToF-SIMS) Imaging
3.13. Preparation of Samples for Quantitative Proteomics Analysis
3.14. Quantitative Proteomics Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin—DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct Cellular Responses to Platinum-Induced DNA Damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef]
- Takahara, P.M.; Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature 1995, 377, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Luo, Q.; Yang, L.; Bing, T.; Li, X.; Guo, W.; Wu, K.; Zhao, Y.; Xiong, S.; Shangguan, D.; et al. Mass Spectrometric Proteomics Reveals that Nuclear Protein Positive Cofactor PC4 Selectively Binds to Cross-Linked DNA by a trans-Platinum Anticancer Complex. J. Am. Chem. Soc. 2014, 136, 2948–2951. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, K.; Jia, F.; Chen, L.; Wang, Z.; Zhang, Y.; Luo, Q.; Liu, S.; Qi, L.; Li, N.; et al. Single cell imaging reveals cisplatin regulating interactions between transcription (co)factors and DNA. Chem. Sci. 2021, 12, 5419–5429. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Structure of Chromatin. Annu. Rev. Biochem. 1977, 46, 931–954. [Google Scholar] [CrossRef]
- McGhee, J.D.; Felsenfeld, G. Nucleosome Structure. Annu. Rev. Biochem. 1980, 49, 1115–1156. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Williams, R.J. Trichostatin A, an inhibitor of histone deacetylase, inhibits hypoxia-induced angiogenesis. Expert Opin. Investig. Drugs 2001, 10, 1571–1573. [Google Scholar] [CrossRef]
- Schölz, C.; Weinert, B.T.; Wagner, S.A.; Beli, P.; Miyake, Y.; Qi, J.; Jensen, L.J.; Streicher, W.; McCarthy, A.R.; Westwood, N.J.; et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 2015, 33, 415–423. [Google Scholar] [CrossRef]
- Asgar, M.A.; Senawong, G.; Sripa, B.; Senawong, T. Synergistic anticancer effects of cisplatin and histone deacetylase inhibitors (SAHA and TSA) on cholangiocarcinoma cell lines. Int. J. Oncol. 2016, 48, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of Histone Deacetylase Increases Cytotoxicity to Anticancer Drugs Targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Jin, K.L.; Park, J.Y.; Noh, E.J.; Hoe, K.L.; Lee, J.H.; Kim, J.H.; Nam, J.H. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J. Gynecol. Oncol. 2010, 21, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA Fragmentation during Apoptosis Induces Phosphorylation of H2AX Histone at Serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Chang, P.V.; Lippard, S.J. Identification of Nuclear Proteins that Interact with Platinum-Modified DNA by Photoaffinity Labeling. J. Am. Chem. Soc. 2004, 126, 6536–6537. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Lazebnik, Y.A.; Kaufmann, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 1994, 371, 346–347. [Google Scholar] [CrossRef]
- Oliver, F.J.; de la Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.C.; de Murcia, G.; Murcia, J.M.-d. Importance of Poly(ADP-ribose) Polymerase and Its Cleavage in Apoptosis: Lesson from an uncleavable mutant. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar] [CrossRef]
