
Cell-Free Protein Expression by a Reconstituted Transcription–Translation System Energized by Sugar Catabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
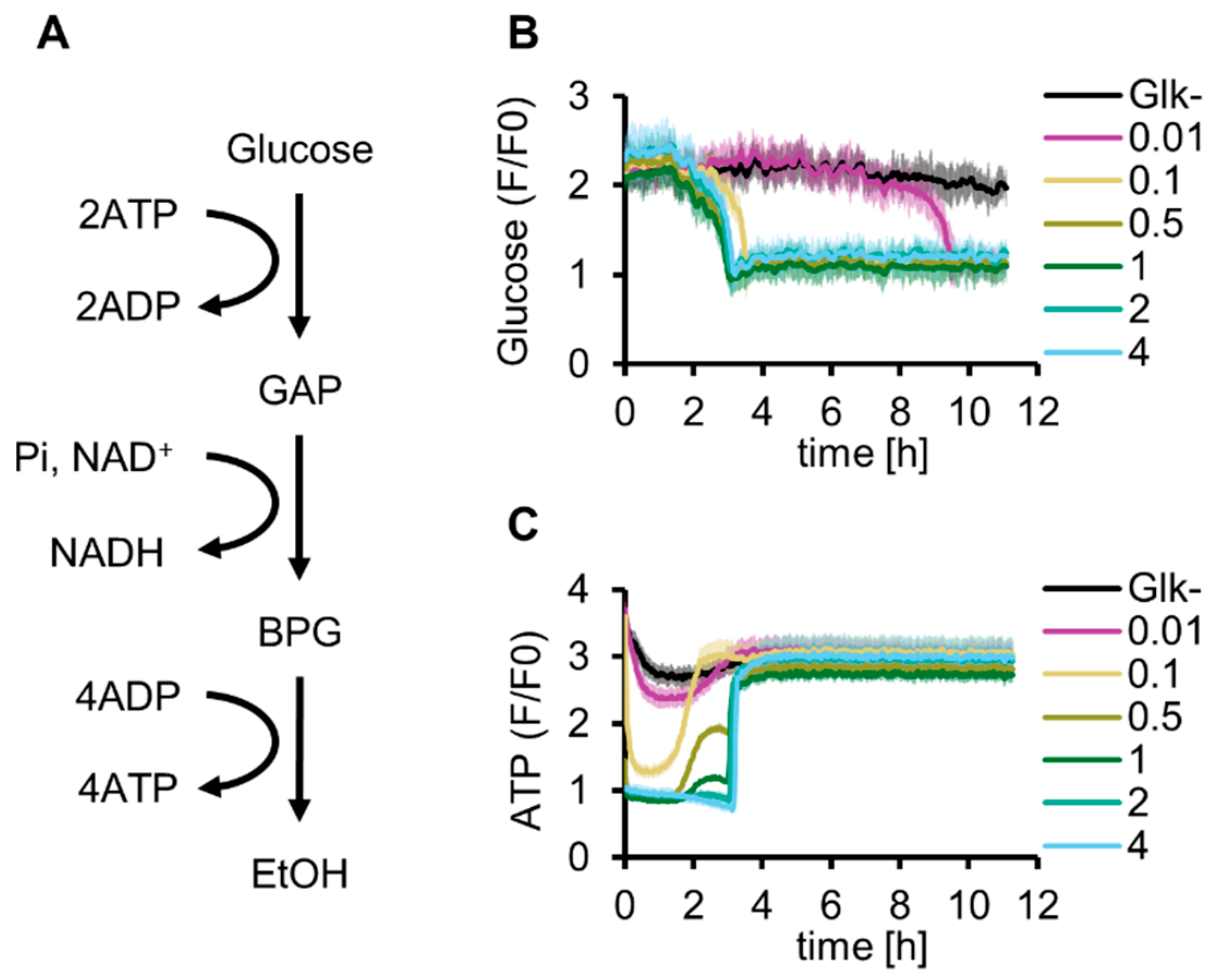
2.1. In Vitro Reconstitution of Glycolysis Using Glucose as an Initial Substrate
2.2. Construction of G-PURE Energized by Sugar
2.3. Optimization of Cell-Free Protein Expression by SG-PURE
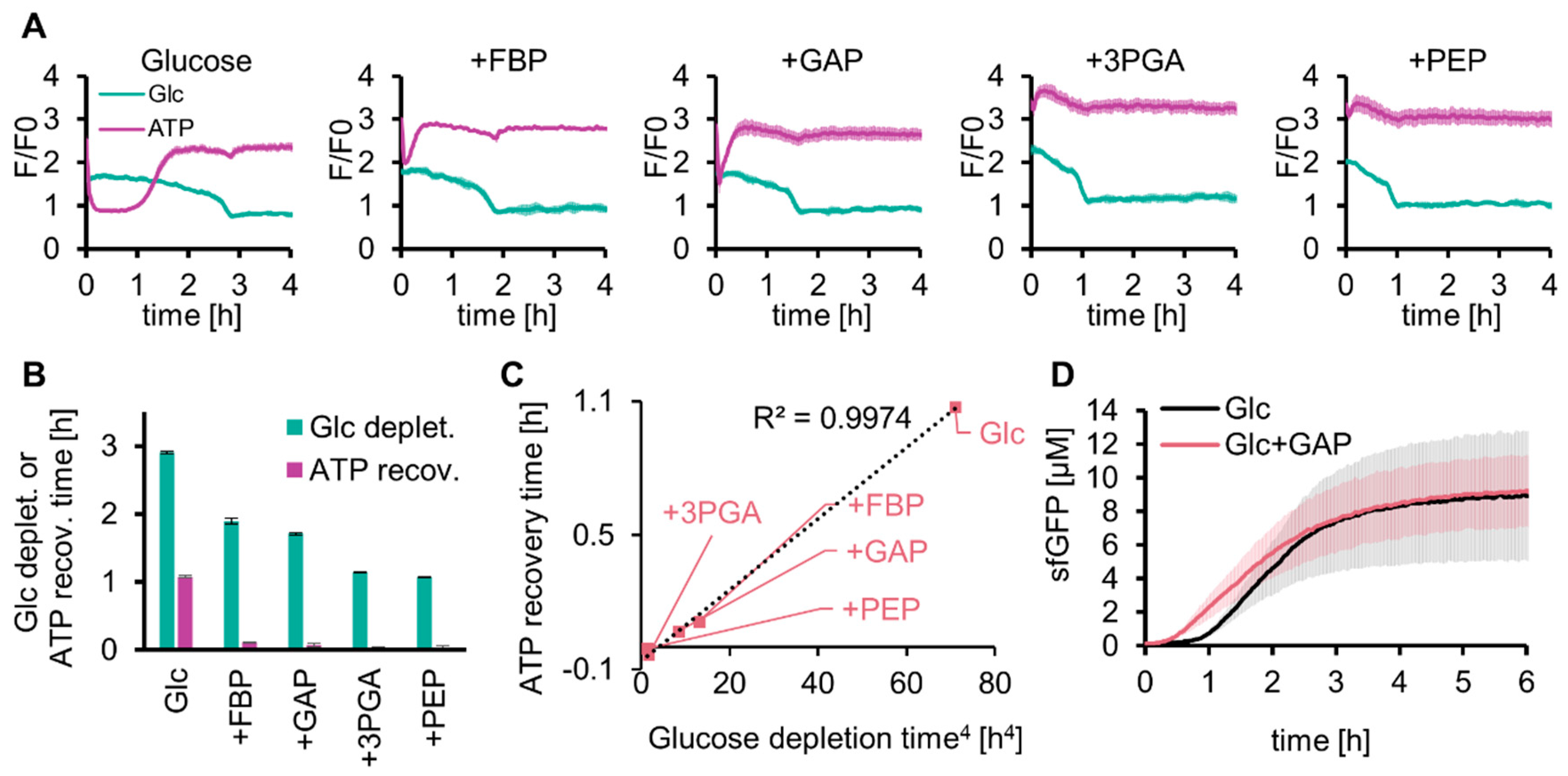
2.4. Effect of Glycolytic Intermediates on Glycolysis and SG-PURE
2.5. Expression of Various Proteins by SG-PURE as a Step toward Self-Replication
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Expression and Purification of Glycolytic Enzymes and PDC
4.3. Expression and Purification of Green Glifon4000 and MaLionR
4.4. In Vitro Reconstitution of Glycolysis
4.5. G-PURE and SG-PURE Reactions
4.6. Estimation of Glucose Depletion Time and ATP Recovery Time of Glycolysis
4.7. Quantification of Ethanol
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boiteux, A.; Hess, B. Design of Glycolysis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1981, 293, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Carreón-Rodríguez, O.E.; Gosset, G.; Escalante, A.; Bolívar, F. Glucose Transport in Escherichia Coli: From Basics to Transport Engineering. Microorganisms 2023, 11, 1588. [Google Scholar] [CrossRef]
- Karp, P.D.; Paley, S.; Caspi, R.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Moore, L.R.; Subhraveti, P.; Gama-Castro, S.; Tierrafria, V.H.; et al. The EcoCyc Database (2023). EcoSal Plus 2023, 11, eesp00022023. [Google Scholar] [CrossRef]
- Locasale, J.W. New Concepts in Feedback Regulation of Glucose Metabolism. Curr. Opin. Syst. Biol. 2018, 8, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Fothergill-Gilmore, L.A. The Evolution of the Glycolytic Pathway. Trends Biochem. Sci. 1986, 11, 47–51. [Google Scholar] [CrossRef]
- Sato, T.; Atomi, H. Novel Metabolic Pathways in Archaea. Curr. Opin. Microbiol. 2011, 14, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Deutscher, J. The Mechanisms of Carbon Catabolite Repression in Bacteria. Curr. Opin. Microbiol. 2008, 11, 87–93. [Google Scholar] [CrossRef]
- Calhoun, K.A.; Swartz, J.R. Energizing Cell-Free Protein Synthesis with Glucose Metabolism. Biotechnol. Bioeng. 2005, 90, 606–613. [Google Scholar] [CrossRef]
- Calhoun, K.A.; Swartz, J.R. An Economical Method for Cell-Free Protein Synthesis Using Glucose and Nucleoside Monophosphates. Biotechnol. Prog. 2005, 21, 1146–1153. [Google Scholar] [CrossRef]
