
Biology and Total Synthesis of n-3 Docosapentaenoic Acid-Derived Specialized Pro-Resolving Mediators



Abstract
:
1. Introduction
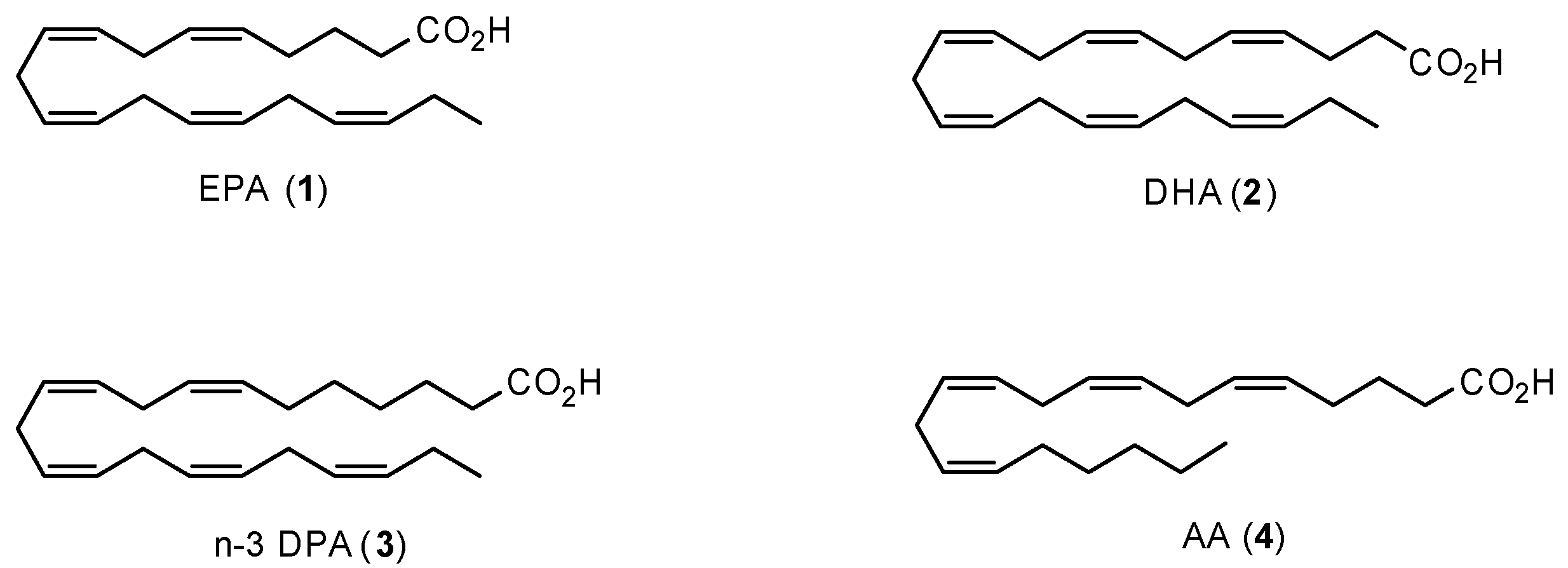
1.1. Polyunsaturated Fatty Acids and Health Effects
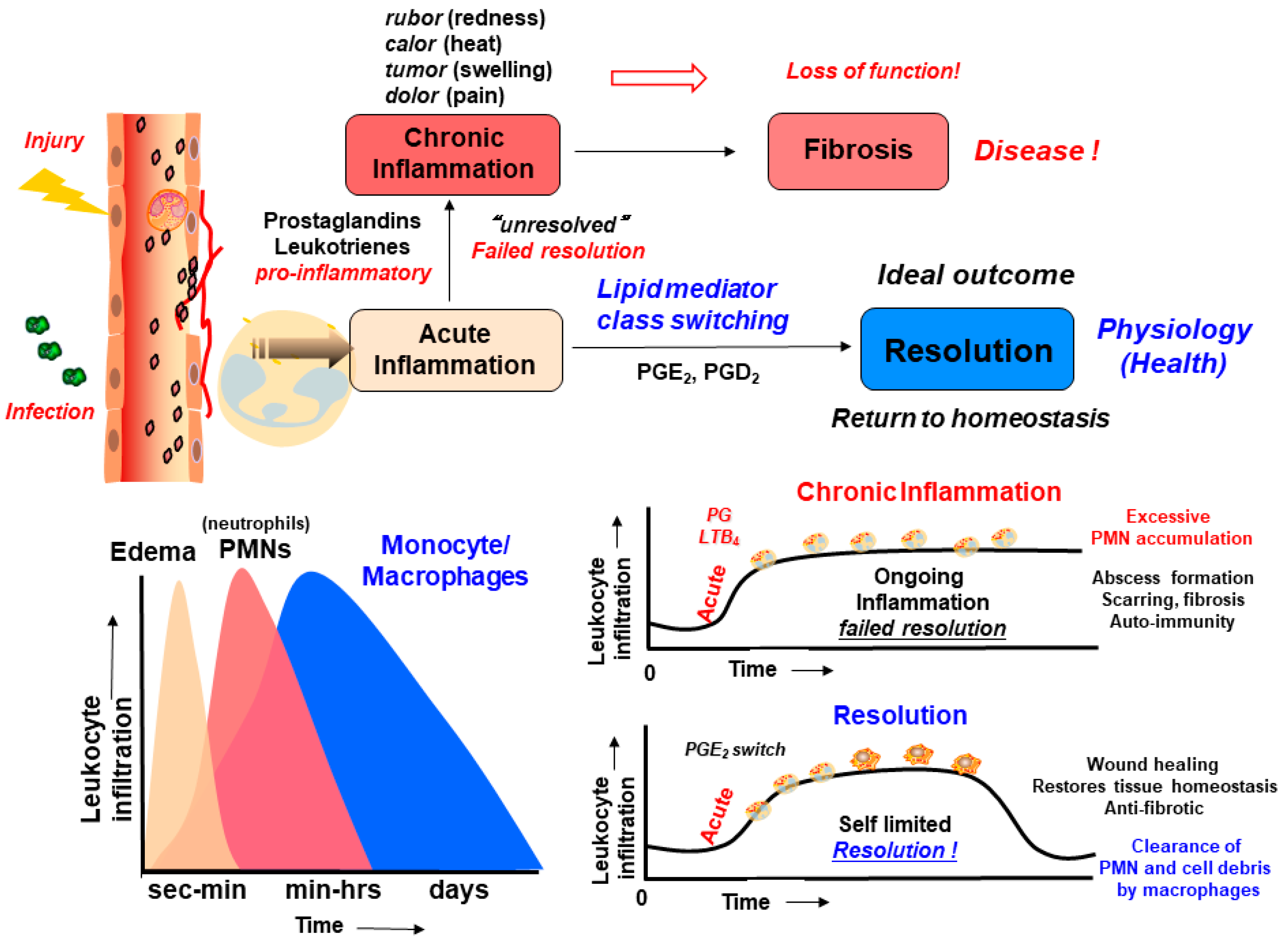
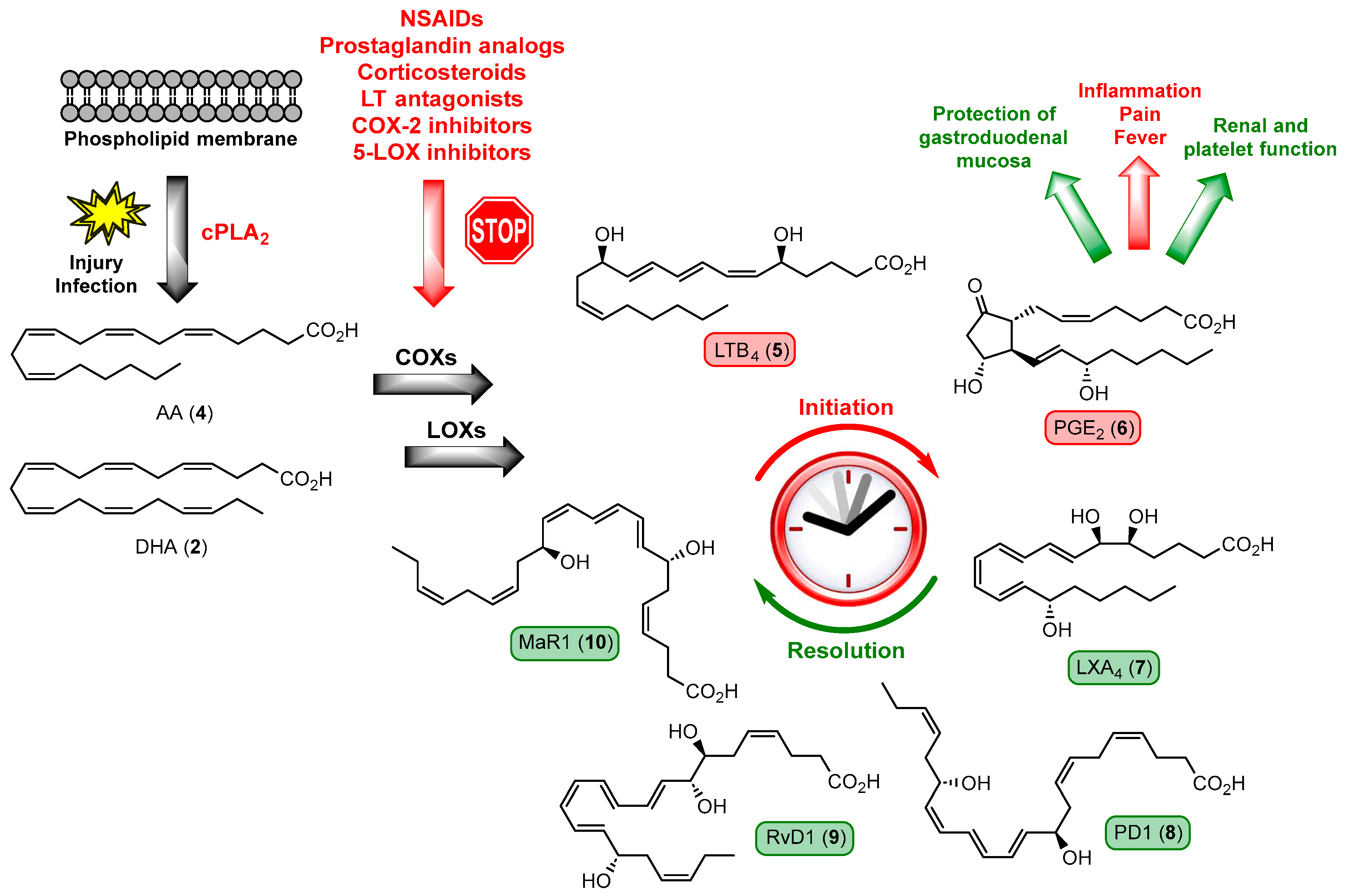
1.2. Inflammation, Resolution of Inflammation, and Lipid Mediators in Inflammation
1.3. Overview of Specialized Pro-Resolving Mediators
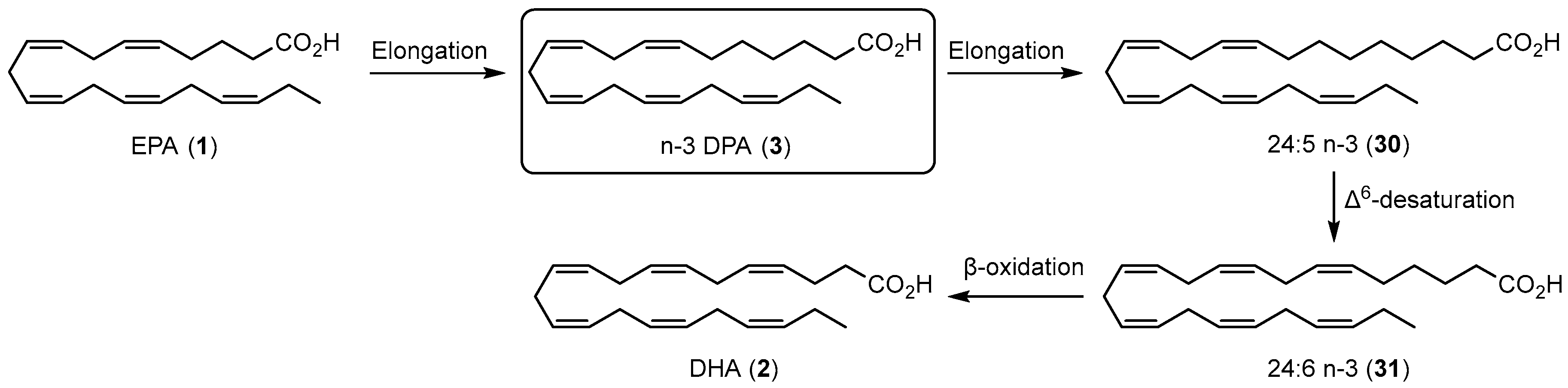
1.4. Specialized Pro-Resolving Mediators Derived from n-3 DPA
1.4.1. Biosynthesis of n-3 DPA Protectins
1.4.2. Biosynthesis of n-3 DPA Maresins
1.4.3. Biosynthesis of n-3 DPA Resolvins
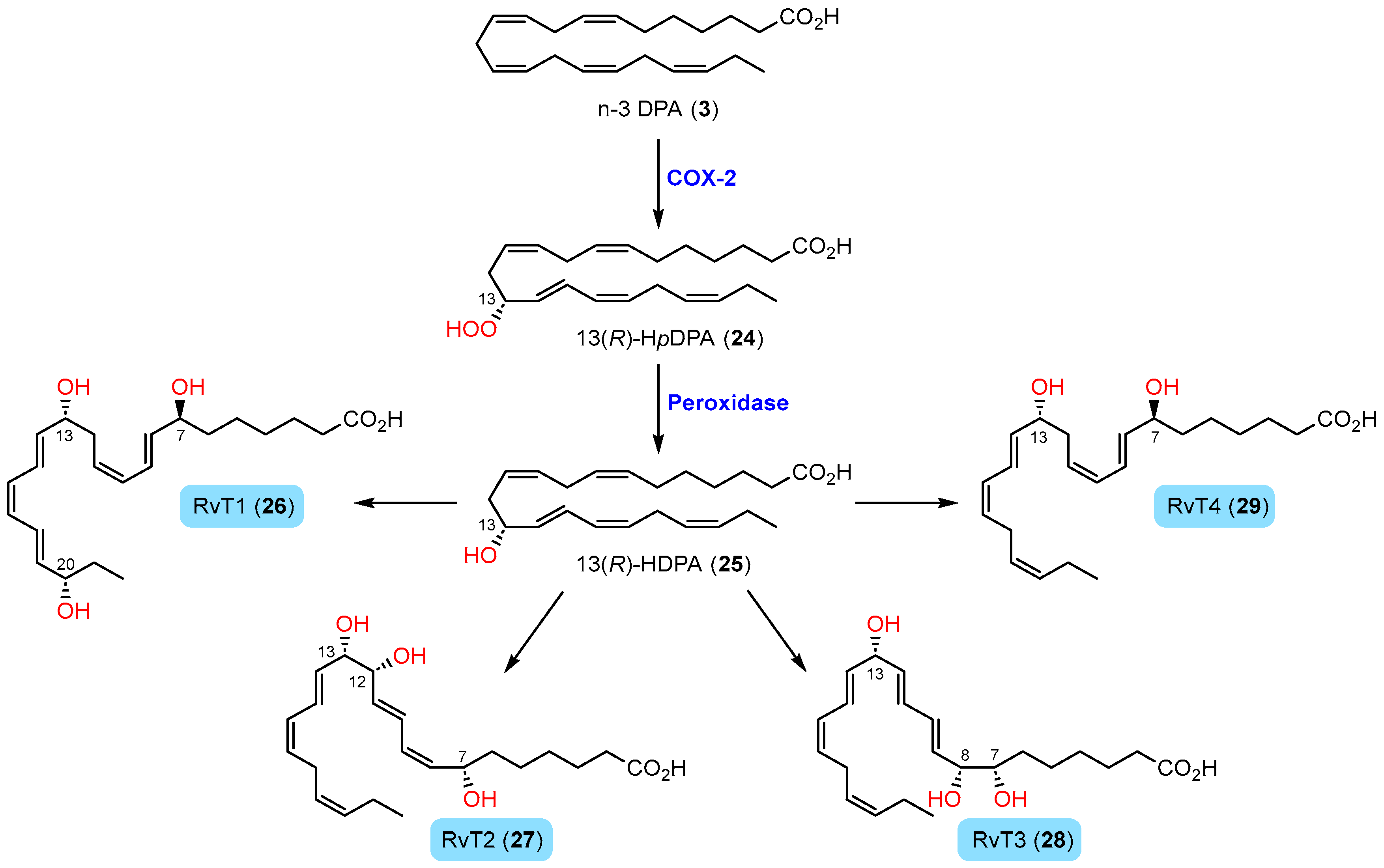
1.4.4. Biosynthesis of 13-Series Resolvins
2. Stereoselective Syntheses of n-3 DPA-Derived SPMs
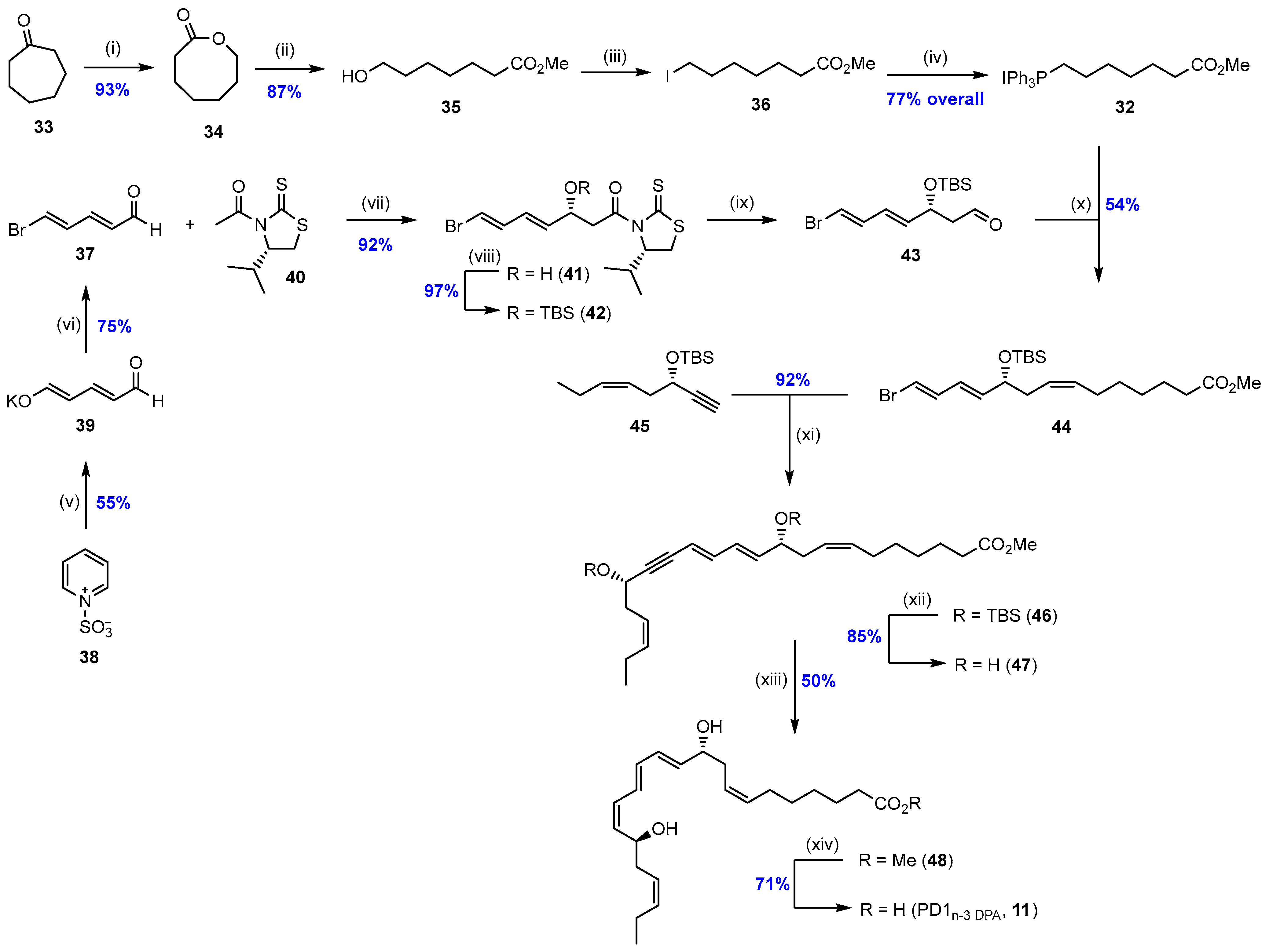
2.1. Synthesis and Biological Studies of PD1n-3 DPA (11)
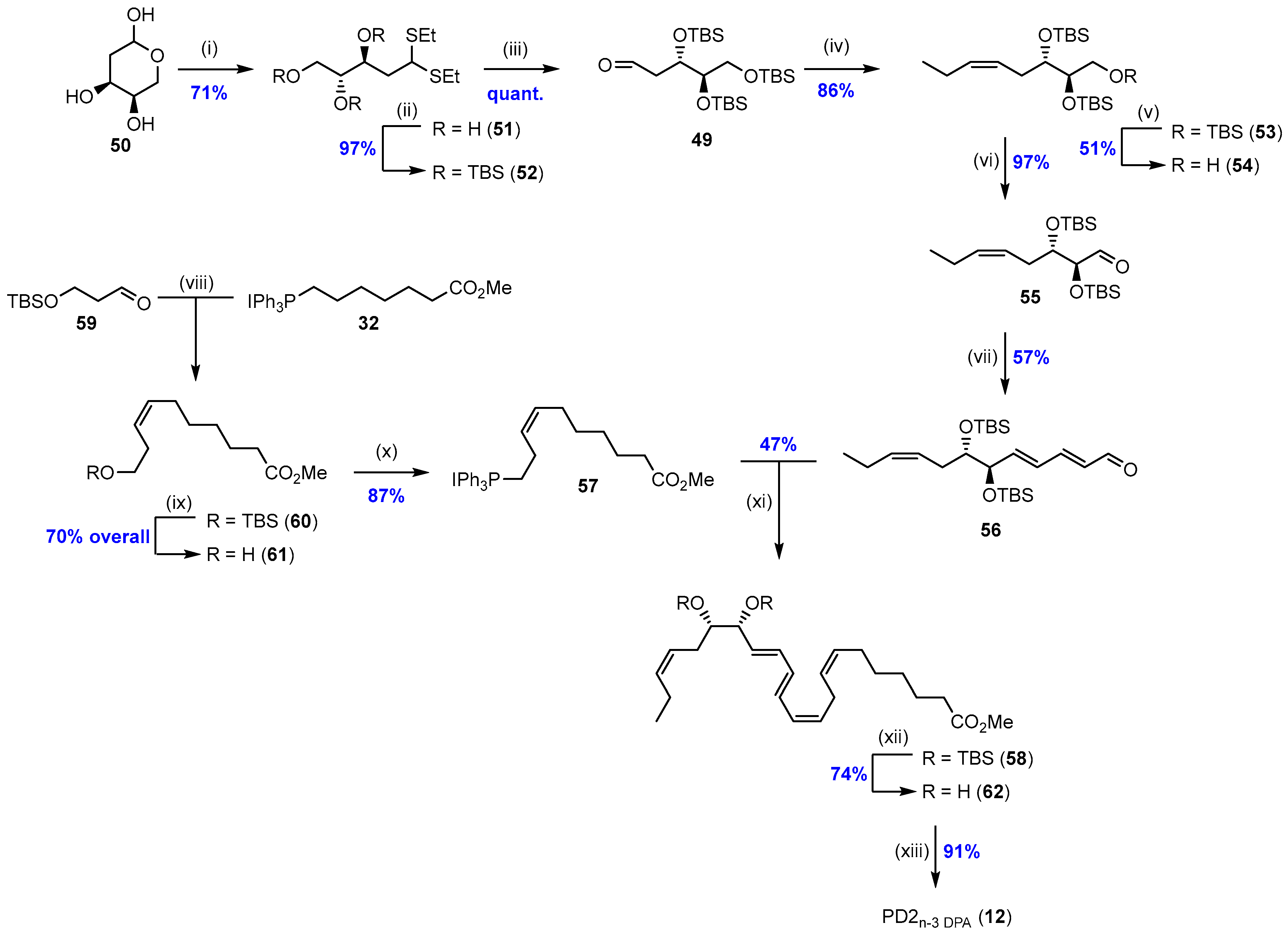
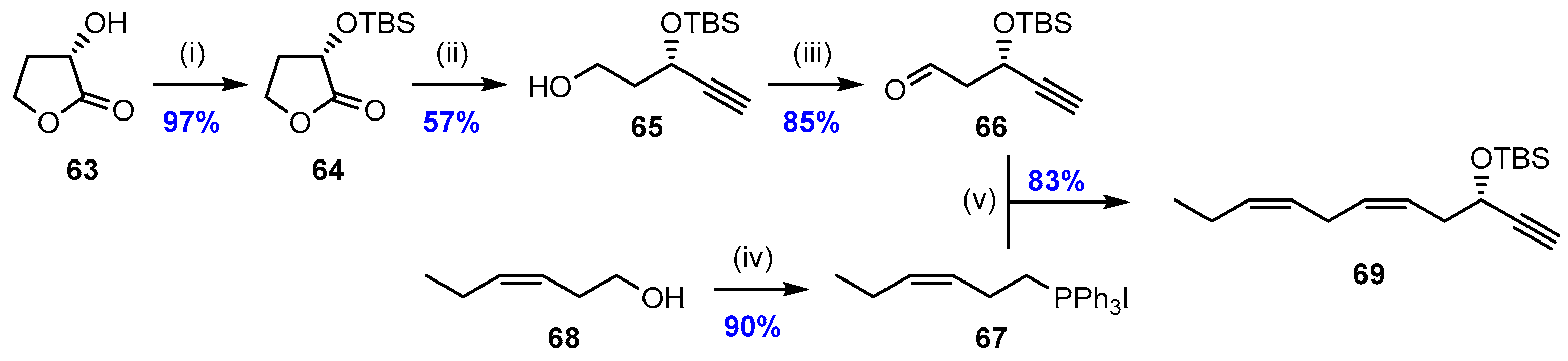
2.2. Synthesis of PD2n-3 DPA (12)
2.3. Synthesis and Biological Evaluations of MaR1n-3 DPA (15)
2.4. Synthesis of MaR2n-3 DPA (16)
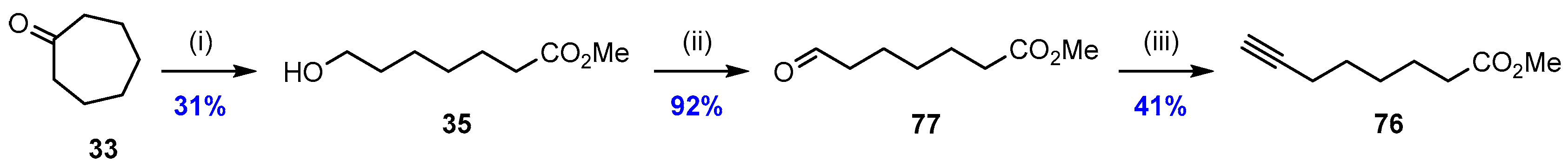
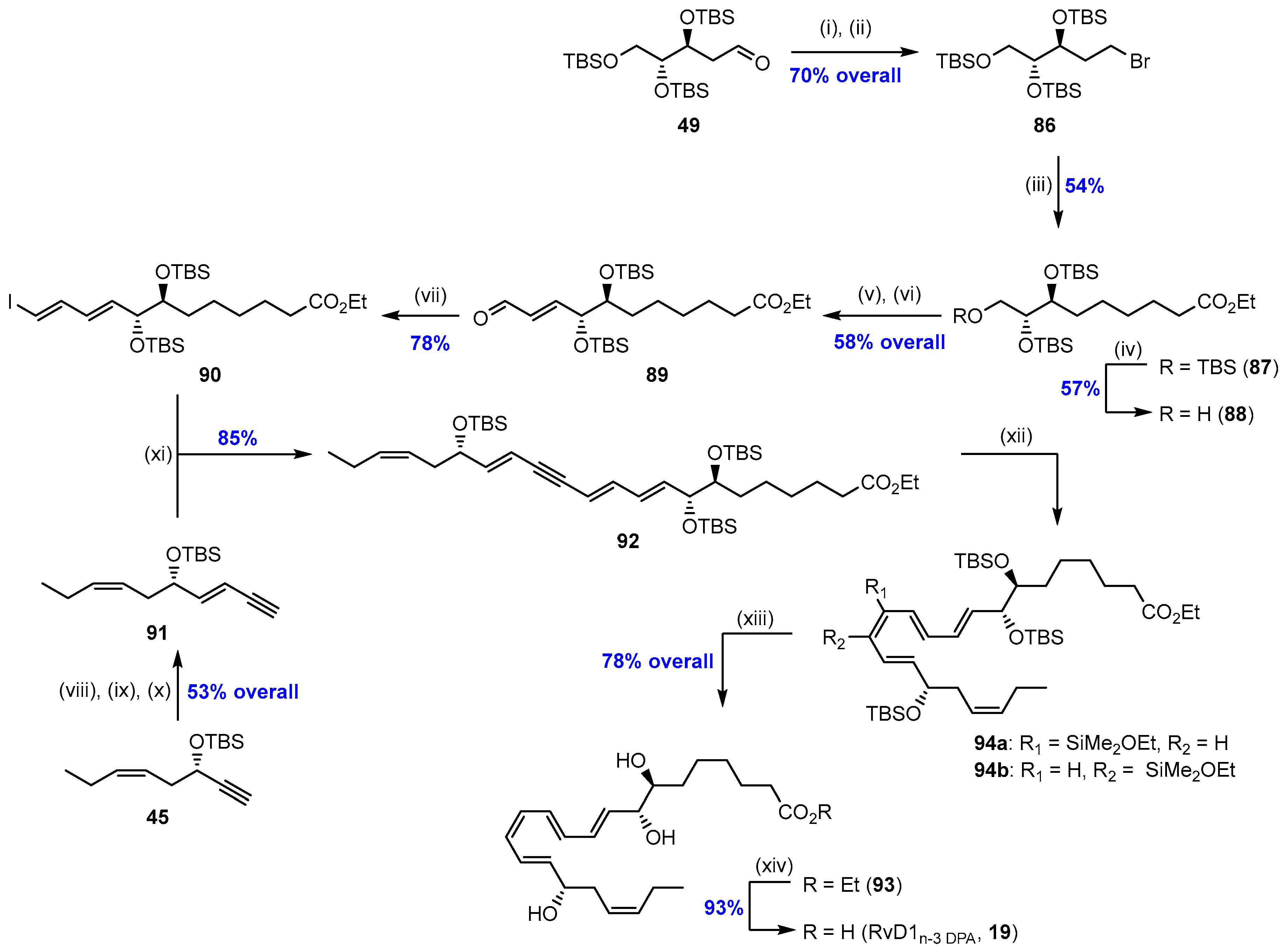
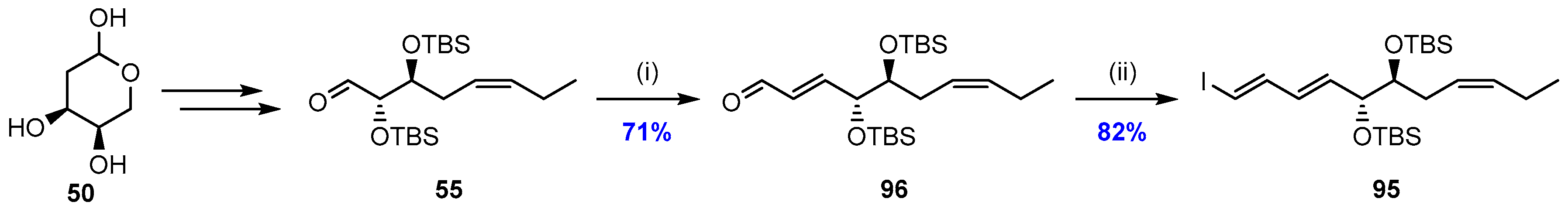
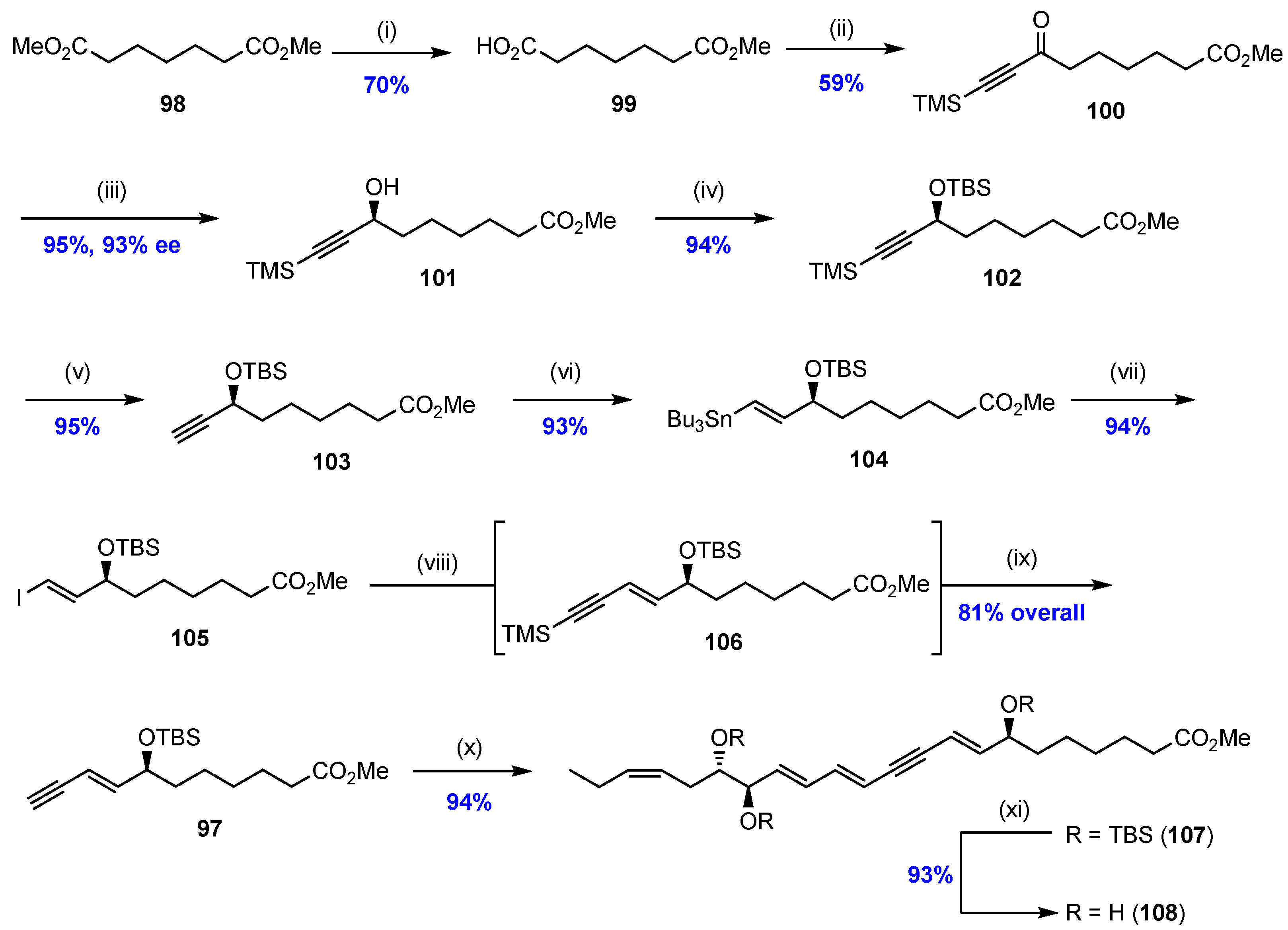
2.5. Synthesis and Biological Evaluations of RvD1n-3 DPA (19)
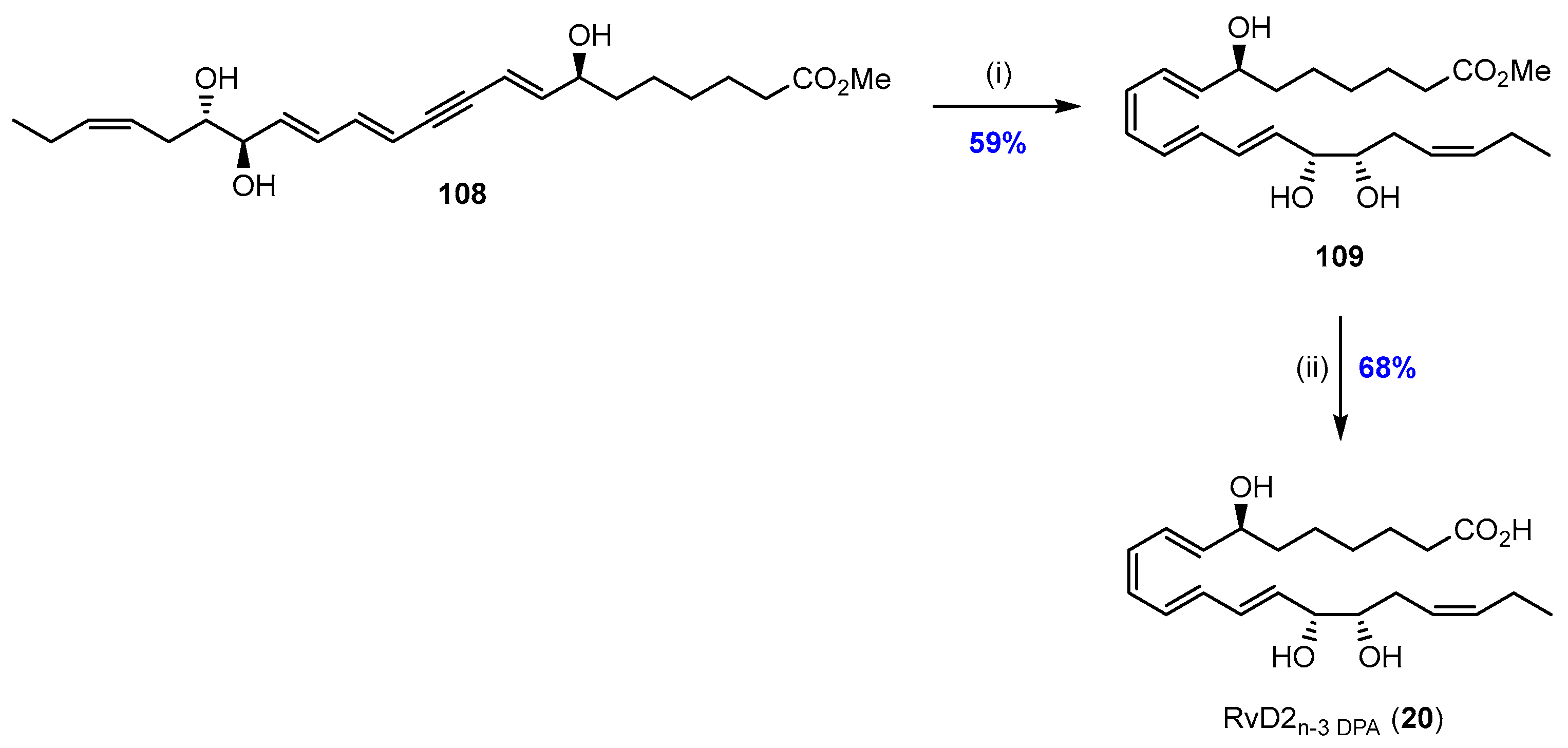
2.6. Synthesis and Biological Evaluations of RvD2n-3 DPA (20)
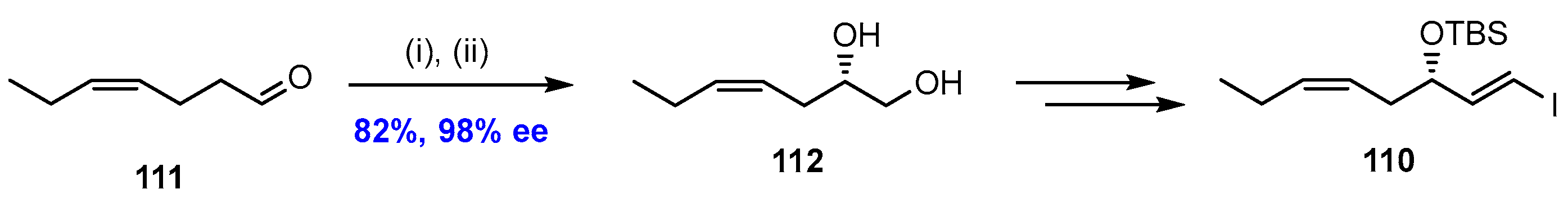
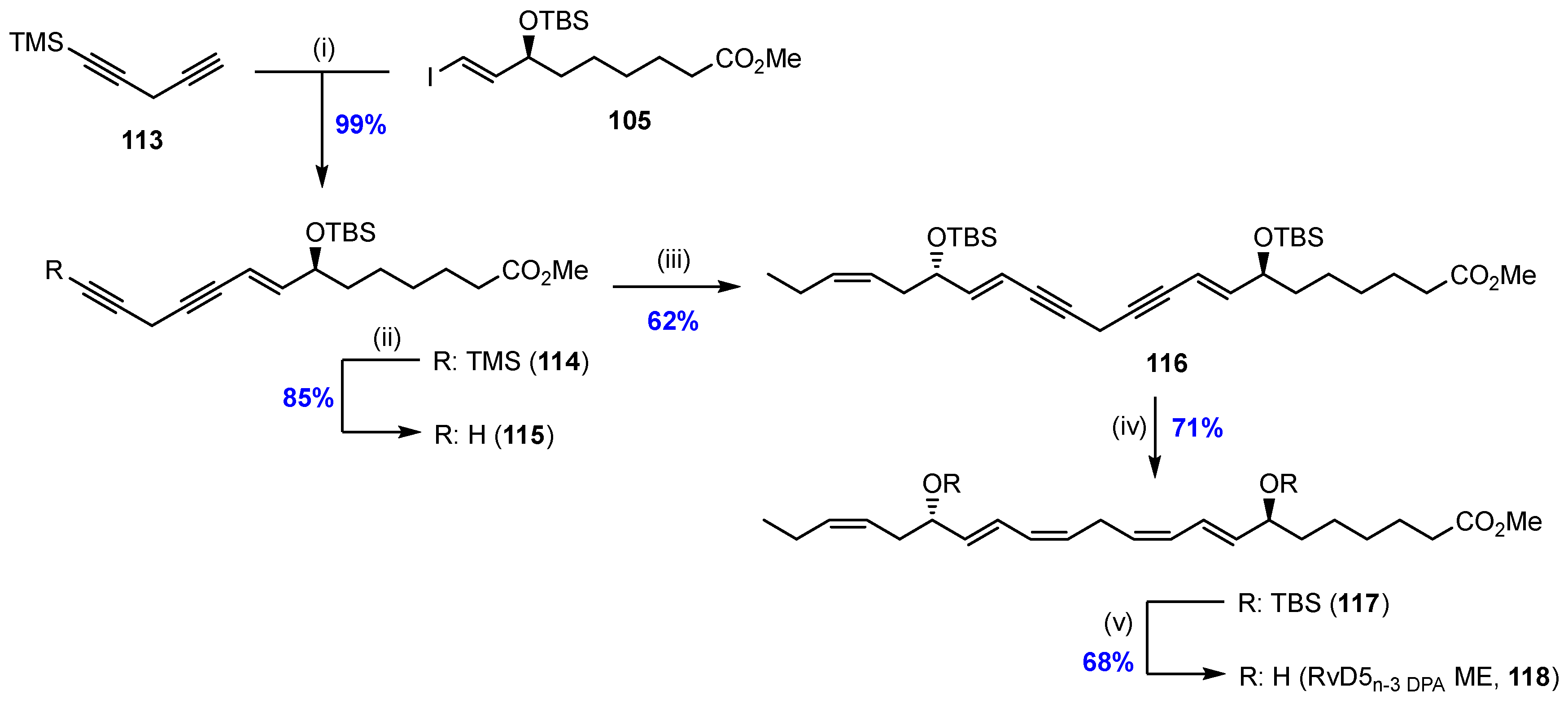
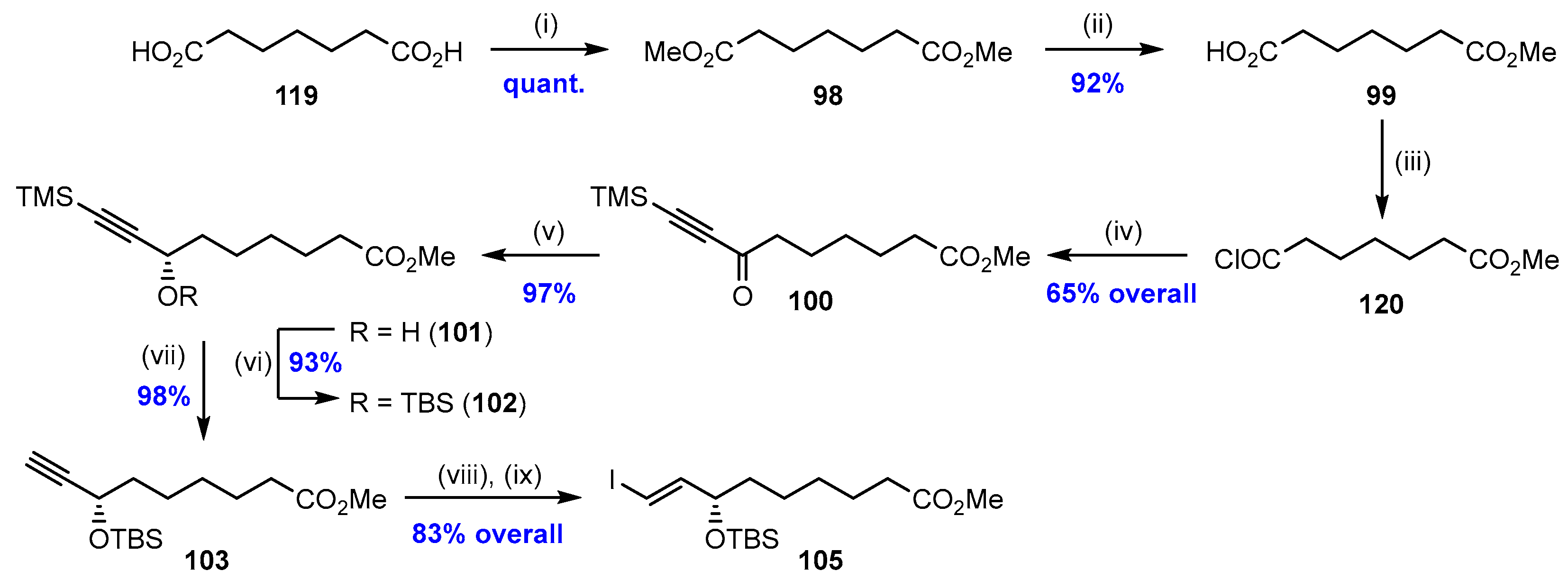
2.7. Synthesis and Biological Evaluations of RvD5n-3 DPA (21)
2.8. Synthesis and Biological Actions of RvT1 (26) and RvT4 (29)
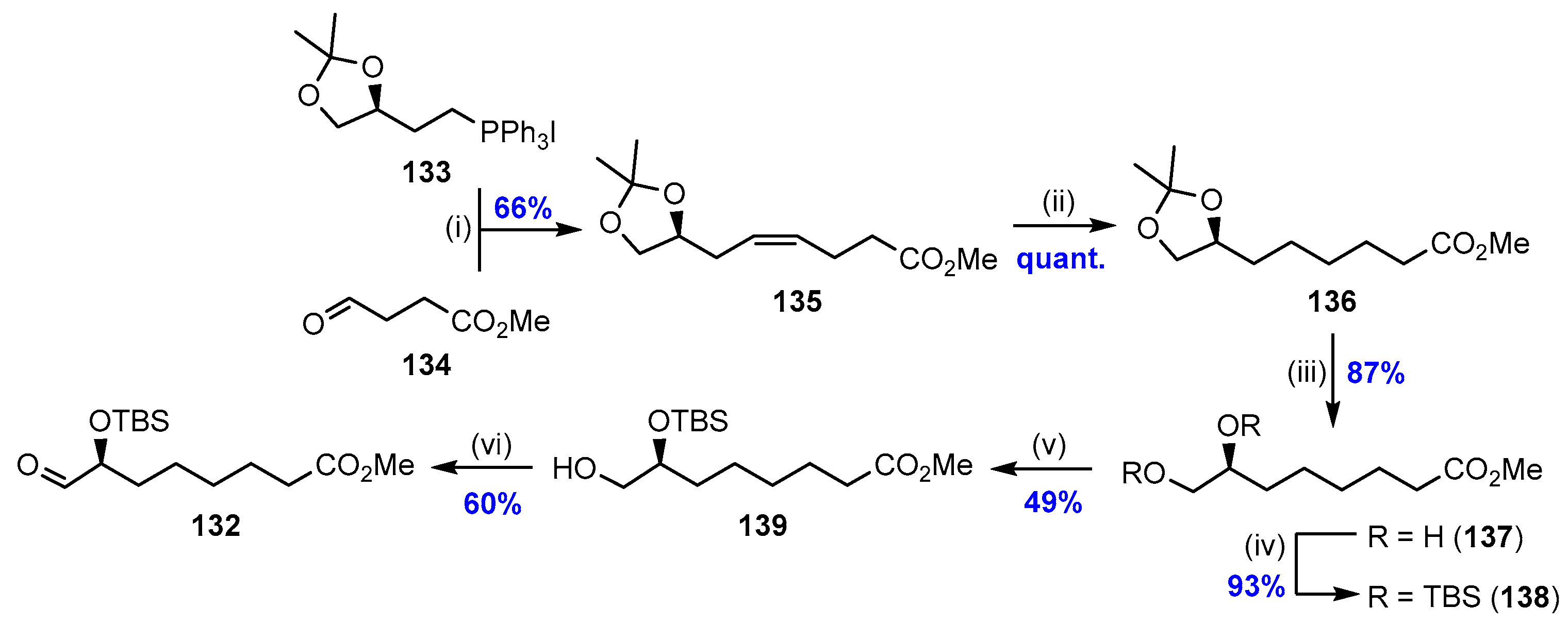
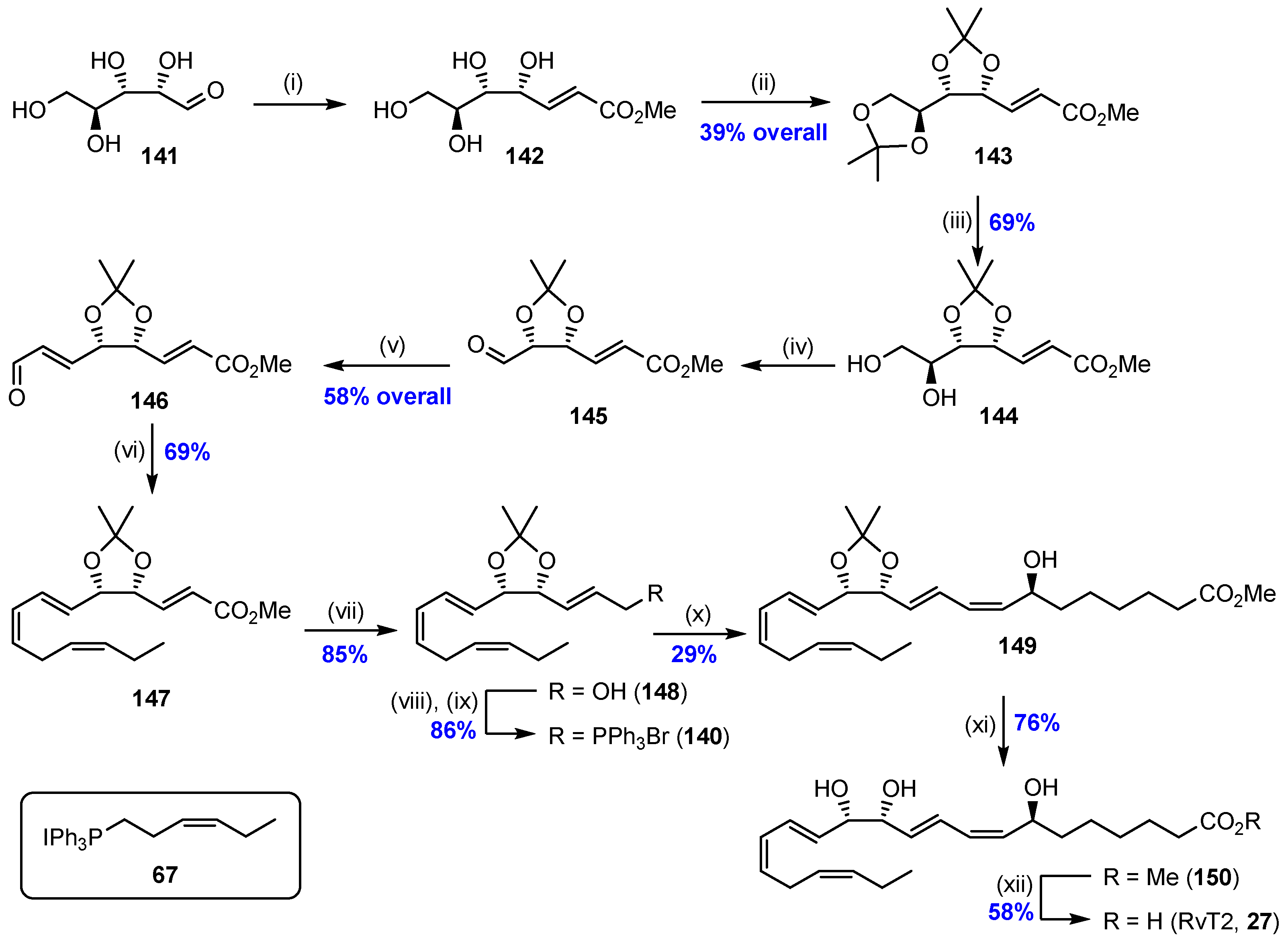
2.9. Synthesis of RvT2 (27) and Biological Actions of the RvTs
2.10. Synthesis and Biological Actions of the ω-22 Monohydroxylated Metabolite 22-OH-PD1 (151)
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calder, P.C.; Yaqoob, P. Understanding Omega-3 Polyunsaturated Fatty Acids. Postgrad. Med. 2009, 121, 148–157. [Google Scholar] [CrossRef]
