
Pharmacophore Modeling and Binding Affinity of Secondary Metabolites from Angelica keiskei to HMG Co-A Reductase

 , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Visualization Results of the HMG-CoA Reductase Pharmacophore Feature
2.2. The Pharmacophore Model Validation Results
2.3. Hit Compound Screening Results
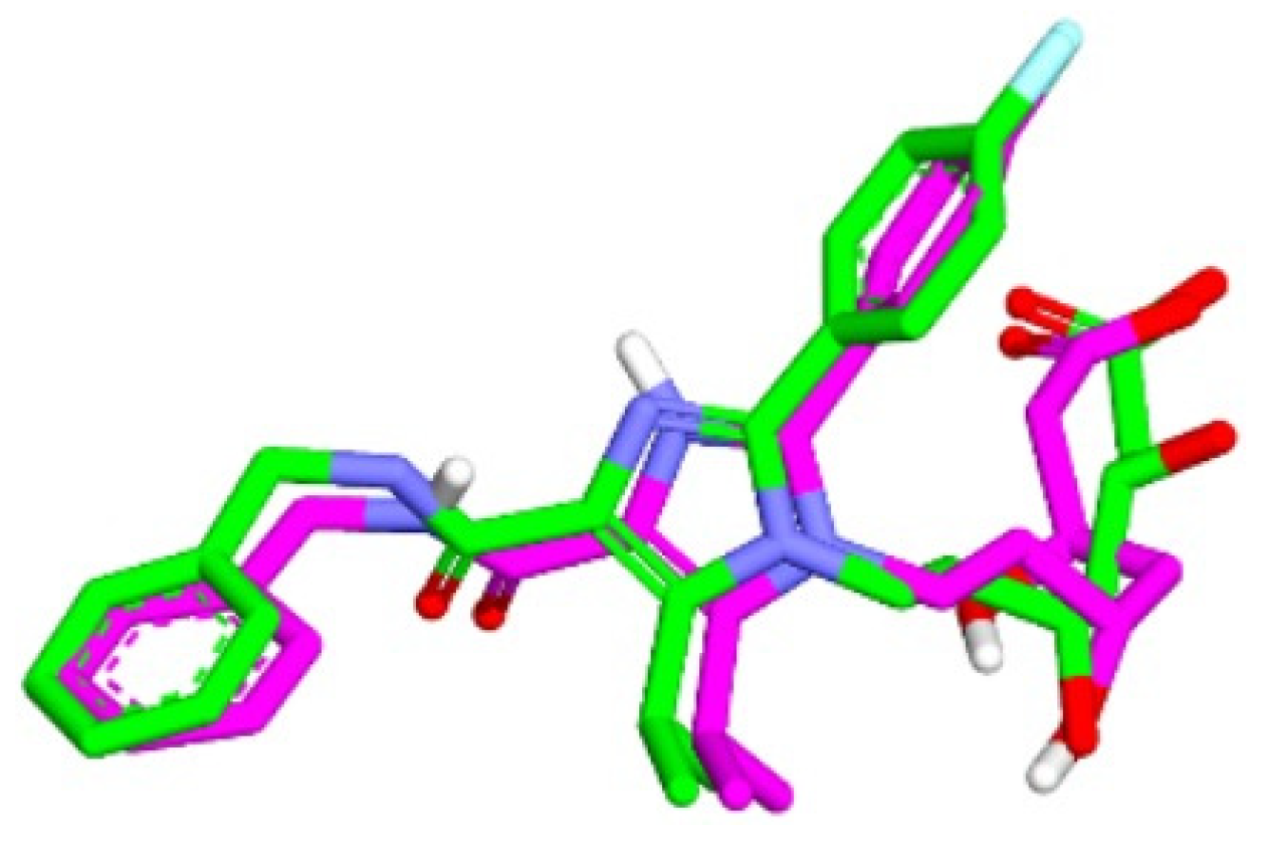
2.4. The Results of Molecular Docking Validation
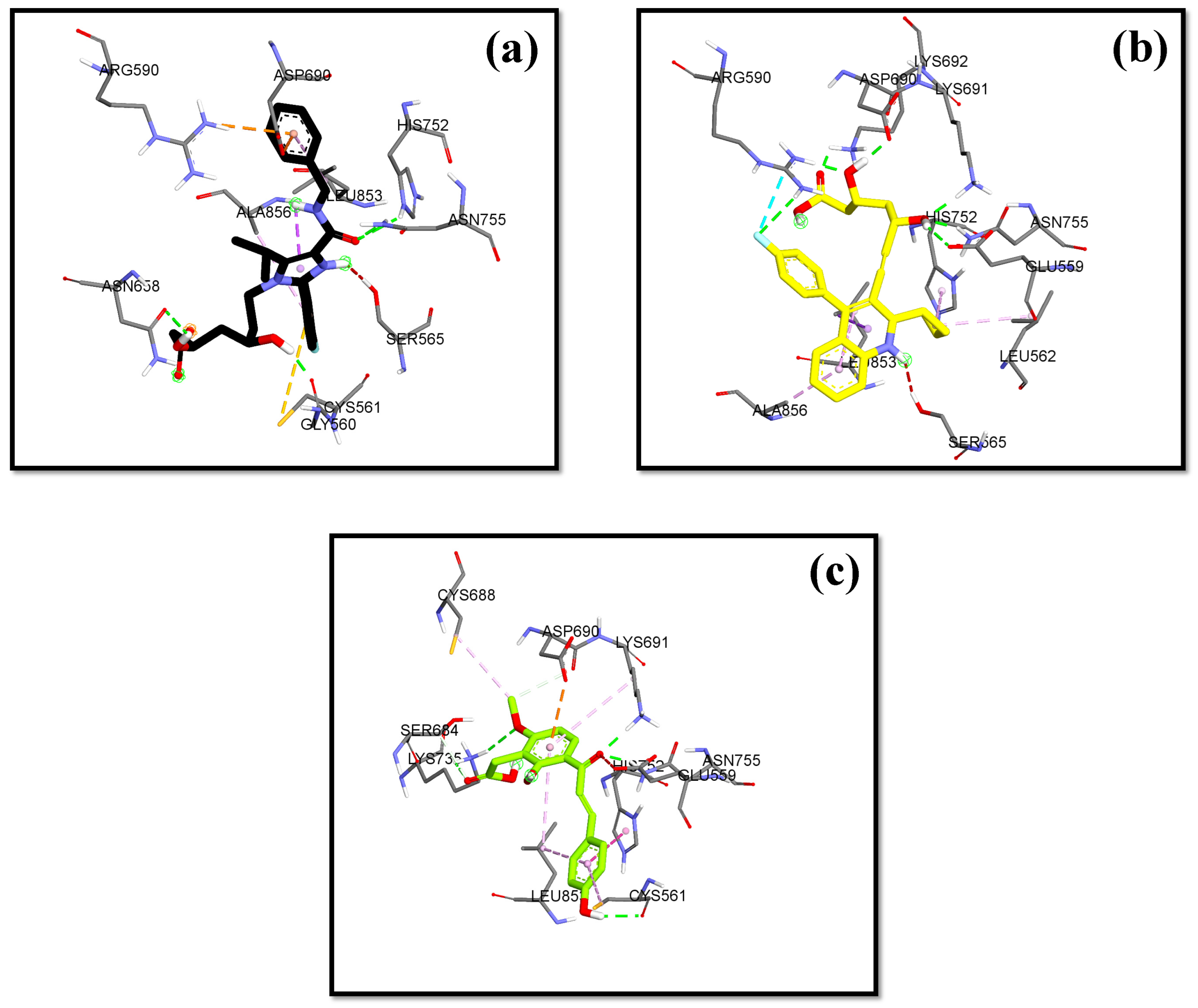
2.5. The Molecular Docking Result of Hit Compounds to HMG-CoA Reductase
3. Discussion
4. Materials and Methods
4.1. Instruments
4.2. Materials
4.3. Methods
4.4. Pharmacophore Modeling
4.5. Pharmacophore Validation
4.6. Screening of Hit Compounds
4.7. Molecular Docking Simulation
4.7.1. Separation of Native Ligands and Receptors
4.7.2. Ligand Preparation
4.7.3. Macromolecule Preparation
4.7.4. Molecular Docking
4.7.5. Overview of Lipinski’s Rule of Five (RO5)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NIH. HMGCR 3-Hydroxy-3-Methylglutaryl-CoA Reductase [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=3156 (accessed on 12 January 2023).