- Kim, T.-W.; Oh, I.-S.; Keum, J.-W.; Kwon, Y.-C.; Byun, J.-Y.; Lee, K.-H.; Choi, C.-Y.; Kim, D.-M. Prolonged Cell-Free Protein Synthesis Using Dual Energy Sources: Combined Use of Creatine Phosphate and Glucose for the Efficient Supply of ATP and Retarded Accumulation of Phosphate. Biotechnol. Bioeng. 2007, 97, 1510–1515. [Google Scholar] [CrossRef]
- Caschera, F.; Noireaux, V. Synthesis of 2.3 Mg/Ml of Protein with an All Escherichia Coli Cell-Free Transcription-Translation System. Biochimie 2014, 99, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-Free Translation Reconstituted with Purified Components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Sato, G.; Kinoshita, S.; Yamada, T.G.; Arai, S.; Kitaguchi, T.; Funahashi, A.; Doi, N.; Fujiwara, K. Metabolic Tug-of-War between Glycolysis and Translation Revealed by Biochemical Reconstitution. ACS Synth. Biol. 2024, 13, 1572–1581. [Google Scholar] [CrossRef]
- Saier, M.H., Jr. The Bacterial Phosphotransferase System: New Frontiers 50 Years after Its Discovery. J. Mol. Microbiol. Biotechnol. 2015, 25, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Völker, B.; Boles, E.; Fuhrmann, G.F. Characterisation of Glucose Transport in Saccharomyces Cerevisiae with Plasma Membrane Vesicles (Countertransport) and Intact Cells (Initial Uptake) with Single Hxt1, Hxt2, Hxt3, Hxt4, Hxt6, Hxt7 or Gal2 Transporters. FEMS Yeast Res. 2002, 2, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Koning, S.M.; Albers, S.-V.; Konings, W.N.; Driessen, A.J.M. Sugar Transport in (Hyper)Thermophilic Archaea. Res. Microbiol. 2002, 153, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Mueckler, M. Glucose Transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed]
- Teusink, B.; Walsh, M.C.; van Dam, K.; Westerhoff, H.V. The Danger of Metabolic Pathways with Turbo Design. Trends Biochem. Sci. 1998, 23, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Welch, P.; Scopes, R.K. Studies on Cell-Free Metabolism: Ethanol Production by a Yeast Glycolytic System Reconstituted from Purified Enzymes. J. Biotechnol. 1985, 2, 257–273. [Google Scholar] [CrossRef]
- Thevelein, J.M.; Hohmann, S. Trehalose Synthase: Guard to the Gate of Glycolysis in Yeast? Trends Biochem. Sci. 1995, 20, 3–10. [Google Scholar] [CrossRef]