- Fetterman, J.W., Jr.; Zdanowicz, M.M. Therapeutic potential of n-3 polyunsaturated fatty acids in disease. Am. J. Health-Syst. Pharm. 2009, 66, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.R.; Jacobson, T.A. The Fats of Life: The Role of Omega-3 Fatty Acids in the Prevention of Coronary Heart Disease. Arch. Intern. Med. 2001, 161, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Cleland, L.G.; James, M.J.; Proudman, S.M. The Role of Fish Oils in the Treatment of Rheumatoid Arthritis. Drugs 2003, 63, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Glass, C.K. Anti-Inflammatory Therapy in Chronic Disease: Challenges and Opportunities. Science 2013, 339, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Burkett, P.R.; Dalli, J.; Abdulnour, R.-E.E.; Colas, R.; Ramon, S.; Phipps, R.P.; Petasis, N.A.; Kuchroo, V.K.; Serhan, C.N.; et al. Cutting Edge: Maresin-1 Engages Regulatory T Cells to Limit Type 2 Innate Lymphoid Cell Activation and Promote Resolution of Lung Inflammation. J. Immunol. 2015, 194, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, J.A.; Katz, R. n-3 long-chain polyunsaturated fatty acids in type 2 diabetes: A review. J. Am. Diet. Assoc. 2005, 105, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Petasis, N.A. Resolvins and Protectins in Inflammation-Resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef]
- Dalli, J. Does promoting resolution instead of inhibiting inflammation represent the new paradigm in treating infections? Mol. Asp. Med. 2017, 58, 12–20. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Serhan, C.N. Protectins: Their biosynthesis, metabolism and structure-functions. Biochem. Pharmacol. 2022, 206, 115330. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Ward, P.A.; Gilroy, D.W. Fundamentals of Inflammation, 1st ed.; Cambridge University Press: New York, NY, USA, 2010; pp. 1–27. [Google Scholar]
- Heidland, A.; Klassen, A.; Rutkowski, P.; Bahner, U. The contribution of Rudolf Virchow to the concept of inflammation: What is still of importance? J. Nephrol. 2006, 19, S102–S109. [Google Scholar] [PubMed]
- Serhan, C.N. Novel Lipid Mediators and Resolution Mechanisms in Acute Inflammation: To Resolve or Not? Am. J. Pathol. 2010, 177, 1576–1591. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N. Resolution phase lipid mediators of inflammation: Agonists of resolution. Curr. Opin. Pharmacol. 2013, 13, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Gilligan, M.M.; Serhan, C.N.; Kashfi, K. Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol. Ther. 2021, 227, 107879. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Kumlin, M.; Ingelman-Sundberg, M.; Wolk, A. Dietary long-chain n− 3 fatty acids for the prevention of cancer: A review of potential mechanisms. Am. J. Clin. Nutr. 2004, 79, 935–945. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Maderna, P.; Godson, C. Lipoxins: Resolutionary road. Br. J. Pharmacol. 2009, 158, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Leroy, X.; Bland, E.J.; Montero-Melendez, T. Resolution Pharmacology: Opportunities for Therapeutic Innovation in Inflammation. Trends Pharmacol. Sci. 2015, 36, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Serhan, C.N. Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br. J. Pharmacol. 2019, 176, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Z.; Zhang, L.; Liu, T.; Park, J.Y.; Berta, T.; Yang, R.; Serhan, C.N.; Ji, R.-R. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat. Med. 2010, 16, 592–597. [Google Scholar] [CrossRef]
- Devchand, P.R.; Arita, M.; Hong, S.; Bannenberg, G.; Moussignac, R.L.; Gronert, K.; Serhan, C.N. Human ALX receptor regulates neutrophil recruitment in transgenic mice: Roles in inflammation and host defense. FASEB J. 2003, 17, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Wang, C.-W.; Arnardottir, H.H.; Li, Y.; Cheng, C.-Y.C.; Dalli, J.; Serhan, C.N. Maresin Biosynthesis and Identification of Maresin 2, a New Anti-Inflammatory and Pro-Resolving Mediator from Human Macrophages. PLoS ONE 2014, 9, e102362. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.-K.; Xu, Z.-Z.; Ji, R.-R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.-L. Resolvins: A Family of Bioactive Products of Omega-3 Fatty Acid Transformation Circuits Initiated by Aspirin Treatment that Counter Proinflammation Signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Marcheselli, V.L.; Serhan, C.N.; Bazan, N.G. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. USA 2004, 101, 8491–8496. [Google Scholar] [CrossRef]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef]
- Serhan, C.N. Lipoxin biosynthesis and its impact in inflammatory and vascular events. Biochim. Biophys. Acta Lipids Lipid Metab. 1994, 1212, 1–25. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Serhan, C.N. Novel n-3 Immunoresolvents: Structures and Actions. Sci. Rep. 2013, 3, 1940. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015, 21, 1071–1075. [Google Scholar] [CrossRef]