- Harvard Health Publishing. How It’s Made: Cholesterol Production in Your Body—Harvard Health. Available online: https://www.health.harvard.edu/heart-health/how-its-made-cholesterol-production-in-your-body (accessed on 9 February 2023).
- Takei, S.; Nagashima, S.; Takei, A.; Yamamuro, D.; Wakabayashi, T.; Murakami, A.; Isoda, M.; Yamazaki, H.; Ebihara, C.; Takahashi, M.; et al. β-Cell–Specific Deletion of HMG-CoA (3-Hydroxy-3-Methylglutaryl-Coenzyme A) Reductase Causes Overt Diabetes Due to Reduction of β-Cell Mass and Impaired Insulin Secretion. Diabetes 2020, 69, 2352–2363. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Ahmed, K.; Al-Mamun, H.; Hoque, F. Preliminary Assessment of Heavy Metal Contamination in Surface Sediments from a River in Bangladesh. Environ. Earth Sci. 2015, 73, 1837–1848. [Google Scholar] [CrossRef]
- Gesto, D.S.; Pereira, C.M.S.; Cerqueira, N.M.F.S.; Sousa, S.F. An Atomic-Level Perspective of HMG-CoA-Reductase: The Target Enzyme to Treat Hypercholesterolemia. Molecules 2020, 25, 3891. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Jeon, J. Computer-Aided Drug Discovery in Plant Pathology. Plant Pathol. J. 2017, 33, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Sarver, R.W.; Bills, E.; Bolton, G.; Bratton, L.D.; Caspers, N.L.; Dunbar, J.B.; Harris, M.S.; Hutchings, R.H.; Kennedy, R.M.; Larsen, S.D.; et al. Thermodynamic and Structure Guided Design of Statin-Based Inhibitors of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase. J. Med. Chem. 2008, 51, 3804–3813. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Antoszczak, M.; Markowska, J.; Huczyński, A. Statins: Hmg-Coa Reductase Inhibitors as Potential Anticancer Agents against Malignant Neoplasms in Women. Pharmaceuticals 2020, 13, 422. [Google Scholar] [CrossRef]
- Bitzur, R.; Cohen, H.; Kamari, Y.; Harats, D. Intolerance to Statins: Mechanisms and Management. Diabetes Care 2013, 36, S325–S330. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Preiss, D.; Kuchenbaecker, K.B.; Holmes, M.V.; Engmann, J.E.L.; Shah, T.; Sofat, R.; Stender, S.; Johnson, P.C.D.; Scott, R.A.; et al. HMG-Coenzyme A Reductase Inhibition, Type 2 Diabetes, and Bodyweight: Evidence from Genetic Analysis and Randomised Trials. Lancet 2015, 385, 351–361. [Google Scholar] [CrossRef]
- Cho, Y.; Choe, E.; Lee, Y.; Seo, J.W.; Choi, Y.; Yun, Y.; Wang, H.J.; Ahn, C.W.; Cha, B.S.; Lee, H.C.; et al. Risk of Diabetes in Patients Treated with HMG-CoA Reductase Inhibitors. Metabolism 2015, 64, 482–488. [Google Scholar] [CrossRef]
- Hoogwerf, B.J. Statins May Increase Diabetes, but Benefit Still Outweighs Risk. Clevel. Clin. J. Med. 2023, 90, 53–62. [Google Scholar] [CrossRef]
- Kusumawardhany, P.A.; Dewi, A.D.R.; Iswadi, H.; WIdjaja, L.K. Tanaman Malaikat Dari Trawas, Indonesia Ashitaba, 1st ed.; Direktorat Penerbitan & Publikasi Ilmiah, Universitas Surabaya: Surabaya, Indonesia, 2020. [Google Scholar]