- Bujara, M.; Schümperli, M.; Pellaux, R.; Heinemann, M.; Panke, S. Optimization of a Blueprint for in Vitro Glycolysis by Metabolic Real-Time Analysis. Nat. Chem. Biol. 2011, 7, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, B.J.; Liu, J.-W.; Kuchel, P.W.; Ollis, D.L. Fermentative Glycolysis with Purified Escherichia Coli Enzymes for in Vitro ATP Production and Evaluating an Engineered Enzyme. J. Biotechnol. 2012, 157, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Jia, X.; Hu, M.; Deng, Z.; Xu, Y.; Liu, T. In Vitro Reconstitution and Optimization of the Entire Pathway to Convert Glucose into Fatty Acid. ACS Synth. Biol. 2017, 6, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Opgenorth, P.H.; Korman, T.P.; Iancu, L.; Bowie, J.U. A Molecular Rheostat Maintains ATP Levels to Drive a Synthetic Biochemistry System. Nat. Chem. Biol. 2017, 13, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, Z.R.; Bianchi, D.M.; Brier, T.A.; Gilbert, B.R.; Earnest, T.M.; Melo, M.C.R.; Safronova, N.; Sáenz, J.P.; Cook, A.T.; Wise, K.S.; et al. Fundamental Behaviors Emerge from Simulations of a Living Minimal Cell. Cell 2022, 185, 345–360.e28. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.; Ito, M.; Harada, K.; Sugawara, I.; Ueda, H.; Tsuboi, T.; Kitaguchi, T. Green Fluorescent Protein-Based Glucose Indicators Report Glucose Dynamics in Living Cells. Anal. Chem. 2019, 91, 4821–4830. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Kriszt, R.; Harada, K.; Looi, L.-S.; Matsuda, S.; Wongso, D.; Suo, S.; Ishiura, S.; Tseng, Y.-H.; Raghunath, M.; et al. RGB-Color Intensiometric Indicators to Visualize Spatiotemporal Dynamics of ATP in Single Cells. Angew. Chem. Int. Ed. 2018, 57, 10873–10878. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, C.; Huang, P.; Kuru, E.; Forster-Benson, E.T.C.; Li, T.; Church, G.M. Dissecting Limiting Factors of the Protein Synthesis Using Recombinant Elements (PURE) System. Translation 2017, 5, e1327006. [Google Scholar] [CrossRef]
- Wang, P.-H.; Fujishima, K.; Berhanu, S.; Kuruma, Y.; Jia, T.Z.; Khusnutdinova, A.N.; Yakunin, A.F.; McGlynn, S.E. A Bifunctional Polyphosphate Kinase Driving the Regeneration of Nucleoside Triphosphate and Reconstituted Cell-Free Protein Synthesis. ACS Synth. Biol. 2020, 9, 36–42. [Google Scholar] [CrossRef]
- Berhanu, S.; Ueda, T.; Kuruma, Y. Artificial Photosynthetic Cell Producing Energy for Protein Synthesis. Nat. Commun. 2019, 10, 1325. [Google Scholar] [CrossRef]