- Primdahl, K.G.; Tungen, J.E.; De Souza, P.R.S.; Colas, R.A.; Dalli, J.; Hansen, T.V.; Vik, A. Stereocontrolled synthesis and investigation of the biosynthetic transformations of 16(S),17(S)-epoxy-PDn-3 DPA. Org. Biomol. Chem. 2017, 15, 8606–8613. [Google Scholar] [CrossRef] [PubMed]
- Pistorius, K.; Souza, P.R.; De Matteis, R.; Austin-Williams, S.; Primdahl, K.G.; Vik, A.; Mazzacuva, F.; Colas, R.A.; Marques, R.M.; Hansen, T.V.; et al. PDn-3 DPA Pathway Regulates Human Monocyte Differentiation and Macrophage Function. Cell Chem. Biol. 2018, 25, 749–760. [Google Scholar] [CrossRef]
- Ariel, A.; Serhan, C.N. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007, 28, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Pistorius, K.; Walker, M.E. Novel n-3 Docosapentaneoic Acid-Derived Pro-resolving Mediators Are Vasculoprotective and Mediate the Actions of Statins in Controlling Inflammation. Adv. Exp. Med. Biol. 2019, 1161, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Primdahl, K.G.; Aursnes, M.; Walker, M.E.; Colas, R.A.; Serhan, C.N.; Dalli, J.; Hansen, T.V.; Vik, A. Synthesis of 13(R)-Hydroxy-7Z,10Z,13R,14E,16Z,19Z Docosapentaenoic Acid (13R-HDPA) and Its Biosynthetic Conversion to the 13-Series Resolvins. J. Nat. Prod. 2016, 79, 2693–2702. [Google Scholar] [CrossRef]
- Calder, P.C. Fatty acids and inflammation: The cutting edge between food and pharma. Eur. J. Pharmacol. 2011, 668, S50–S58. [Google Scholar] [CrossRef]
- De Caterina, R. n–3 Fatty Acids in Cardiovascular Disease. N. Engl. J. Med. 2011, 364, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, R.N.; Tanaka, T.; Tang, W.; Manichaikul, A.; Foy, M.; Kabagambe, E.K.; Nettleton, J.A.; King, I.B.; Weng, L.-C.; Bhattacharya, S.; et al. Genetic Loci Associated with Plasma Phospholipid n-3 Fatty Acids: A Meta-Analysis of Genome-Wide Association Studies from the CHARGE Consortium. PLoS Genet. 2011, 7, e1002193. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.-L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52, 1702590. [Google Scholar] [CrossRef] [PubMed]
- Aursnes, M.; Tungen, J.E.; Vik, A.; Colas, R.; Cheng, C.-Y.C.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Total Synthesis of the Lipid Mediator PD1n-3 DPA: Configurational Assignments and Anti-inflammatory and Pro-resolving Actions. J. Nat. Prod. 2014, 77, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Becher, J. Glutaconaldehyde sodium salt from hydrolysis of pyridinium-1-sulfonate. Org. Synth. 1979, 59, 79. [Google Scholar] [CrossRef]