- Battenberg, O.A.; Yang, Y.; Verhelst, S.H.L.; Sieber, S.A. Target Profiling of 4-Hydroxyderricin in S. Aureus Reveals Seryl-TRNA Synthetase Binding and Inhibition by Covalent Modification. Mol. Biosyst. 2013, 9, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sawada, K.; Yamamoto, N.; Ashida, H. 4-Hydroxyderricin and Xanthoangelol from Ashitaba (Angelica keiskei) Suppress Differentiation of Preadiopocytes to Adipocytes via AMPK and MAPK Pathways. Mol. Nutr. Food Res. 2013, 57, 1729–1740. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Son, Y.K.; Kim, G.H.; Hwang, K.H. Xanthoangelol and 4-Hydroxyderricin Are the Major Active Principles of the Inhibitory Activities against Monoamine Oxidases on Angelica keiskei K. Biomol. Ther. 2013, 21, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Curtis-Long, M.J.; Yuk, H.J.; Wang, Y.; Song, Y.H.; Jeong, S.H.; Park, K.H. Quantitative Analysis of Phenolic Metabolites from Different Parts of Angelica keiskei by HPLC-ESI MS/MS and Their Xanthine Oxidase Inhibition. Food Chem. 2014, 153, 20–27. [Google Scholar] [CrossRef]
- Chang, H.R.; Lee, H.J.; Ryu, J. Chalcones from Angelica keiskei Attenuate the Inflammatory Responses by Suppressing Nuclear Translocation of NF-κB. J. Med. Food 2014, 17, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Park, Y.O.; Yu, C.; Lee, S.E.; Park, Y.H. Bioassay-Guided Isolation and Identification of Anti-Platelet-Active Compounds from the Root of Ashitaba (Angelica keiskei Koidz.). Nat. Prod. Res. 2014, 28, 2312–2316. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Kawabata, K.; Miyashita, M.; Okumura, M.; Yamamoto, N.; Takahashi, M.; Ashida, H.; Ohigashi, H. Inhibitory Effects of 4-Hydroxyderricin and Xanthoangelol on Lipopolysaccharide-Induced Inflammatory Responses in RAW264 Macrophages. J. Agric. Food Chem. 2014, 62, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Kil, Y.; Choi, S.; Lee, Y.; Jafari, M.; Seo, E. Chalcones from Angelica keiskei: Evaluation of Their Heat Shock Protein Inducing Activities. J. Nat. Prod. 2015, 78, 2481–2487. [Google Scholar] [CrossRef]
- Kil, Y.-S.; Nam, J.-W.; Lee, J.; Seo, E.K. Separation of Two Major Chalcones from Angelica keiskei by High-Speed Counter-Current Chromatography. Arch. Pharm. Res. 2015, 38, 1506–1511. [Google Scholar] [CrossRef]
- Kil, Y.; Kwon, J.; Lee, D.; Seo, E.K. Three New Chalcones from the Aerial Parts of Angelica keiskei. Helv. Chim. Acta 2016, 99, 393–397. [Google Scholar] [CrossRef]
- Li, J.L.; Gao, L.X.; Meng, F.W.; Tang, C.L.; Zhang, R.J.; Li, J.; Luo, C.; Li, J.; Zhao, W.M. PTP1B Inhibitors from Stems of Angelica keiskei (Ashitaba). Bioorganic Med. Chem. Lett. 2015, 25, 2028–2032. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Ko, J.A.; Kim, D.W.; Kim, Y.M.; Kwon, H.J.; Jeong, H.J.; Kim, C.Y.; Park, K.H.; Lee, W.S.; Ryu, Y.B. Chalcones Isolated from Angelica keiskei Inhibit Cysteine Proteases of SARS-CoV. J. Enzym. Inhib. Med. Chem. 2016, 31, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Iimura, K.; Hattan, J.; Misawa, N.; Shindo, K. CDNA Cloning and Functional Analyses of Ashitaba (Angelica keiskei) Sesquiterpene Synthase Genes. J. Oleo Sci. 2020, 69, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liang, C.; Xu, Q.; Zhang, X.; Liang, X.; Huang, Q.Y.; Su, X.J. Study on Chemical Constituents of Angelica keiskei. Chin. J. Exp. Tradit. Med. Formulae 2012, 18, 103–105. [Google Scholar]