- Sugii, S.; Hagino, K.; Mizuuchi, R.; Ichihashi, N. Cell-Free Expression of RuBisCO for ATP Production in the Synthetic Cells. Synth. Biol. 2023, 8, ysad016. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Adam, D.; Giaveri, S.; Barthel, S.; Cestellos-Blanco, S.; Hege, D.; Paczia, N.; Castañeda-Losada, L.; Klose, M.; Arndt, F.; et al. ATP Production from Electricity with a New-to-Nature Electrobiological Module. Joule 2023, 7, 1745–1758. [Google Scholar] [CrossRef]
- Giaveri, S.; Bohra, N.; Diehl, C.; Ballinger, M.; Paczia, N.; Glatter, T.; Erb, T.J. An Interdependent Metabolic and Genetic Network Shows Emergent Properties in Vitro. bioRxiv, 2023; preprint. [Google Scholar]
- Fujiwara, K.; Katayama, T.; Nomura, S.-I.M. Cooperative Working of Bacterial Chromosome Replication Proteins Generated by a Reconstituted Protein Expression System. Nucleic Acids Res. 2013, 41, 7176–7183. [Google Scholar] [CrossRef]
- Yoshida, A.; Kohyama, S.; Fujiwara, K.; Nishikawa, S.; Doi, N. Regulation of Spatiotemporal Patterning in Artificial Cells by a Defined Protein Expression System. Chem. Sci. 2019, 10, 11064–11072. [Google Scholar] [CrossRef] [PubMed]
- Libicher, K.; Hornberger, R.; Heymann, M.; Mutschler, H. In Vitro Self-Replication and Multicistronic Expression of Large Synthetic Genomes. Nat. Commun. 2020, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Doerr, A.; Foschepoth, D.; Forster, A.C.; Danelon, C. In Vitro Synthesis of 32 Translation-Factor Proteins from a Single Template Reveals Impaired Ribosomal Processivity. Sci. Rep. 2021, 11, 1898. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, S.; Merino-Salomón, A.; Schwille, P. In Vitro Assembly, Positioning and Contraction of a Division Ring in Minimal Cells. Nat. Commun. 2022, 13, 6098. [Google Scholar] [CrossRef] [PubMed]
- Eto, S.; Matsumura, R.; Shimane, Y.; Fujimi, M.; Berhanu, S.; Kasama, T.; Kuruma, Y. Phospholipid Synthesis inside Phospholipid Membrane Vesicles. Commun. Biol. 2022, 5, 1016. [Google Scholar] [CrossRef] [PubMed]
- Miyachi, R.; Shimizu, Y.; Ichihashi, N. Transfer RNA Synthesis-Coupled Translation and DNA Replication in a Reconstituted Transcription/Translation System. ACS Synth. Biol. 2022, 11, 2791–2799. [Google Scholar] [CrossRef]
- Hagino, K.; Ichihashi, N. In Vitro Transcription/Translation-Coupled DNA Replication through Partial Regeneration of 20 Aminoacyl-TRNA Synthetases. ACS Synth. Biol. 2023, 12, 1252–1263. [Google Scholar] [CrossRef]