- Soullez, D.; Plé, G.; Duhamel, L. ω-Halogeno polyenals: Preparation and application to a one-pot synthesis of polyenals from carbonyl compounds. J. Chem. Soc. Perkin Trans. 1 1997, 1639–1645. [Google Scholar] [CrossRef]
- Romero-Ortega, M.; Colby, D.A.; Olivo, H.F. Synthesis of the C10–C17 fragment of aurisides and callipeltosides. Tetrahedron Lett. 2002, 43, 6439–6441. [Google Scholar] [CrossRef]
- Corey, E.; Cho, H.; Rücker, C.; Hua, D.H. Studies with trialkylsilyltriflates: New syntheses and applications. Tetrahedron Lett. 1981, 22, 3455–3458. [Google Scholar] [CrossRef]
- Tello-Aburto, R.; Ochoa-Teran, A.; Olivo, H.F. Model studies on the ring construction of the auriside macrolactone. Tetrahedron Lett. 2006, 47, 5915–5917. [Google Scholar] [CrossRef]
- Nicolaou, K.; Webber, S.; Ramphal, J.; Abe, Y. Stereocontrolled Total Synthesis of Lipoxins A5 and B5. Angew. Chem. Int. Ed. 1987, 26, 1019–1021. [Google Scholar] [CrossRef]
- Yadav, J.S.; Deshpande, P.K.; Sharma, G.V.M. Stereoselective Synthesis of (S)-13-Hydroxy Octadeca-(9Z,11E)-di- and (9Z,11E,15Z)-trienoic Acids: Selfdefensive Substances Against Rice Blast Disease. Tetrahedron 1992, 48, 4465–4474. [Google Scholar] [CrossRef]
- Frigerio, F.; Pasqualini, G.; Craparotta, I.; Marchini, S.; van Vliet, E.A.; Foerch, P.; Vandenplas, C.; Leclercq, K.; Aronica, E.; Porcu, L.; et al. n-3 Docosapentaenoic acid-derived protectin D1 promotes resolution of neuroinflammation and arrests epileptogenesis. Brain 2018, 141, 3130–3143. [Google Scholar] [CrossRef] [PubMed]
- Gobbetti, T.; Dalli, J.; Colas, R.A.; Federici Canova, D.; Aursnes, M.; Bonnet, D.; Alric, L.; Vergnolle, N.; Deraison, C.; Hansen, T.V. Protectin D1n-3 DPA and resolvin D5n-3 DPA are effectors of intestinal protection. Proc. Natl. Acad. Sci. USA 2017, 114, 3963–3968. [Google Scholar] [CrossRef]
- Tungen, J.E.; Primdahl, K.G.; Hansen, T.V. The First Total Synthesis of the Lipid Mediator PD2n-3 DPA. J. Nat. Prod. 2020, 83, 2255–2260. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Gerstmann, L.; Vik, A.; De Matteis, R.; Colas, R.A.; Dalli, J.; Chiang, N.; Serhan, C.N.; Kalesse, M.; Hansen, T.V. Resolving Inflammation: Synthesis, Configurational Assignment, and Biological Evaluations of RvD1n-3 DPA. Chem. Eur. J. 2019, 25, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Aursnes, M.; Dalli, J.; Arnardottir, H.; Serhan, C.N.; Hansen, T.V. Total Synthesis of the Anti-inflammatory and Pro-resolving Lipid Mediator MaR1n-3 DPA Utilizing an sp3–sp3 Negishi Cross-Coupling Reaction. Chem. Eur. J. 2014, 20, 14575–14578. [Google Scholar] [CrossRef] [PubMed]