- Luo, L.; Wang, R.; Wang, X.; Ma, Z.; Li, N. Compounds from Angelica keiskei with NQO1 Induction, DPPH Scavenging and α-Glucosidase Inhibitory Activities. Food Chem. 2012, 131, 992–998. [Google Scholar] [CrossRef]
- Zhang, T.; Yamashita, Y.; Yasuda, M.; Yamamoto, N.; Ashida, H. Ashitaba (Angelica keiskei) Extract Prevents Adiposity in High-Fat Diet-Fed C57BL/6 Mice. Food Funct. 2015, 6, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Yuliantini, A.; Nurnahari, N.; Jamil, M.F.; Asnawi, A. Skrininig Fitokimia Ashitaba (Angelica keiskei) Terhadap Enoil ACP Reduktase (InhA) Mycobacterium Tuberculosis Sebagai Senyawa Potensial Anti-TB. Indones. Nat. Res. Pharm. J. 2022, 7, 1–10. [Google Scholar] [CrossRef]
- Muchtaridi; Yanuar, A.; Megantara, S.; Purnomo, H. Teknik Perancangan Obat Berbantukan Komputer; UNPAD Press: Sumedang, Indonesia, 2020. [Google Scholar]
- Nur, M.R.F.; Oktora, S.I. Analisis Kurva Roc Pada Model Logit Dalam Pemodelan Determinan Lansia Bekerja Di Kawasan Timur Indonesia. Indones. J. Stat. Its Appl. 2020, 4, 116–135. [Google Scholar] [CrossRef]
- Arba, M.; Arfan; Trisnawati, A.; Kurniawati, D. Pemodelan Farmakofor Untuk Identifikasi Inhibitor Heat Shock Proteins-90 (HSP-90). J. Farm. Galen. (Galen. J. Pharm.) (e-J.) 2020, 6, 229–236. [Google Scholar]
- Rachmania, R.A. Validasi Protokol Skrining Virtual Dan Analisis Interaksi Inhibitor Antiproliferasi Sel Kanker Berbasis Bahan Alam Terhadap Reseptor Cyclin-Dependent Kinase 4 (Cdk 4). Media Farm. J. Ilmu Farm. 2019, 16, 21. [Google Scholar] [CrossRef]
- Istvan, E.S.; Deisenhofer, J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science 2001, 292, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Palnitkar, M.; Buchanan, S.K.; Deisenhofer, J. Crystal Structure of the Catalytic Portion of Human HMG-CoA Reductase: Insights into Regulation of Activity and Catalysis. EMBO J. 2000, 19, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.H.S.; Oliveira, A.R.S.; Dos Santos, A.M.; da Costa, K.S.; Lima, A.H.L.E.; Alves, C.N.; Lameira, J. Computational Study of Conformational Changes in Human 3-Hydroxy-3-Methylglutaryl Coenzyme Reductase Induced by Substrate Binding. J. Biomol. Struct. Dyn. 2019, 37, 4374–4383. [Google Scholar] [CrossRef]
- Carter, A.A.; Gomes, T.; Camacho, X.; Juurlink, D.N.; Shah, B.R.; Mamdani, M.M. Risk of Incident Diabetes among Patients Treated with Statins: Population-Based Study. BMJ 2013, 346, f2610. [Google Scholar] [CrossRef] [PubMed]