- Bonne, G.; Barletta, M.R.D.; Varnous, S.; Bécane, H.-M.; Hammouda, E.-H.; Merlini, L.; Muntoni, F.; Greenberg, C.R.; Gary, F.; Urtizberea, J.-A.; et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 1999, 21, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Oberhammer, F.A.; Hochegger, K.; Fröschl, G.; Tiefenbacher, R.; Pavelka, M. Chromatin condensation during apoptosis is accompanied by degradation of lamin A+B, without enhanced activation of cdc2 kinase. J. Cell Biol. 1994, 126, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Perez, D.; White, E. Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 1996, 135, 1441–1455. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Kang, J.; Hwang, J.S.; Li, J.; Boese, A.C.; Wang, X.; Yang, L.; Boggon, T.J.; Chen, G.Z.; Saba, N.F.; et al. Cisplatin-mediated activation of glucocorticoid receptor induces platinum resistance via MAST1. Nat. Commun. 2021, 12, 4960. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wang, J.; Zhao, Y.; Zhang, Y.; Luo, Q.; Qi, L.; Hou, Y.; Du, J.; Wang, F. In Situ Visualization of Proteins in Single Cells by Time-of-Flight-Secondary Ion Mass Spectrometry Coupled with Genetically Encoded Chemical Tags. Anal. Chem. 2020, 92, 15517–15525. [Google Scholar] [CrossRef]
- Hou, Y.; Gan, T.; Fang, T.; Zhao, Y.; Luo, Q.; Liu, X.; Qi, L.; Zhang, Y.; Jia, F.; Han, J.; et al. G-quadruplex inducer/stabilizer pyridostatin targets SUB1 to promote cytotoxicity of a transplatinum complex. Nucleic Acids Res. 2022, 50, 3070–3082. [Google Scholar] [CrossRef]
- Hlavanda, E.; Klement, E.; Kókai, E.; Kovács, J.; Vincze, O.; Tőkési, N.; Orosz, F.; Medzihradszky, K.F.; Dombrádi, V.; Ovádi, J. Phosphorylation Blocks the Activity of Tubulin Polymerization-promoting Protein (TPPP): Identification of sites targeted by different kinases. J. Biol. Chem. 2007, 282, 29531–29539. [Google Scholar] [CrossRef] [PubMed]
- Inokawa, Y.; Sonohara, F.; Kanda, M.; Hayashi, M.; Nishikawa, Y.; Sugimoto, H.; Kodera, Y.; Nomoto, S. Correlation Between Poor Prognosis and Lower TPPP Gene Expression in Hepatocellular Carcinoma. Anticancer Res. 2016, 36, 4639–4645. [Google Scholar] [CrossRef] [PubMed]
- Bradney, C.; Hjelmeland, M.; Komatsu, Y.; Yoshida, M.; Yao, T.-P.; Zhuang, Y. Regulation of E2A Activities by Histone Acetyltransferases in B Lymphocyte Development. J. Biol. Chem. 2003, 278, 2370–2376. [Google Scholar] [CrossRef]
- Slyper, M.; Shahar, A.; Bar-Ziv, A.; Granit, R.Z.; Hamburger, T.; Maly, B.; Peretz, T.; Ben-Porath, I. Control of Breast Cancer Growth and Initiation by the Stem Cell–Associated Transcription Factor TCF3. Cancer Res. 2012, 72, 5613–5624. [Google Scholar] [CrossRef]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.G.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef]
- Ethiraj, P.; Veerappan, K.; Samuel, S.; Sivapatham, S. Interferon β improves the efficacy of low dose cisplatin by inhibiting NF-κB/p-Akt signaling on HeLa cells. Biomed. Pharmacother. 2016, 82, 124–132. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, Q.; Liu, W.; Guo, Y.; Xu, N.; Xu, L.; Liu, S.; Li, S.; Xu, Y.; Sun, L. Increased intracellular Ca2+ decreases cisplatin resistance by regulating iNOS expression in human ovarian cancer cells. Biomed. Pharmacother. 2017, 86, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bonifati, S.; Qin, Z.; St. Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, M.; Shi, B.; Di, S.; Sun, R.; Jiang, H.; Li, Z. Radiation enhances the efficacy of EGFR-targeted CAR-T cells against triple-negative breast cancer by activating NF-κB/Icam1 signaling. Mol. Ther. 2022, 30, 3379–3393. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tabanera, E.; García-García, L.; Rodríguez-Martín, C.; Cervera, S.T.; González-González, L.; Robledo, C.; Josa, S.; Martínez, S.; Chapado, L.; Monzón, S.; et al. CD44 Modulates Cell Migration and Invasion in Ewing Sarcoma Cells. Int. J. Mol. Sci. 2023, 24, 11774. [Google Scholar] [CrossRef]
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, Y.; Shen, J.; Cai, Z.; Zhao, C.; Chen, H.; Luo, X.; Hu, N.; Cui, W.; Huang, W. Injectable hydrogel microspheres with self-renewable hydration layers alleviate osteoarthritis. Sci. Adv. 2022, 8, eabl6449. [Google Scholar] [CrossRef]
- Parker, J.L.; Deme, J.C.; Kolokouris, D.; Kuteyi, G.; Biggin, P.C.; Lea, S.M.; Newstead, S. Molecular basis for redox control by the human cystine/glutamate antiporter system xc−. Nat. Commun. 2021, 12, 7147. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Endale, H.T.; Tesfaye, W.; Mengstie, T.A. ROS induced lipid peroxidation and their role in ferroptosis. Front. Cell Dev. Biol. 2023, 11, 1226044. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Schroth, G.P.; Matthews, H.R.; Yau, P.; Bradbury, E.M. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. J. Biol. Chem. 1993, 268, 305–314. [Google Scholar] [CrossRef]
- Guha, M.; Srinivasan, S.; Johnson, F.B.; Ruthel, G.; Guja, K.; Garcia-Diaz, M.; Kaufman, B.A.; Glineburg, M.R.; Fang, J.; Nakagawa, H.; et al. hnRNPA2 mediated acetylation reduces telomere length in response to mitochondrial dysfunction. PLoS ONE 2018, 13, e0206897. [Google Scholar] [CrossRef]
- Savic, V.; Sanborn, K.B.; Orange, J.S.; Bassing, C.H. Chipping away at γ-H2AX foci. Cell Cycle 2009, 8, 3285–3290. [Google Scholar] [CrossRef]
- Price, B.D.; D’Andrea, A.D. Chromatin Remodeling at DNA Double-Strand Breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef]
- Xu, Y.; Price, B.D. Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle 2011, 10, 261–267. [Google Scholar] [CrossRef]
- Ogiwara, H.; Ui, A.; Otsuka, A.; Satoh, H.; Yokomi, I.; Nakajima, S.; Yasui, A.; Yokota, J.; Kohno, T. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene 2011, 30, 2135–2146. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Dion, M.F.; Altschuler, S.J.; Wu, L.F.; Rando, O.J. Genomic characterization reveals a simple histone H4 acetylation code. Proc. Natl. Acad. Sci. USA 2005, 102, 5501–5506. [Google Scholar] [CrossRef]
- Penicud, K.; Behrens, A. DMAP1 is an essential regulator of ATM activity and function. Oncogene 2014, 33, 525–531. [Google Scholar] [CrossRef] [PubMed]
- García-González, R.; Morejón-García, P.; Campillo-Marcos, I.; Salzano, M.; Lazo, P.A. VRK1 Phosphorylates Tip60/KAT5 and Is Required for H4K16 Acetylation in Response to DNA Damage. Cancers 2020, 12, 2986. [Google Scholar] [CrossRef]