- Detterbeck, R.; Guggisberg, A.; Popaj, K.; Hesse, M. (−)-(3S)-3-(Tosylamino) butano-4-lactone, a Versatile Chiral Synthon for the Enantioselective Synthesis of Different Types of Polyamine Macrocycles: Determination of the Absolute Configuration of (−)-(R)-Budmunchiamine A. Helv. Chim. Acta 2002, 85, 1742–1758. [Google Scholar] [CrossRef]
- Sønderskov, J.; Tungen, J.E.; Palmas, F.; Dalli, J.; Serhan, C.N.; Stenstrøm, Y.; Hansen, T.V. Stereoselective synthesis of MaR2n-3 DPA. Tetrahedron Lett. 2020, 61, 151510. [Google Scholar] [CrossRef]
- Rooke, D.A.; Ferreira, E.M. Platinum-Catalyzed Hydrosilylations of Internal Alkynes: Harnessing Substituent Effects to Achieve High Regioselectivity. Angew. Chem. Int. Ed. 2012, 51, 3225–3230. [Google Scholar] [CrossRef]
- Reinertsen, A.F.; Primdahl, K.G.; De Matteis, R.; Dalli, J.; Hansen, T.V. Stereoselective Synthesis, Configurational Assignment and Biological Evaluations of the Lipid Mediator RvD2n-3 DPA. Chem. Eur. J. 2022, 28, e202103857. [Google Scholar] [CrossRef]
- Rodriguez, A.R.; Spur, B.W. First total syntheses of the pro-resolving lipid mediators 7(S),13(R),20(S)-Resolvin T1 and 7(S),13(R)-Resolvin T4. Tetrahedron Lett. 2020, 61, 151473. [Google Scholar] [CrossRef] [PubMed]
- Boland, W.; Schroer, N.; Sieler, C.; Feigel, M. Sterospecific Syntheses and Spectroscopic Properties of Isomeric 2,4,6,8-Undecatetraenes. New Hydrocarbons from the Marine Brown Alga Giffordia mitchellae. Part IV. Helv. Chim. Acta 1987, 70, 1025–1040. [Google Scholar] [CrossRef]
- Mohamed, Y.M.A.; Hansen, T.V. Z-Stereoselective semi-reduction of alkynes: Modification of the Boland protocol. Tetrahedron 2013, 69, 3872–3877. [Google Scholar] [CrossRef]
- Näf, F.; Decorzant, R.; Thommen, W.; Willhalm, B.; Ohloff, G. The Four Isomeric 1,3,5-Undecatrienes. Synthesis and configurational assignment. Helv. Chim. Acta 1975, 58, 1016–1037. [Google Scholar] [CrossRef]
- Koenis, D.S.; Beegun, I.; Jouvene, C.C.; Aguirre, G.A.; Souza, P.R.; Gonzalez-Nunez, M.; Ly, L.; Pistorius, K.; Kocher, H.M.; Ricketts, W.; et al. Disrupted Resolution Mechanisms Favor Altered Phagocyte Responses in COVID-19. Circ. Res. 2021, 129, e54–e71. [Google Scholar] [CrossRef] [PubMed]