- Fujiwara, K.; Sawamura, T.; Niwa, T.; Deyama, T.; Nomura, S.-I.M.; Taguchi, H.; Doi, N. In Vitro Transcription-Translation Using Bacterial Genome as a Template to Reconstitute Intracellular Profile. Nucleic Acids Res. 2017, 45, 11449–11458. [Google Scholar] [CrossRef] [PubMed]
- Deyama, T.; Matsui, Y.; Chadani, Y.; Sekine, Y.; Doi, N.; Fujiwara, K. Transcription-Translation of the Escherichia Coli Genome within Artificial Cells. Chem. Commun. 2021, 57, 10367–10370. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Yu, W.-C.; Jia, T.Z.; He, M.-J.; Khusnutdinova, A.; Yakunin, A.F.; Chiang, Y.-R.; Fujishima, K.; Wang, P.-H. Amino Acid Self-Regenerating Cell-Free Protein Synthesis System That Feeds on PLA Plastics, CO2, Ammonium, and α-Ketoglutarate. ACS Catal. 2024, 14, 7696–7706. [Google Scholar] [CrossRef]
- Stevenson, B.J.; Liu, J.-W.; Ollis, D.L. Directed Evolution of Yeast Pyruvate Decarboxylase 1 for Attenuated Regulation and Increased Stability. Biochemistry 2008, 47, 3013–3025. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Sato, G.; Takada, S.; Kohyama, S.; Honda, G.; Yanagisawa, M.; Hori, Y.; Doi, N.; Yoshinaga, N.; Fujiwara, K. Multimolecular Competition Effect as a Modulator of Protein Localization and Biochemical Networks in Cell-Size Space. Adv. Sci. 2024, 11, e2308030. [Google Scholar] [CrossRef]
- Fujiwara, K.; Adachi, T.; Doi, N. Artificial Cell Fermentation as a Platform for Highly Efficient Cascade Conversion. ACS Synth. Biol. 2018, 7, 363–370. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, G.; Miyazawa, S.; Doi, N.; Fujiwara, K. Cell-Free Protein Expression by a Reconstituted Transcription–Translation System Energized by Sugar Catabolism. Molecules 2024, 29, 2956. https://doi.org/10.3390/molecules29132956
Sato G, Miyazawa S, Doi N, Fujiwara K. Cell-Free Protein Expression by a Reconstituted Transcription–Translation System Energized by Sugar Catabolism. Molecules. 2024; 29(13):2956. https://doi.org/10.3390/molecules29132956
Chicago/Turabian StyleSato, Gaku, Shintaro Miyazawa, Nobuhide Doi, and Kei Fujiwara. 2024. "Cell-Free Protein Expression by a Reconstituted Transcription–Translation System Energized by Sugar Catabolism" Molecules 29, no. 13: 2956. https://doi.org/10.3390/molecules29132956
APA StyleSato, G., Miyazawa, S., Doi, N., & Fujiwara, K. (2024). Cell-Free Protein Expression by a Reconstituted Transcription–Translation System Energized by Sugar Catabolism. Molecules, 29(13), 2956. https://doi.org/10.3390/molecules29132956