- Yu, Y.; Teng, Y.; Liu, H.; Reed, S.H.; Waters, R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 2005, 102, 8650–8655. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.-R.; Smerdon, M.J. Histone H3 Lysine 14 (H3K14) Acetylation Facilitates DNA Repair in a Positioned Nucleosome by Stabilizing the Binding of the Chromatin Remodeler RSC (Remodels Structure of Chromatin). J. Biol. Chem. 2014, 289, 8353–8363. [Google Scholar] [CrossRef]
- Chang, C.S.; Pillus, L. Collaboration Between the Essential Esa1 Acetyltransferase and the Rpd3 Deacetylase Is Mediated by H4K12 Histone Acetylation in Saccharomyces cerevisiae. Genetics 2009, 183, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Rayan, N.A.; Muratani, M.; Lim, S.; Elanggovan, B.; Xin, L.; Lu, T.; Makhija, H.; Poschmann, J.; Lufkin, T.; et al. Comprehensive benchmarking reveals H2BK20 acetylation as a distinctive signature of cell-state-specific enhancers and promoters. Genome Res. 2016, 26, 612–623. [Google Scholar] [CrossRef]
- Cheng, D.; Dong, Z.; Lin, P.; Shen, G.; Xia, Q. Transcriptional Activation of Ecdysone-Responsive Genes Requires H3K27 Acetylation at Enhancers. Int. J. Mol. Sci. 2022, 23, 10791. [Google Scholar] [CrossRef]
- Beck, A.; Eberherr, C.; Hagemann, M.; Cairo, S.; Häberle, B.; Vokuhl, C.; von Schweinitz, D.; Kappler, R. Connectivity map identifies HDAC inhibition as a treatment option of high-risk hepatoblastoma. Cancer Biol. Ther. 2016, 17, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bao, X.; Ren, Y.; Jia, L.; Zou, S.; Han, J.; Zhao, M.; Han, M.; Li, H.; Hua, Q.; et al. Targeting HDAC/OAZ1 axis with a novel inhibitor effectively reverses cisplatin resistance in non-small cell lung cancer. Cell Death Dis. 2019, 10, 400. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Yang, P.; Sun, C.; Zhang, Y. Comprehensive comparison of sample preparation workflows for proteomics. Mol. Omics 2022, 18, 555–567. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (μM) of Cisplatin | SI 1 | |
|---|---|---|---|
| −TSA | +TSA | ||
| A549 | 7.8 ± 0.6 | 2.3 ± 0.4 | 3.4 |
| A549/DDP | 46.7 ± 2.5 | 35.2 ± 2.2 | 1.3 |
| GIST | 11.8 ± 0.9 | 8.1 ± 0.8 | 1.5 |
| HeLa | 10.2 ± 0.8 | 7.2 ± 1.0 | 1.4 |
| HepG2 | 17.3 ± 2.2 | 13.1 ± 1.2 | 1.3 |
| MCF7 | 14.3 ± 2.9 | 11.1 ± 0.8 | 1.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Luo, Q.; Zeng, F.; Liu, X.; Han, J.; Gu, L.; Tian, X.; Zhang, Y.; Zhao, Y.; Wang, F. Trichostatin A Promotes Cytotoxicity of Cisplatin, as Evidenced by Enhanced Apoptosis/Cell Death Markers. Molecules 2024, 29, 2623. https://doi.org/10.3390/molecules29112623
Zhou Y, Luo Q, Zeng F, Liu X, Han J, Gu L, Tian X, Zhang Y, Zhao Y, Wang F. Trichostatin A Promotes Cytotoxicity of Cisplatin, as Evidenced by Enhanced Apoptosis/Cell Death Markers. Molecules. 2024; 29(11):2623. https://doi.org/10.3390/molecules29112623
Chicago/Turabian StyleZhou, Yang, Qun Luo, Fangang Zeng, Xingkai Liu, Juanjuan Han, Liangzhen Gu, Xiao Tian, Yanyan Zhang, Yao Zhao, and Fuyi Wang. 2024. "Trichostatin A Promotes Cytotoxicity of Cisplatin, as Evidenced by Enhanced Apoptosis/Cell Death Markers" Molecules 29, no. 11: 2623. https://doi.org/10.3390/molecules29112623
APA StyleZhou, Y., Luo, Q., Zeng, F., Liu, X., Han, J., Gu, L., Tian, X., Zhang, Y., Zhao, Y., & Wang, F. (2024). Trichostatin A Promotes Cytotoxicity of Cisplatin, as Evidenced by Enhanced Apoptosis/Cell Death Markers. Molecules, 29(11), 2623. https://doi.org/10.3390/molecules29112623