- Ervik, K.; Reinertsen, A.F.; Koenis, D.S.; Dalli, J.; Hansen, T.V. Stereoselective Synthesis, Pro-resolution, and Anti-inflammatory Actions of RvD5n-3 DPA. J. Nat. Prod. 2023, 86, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Reinertsen, A.F.; Primdahl, K.G.; Shay, A.E.; Serhan, C.N.; Hansen, T.V.; Aursnes, M. Stereoselective Synthesis and Structural Confirmation of the Specialized Pro-Resolving Mediator Resolvin E4. J. Org. Chem. 2021, 86, 3535–3545. [Google Scholar] [CrossRef]
- Flak, M.B.; Koenis, D.S.; Sobrino, A.; Smith, J.; Pistorius, K.; Palmas, F.; Dalli, J. GPR101 mediates the pro-resolving actions of RvD5n-3 DPA in arthritis and infections. J. Clin. Investig. 2020, 130, 359–373. [Google Scholar] [CrossRef]
- Walker, M.E.; De Matteis, R.; Perretti, M.; Dalli, J. Resolvin T4 enhances macrophage cholesterol efflux to reduce vascular disease. Nat. Commun. 2024, 15, 975. [Google Scholar] [CrossRef]
- Rodriguez, A.R.; Spur, B.W. First total synthesis of the pro-resolving lipid mediator 7(S),12(R),13(S)-Resolvin T2 and its 13(R)-epimer. Tetrahedron Lett. 2020, 61, 151857. [Google Scholar] [CrossRef]
- Chiang, N.; Sakuma, M.; Rodriguez, A.R.; Spur, B.W.; Irimia, D.; Serhan, C.N. Resolvin T-series reduce neutrophil extracellular traps. Blood 2022, 139, 1222–1233. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed]
- Azcutia, V.; Parkos, C.A.; Brazil, J.C. Role of negative regulation of immune signaling pathways in neutrophil function. J. Leukoc. Biol. 2018, 103, 1029–1041. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.W.; Berg, N.K.; Reskallah, A.; Yuan, X.; Eltzschig, H.K. Acute Respiratory Distress Syndrome: Contemporary Management and Novel Approaches during COVID-19. Anesthesiology 2021, 134, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018, 32, 6358–6370. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and COVID-19: Nots, NETs, and knots. J. Exp. Med. 2020, 217, e20201439. [Google Scholar] [CrossRef]
- Vik, A.; Dalli, J.; Hansen, T.V. Recent advances in the chemistry and biology of anti-inflammatory and specialized pro-resolving mediators biosynthesized from n-3 docosapentaenoic acid. Bioorg. Med. Chem. Lett. 2017, 27, 2259–2266. [Google Scholar] [CrossRef]