- Aziz, F.K.; Nukitasari, C.; Oktavianingrum, F.A.; Aryati, L.W.; Santoso, B. Hasil In Silico Senyawa Z12501572, Z00321025, SCB5631028 Dan SCB13970547 Dibandingkan Turunan Zerumbon Terhadap Human Liver Glycogen Phosphorylase (1l5Q) Sebagai Antidiabetes. J. Kim. Val. 2016, 2, 120–124. [Google Scholar] [CrossRef]
- Santoso, B. Profil Sebaran Hasil Idock Untuk Kelompok Senyawa Aktif, Decoys, Dan ZINC-Induser Terhadap Protein PPAR-Gamma. In Proceedings of the 5th University Research Colloquium (URECOL), Yogyakarta, Indonesia, 18 February 2017; pp. 1254–1259. [Google Scholar]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Liliana; Istyastono, E.P. Uji in silico senyawa 2,6-dihidroksiantraquinon sebagai ligan pada reseptor estrogen alfa. J. Famasi Sains Dan Komunitas 2015, 12, 76–79. [Google Scholar]
- Jiang, S.-Y.; Li, H.; Tang, J.-J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.-K.; Shi, X.-J.; Cui, H.-W.; Tang, J.; et al. Discovery of a Potent HMG-CoA Reductase Degrader That Eliminates Statin-Induced Reductase Accumulation and Lowers Cholesterol. Nat. Commun. 2018, 9, 5138. [Google Scholar] [CrossRef]
- Vögeli, B.; Shima, S.; Erb, T.J.; Wagner, T. Crystal Structure of Archaeal HMG-CoA Reductase: Insights into Structural Changes of the C-Terminal Helix of the Class-I Enzyme. FEBS Lett. 2019, 593, 543–553. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and Drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Kilo, A.L.; Aman, L.; Sabihi, I.; Kilo, J. Studi Potensi Prazolin Tersubstitusi 1-N Dari Tiosemikarbazon Sebagai Agen Antiamuba Melalui Uji in-Silico. Indones. J. Chem. 2019, 7, 9–16. [Google Scholar]
- Naufa, F.; Mutiah, R.; Yen, Y.; Indrawijaya, A. Studi in Silico Potensi Senyawa Katekin Teh Hijau (Camellia Sinensis) Sebagai Antivirus SARS CoV-2 Terhadap Spike Glycoprotein (6LZG) Dan Main Protease (5R7Y). J. Food Pharm. Sci. 2022, 2022, 584–596. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Result |
|---|---|
| Total compounds in database (D) | 9183 |
| Active total in database (A) | 299 |
| Total hits (Ht) | 970 |
| Active hits (Ha)/True Positive (TP) | 201 |
| False Positives (FP) | 769 |
| True Negatives (TN = D − FP) | 8414 |
| False Negatives (FN = A − Ha) | 98 |
| Area Under ROC Curve (AUC) | 0.8 |
| Enrichment Factor (EF = [Ha × D]/[Ht × A]) | 6.4 |
| Goodness of Hit score | 0.3 |
| Sensitivity (TPR = TP/A) | 0.67 |
| Specificity (TNR = TN/D) | 0.92 |
| Accuracy (ACC = (TP + TN)/(A + D)) | 0.91 |
| No. | Compound | Fit-Pharmacophore Score (%) |
|---|---|---|
| 1 | 4HI | 47.05 |
| 2 | Pitavastatin | 48.14 |
| 3 | Atorvastatin | 48.07 |
| 4 | Lovastatin | 47.55 |
| 5 | Simvastatin | 47.44 |
| 6 | Mevastatin (Compactin) | 47.39 |
| 7 | 4′-O-Geranylnaringenin | 47.98 |
| 8 | Luteolin | 47.70 |
| 9 | Cynaroside | 47.47 |
| 10 | 7-O-Methyl prostratol F | 47.40 |
| 11 | Xanthokeismin A | 47.31 |
| 12 | Daucosterol | 46.92 |
| 13 | Isobavachalcone | 46.92 |
| 14 | Dorsmannin A | 46.91 |
| 15 | 3′-Carboxymethyl-4,2′-dihydroxy-4′-methoxy chalcone | 46.90 |
| 16 | Xanthokeistal A | 46.71 |