- Nesman, J.I.; Primdahl, K.G.; Tungen, J.E.; Palmas, F.; Dalli, J.; Hansen, T.V. Synthesis, structural confirmation, and biosynthesis of 22-OH-PD1n-3 DPA. Molecules 2019, 24, 3228. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Cameron-Smith, D.; Garg, M.; Sinclair, A.J. Docosapentaenoic acid (22: 5n-3): A review of its biological effects. Prog. Lipid Res. 2011, 50, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Haatveit, Å.S.; Hansen, T.V. The Biosynthetic Pathways of the Protectins. Prostaglandins Other Lipid Mediat. 2023, 169, 106787. [Google Scholar] [CrossRef]
- Nesman, J.I.; Chen, O.; Luo, X.; Ji, R.-R.; Serhan, C.N.; Hansen, T.V. A new synthetic protectin D1 analog 3-oxa-PD1n-3 DPA reduces neuropathic pain and chronic itch in mice. Org. Biomol. Chem. 2021, 19, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Furutani, K.; Chen, O.; McGinnis, A.; Wang, Y.; Serhan, C.N.; Hansen, T.V.; Ji, R.-R. Novel pro-resolving lipid mediator mimetic 3-oxa-PD1n-3 DPA reduces acute and chronic itch by modulating excitatory and inhibitory synaptic transmission and astroglial secretion of lipocalin-2 in mice. Pain 2023, 164, 1340–1354. [Google Scholar] [CrossRef] [PubMed]
- Dayaker, G.; Durand, T.; Balas, L. A versatile and stereocontrolled total synthesis of dihydroxylated docosatrienes containing a conjugated E, E, Z-Triene. Chem. Eur. J. 2014, 20, 2879–2887. [Google Scholar] [CrossRef]
- Hansen, T.V.; Dalli, J.; Serhan, C.N. The novel lipid mediator PD1n-3 DPA: An overview of the structural elucidation, synthesis, biosynthesis and bioactions. Prostaglandins Other Lipid Mediat. 2017, 133, 103–110. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinertsen, A.F.; Vik, A.; Hansen, T.V. Biology and Total Synthesis of n-3 Docosapentaenoic Acid-Derived Specialized Pro-Resolving Mediators. Molecules 2024, 29, 2833. https://doi.org/10.3390/molecules29122833
Reinertsen AF, Vik A, Hansen TV. Biology and Total Synthesis of n-3 Docosapentaenoic Acid-Derived Specialized Pro-Resolving Mediators. Molecules. 2024; 29(12):2833. https://doi.org/10.3390/molecules29122833
Chicago/Turabian StyleReinertsen, Amalie Føreid, Anders Vik, and Trond Vidar Hansen. 2024. "Biology and Total Synthesis of n-3 Docosapentaenoic Acid-Derived Specialized Pro-Resolving Mediators" Molecules 29, no. 12: 2833. https://doi.org/10.3390/molecules29122833
APA StyleReinertsen, A. F., Vik, A., & Hansen, T. V. (2024). Biology and Total Synthesis of n-3 Docosapentaenoic Acid-Derived Specialized Pro-Resolving Mediators. Molecules, 29(12), 2833. https://doi.org/10.3390/molecules29122833