| No. | Compound | ΔG (kcal/mol) | Ki (µM) | Important Amino Acid Residues [7] | Other Amino Acid Residues |
|---|---|---|---|---|---|
| Reference Drugs | |||||
| 1 | Pitavastatin | −8.24 | 2.11 | ARG590C, ASN755D, ASP690C, GLU559D, LYS691D, SER565D | LEU562D, LYS692C, HIS752D, LEU853D, ALA856D |
| 2 | Atorvastatin | −5.49 | 1148.17 | ARG590C, GLU559D, LYS735D, SER684C | CYS561D, ALA564D, LEU853D |
| 3 | Lovastatin | −6.88 | 10.65 | ARG590C, ASP690C, LYS691C | LEU562D, HIS752D, LEU853D, ALA856D |
| 4 | Simvastatin | −6.50 | 22.34 | ARG590C, ASP690C, LYS735D, SER684C | SER661C, VAL683C, LYS692C, HIS752D, LEU853D, LEU857D |
| 5 | Mevastatin (Compactin) | −6.86 | 11.82 | ARG590C, LYS691C, SER565D | CYS561D, LEU562D, MET657C, LEU853D, LEU857D |
| Ashitaba’s Compounds | |||||
| 1 | 4′-O-Geranylnaringenin | −6.48 | 20.24 | ARG590C, ASN755D, ASP690C, LYS691C, SER684C | CYS561D, LEU562D, ASN686C, ALA751D, HIS752D, LEU853D, ALA856D |
| 2 | Luteolin | −6.03 | 40.69 | ASP690C, GLU559D, LYS735D, SER565D, SER684C | LEU562D, ALA751D, HIS752D, LEU853D |
| 3 | Cynaroside | −5.43 | 153.65 | ARG590C, ASP690C, ASN755D, GLU559D | LYS692C, ALA751D, LEU853D, ALA856D |
| 4 | 7-O-Methyl prostratol F | −5.84 | 70.70 | ASN755D, ASP690C, GLU559D, LYS735D, LYS691C, SER684C | CYS561D, CYS688C, HIS752D, LEU853D |
| 5 | Xanthokeismin A | −5.11 | 202.71 | ARG590C, ASN755D, ASP690C, LYS691C, SER565D, SER684C | CYS561D, LEU562D, VAL683C, LEU853D, ALA856D, LEU857D |
| 6 | Daucosterol | −5.41 | 203.28 | ARG590C, ASN755D, ASP690C, GLU559D, LYS735D, LYS691C | CYS561D, ALA564D, ALA751D, ALA856D, LEU853D |
| 7 | Isobavachalcone | −6.00 | 42.71 | ARG590C, ASP690C, GLU559D, LYS691C, SER565D | MET657C, ALA751D, SER852D, LEU853D, ALA856D |
| 8 | Dorsmannin A | −6.65 | 15.78 | ARG590C, ASP690C | CYS561D, LEU562D, LEU853D, ALA856D |
| 9 | 3′-Carboxymethyl-4,2′-dihydroxy-4′-methoxy chalcone | −6.67 | 16.66 | ASN755D, ASP690C, GLU559D, LYS735D, LYS691C. SER684C | CYS561D, CYS688C, HIS752D, LEU853D |
| 10 | Xanthokeistal A | −4.77 | 487.55 | ASP690C, LYS691C, SER565D | CYS561D, LYS692C, ALA751D, HIS752D, LEU853D, ALA856D |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aulifa, D.L.; Amirah, S.R.; Rahayu, D.; Megantara, S.; Muchtaridi, M. Pharmacophore Modeling and Binding Affinity of Secondary Metabolites from Angelica keiskei to HMG Co-A Reductase. Molecules 2024, 29, 2983. https://doi.org/10.3390/molecules29132983
Aulifa DL, Amirah SR, Rahayu D, Megantara S, Muchtaridi M. Pharmacophore Modeling and Binding Affinity of Secondary Metabolites from Angelica keiskei to HMG Co-A Reductase. Molecules. 2024; 29(13):2983. https://doi.org/10.3390/molecules29132983
Chicago/Turabian StyleAulifa, Diah Lia, Siti Rafa Amirah, Driyanti Rahayu, Sandra Megantara, and Muchtaridi Muchtaridi. 2024. "Pharmacophore Modeling and Binding Affinity of Secondary Metabolites from Angelica keiskei to HMG Co-A Reductase" Molecules 29, no. 13: 2983. https://doi.org/10.3390/molecules29132983