
Diffusion Nuclear Magnetic Resonance Measurements on Cationic Gold (I) Complexes in Catalytic Conditions: Counterion and Solvent Effects




Abstract
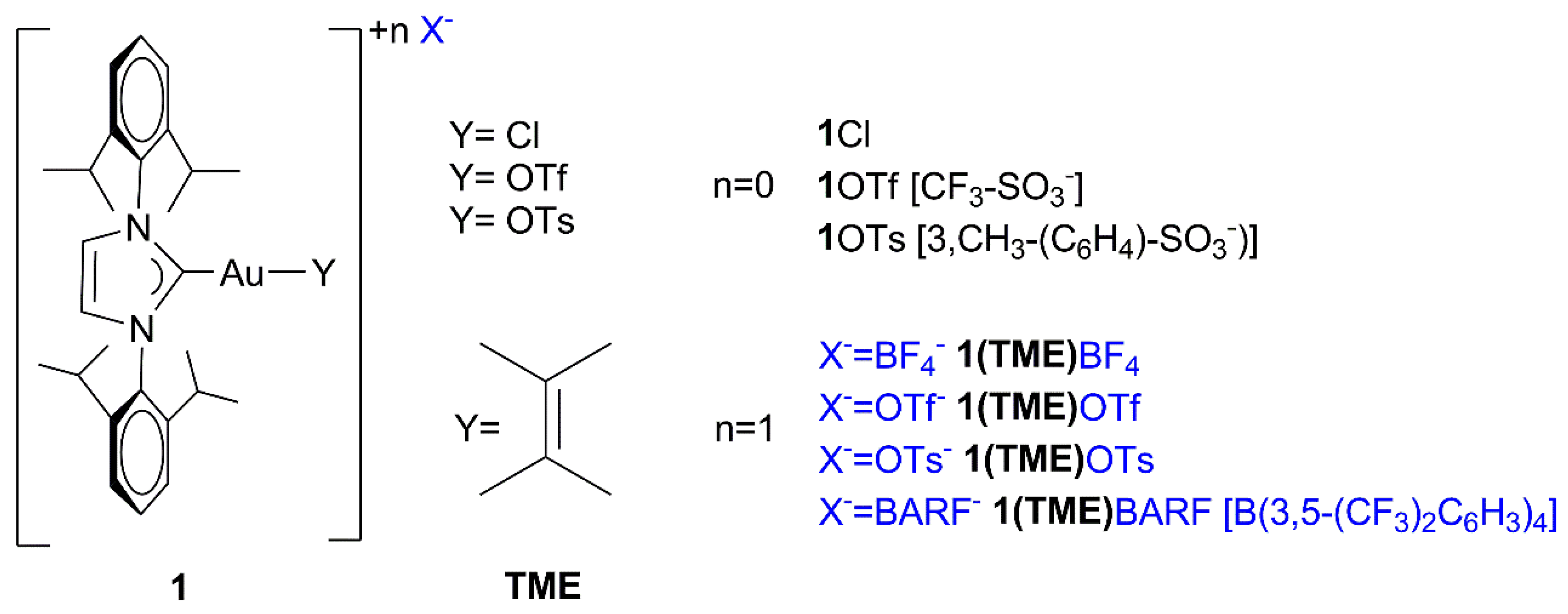
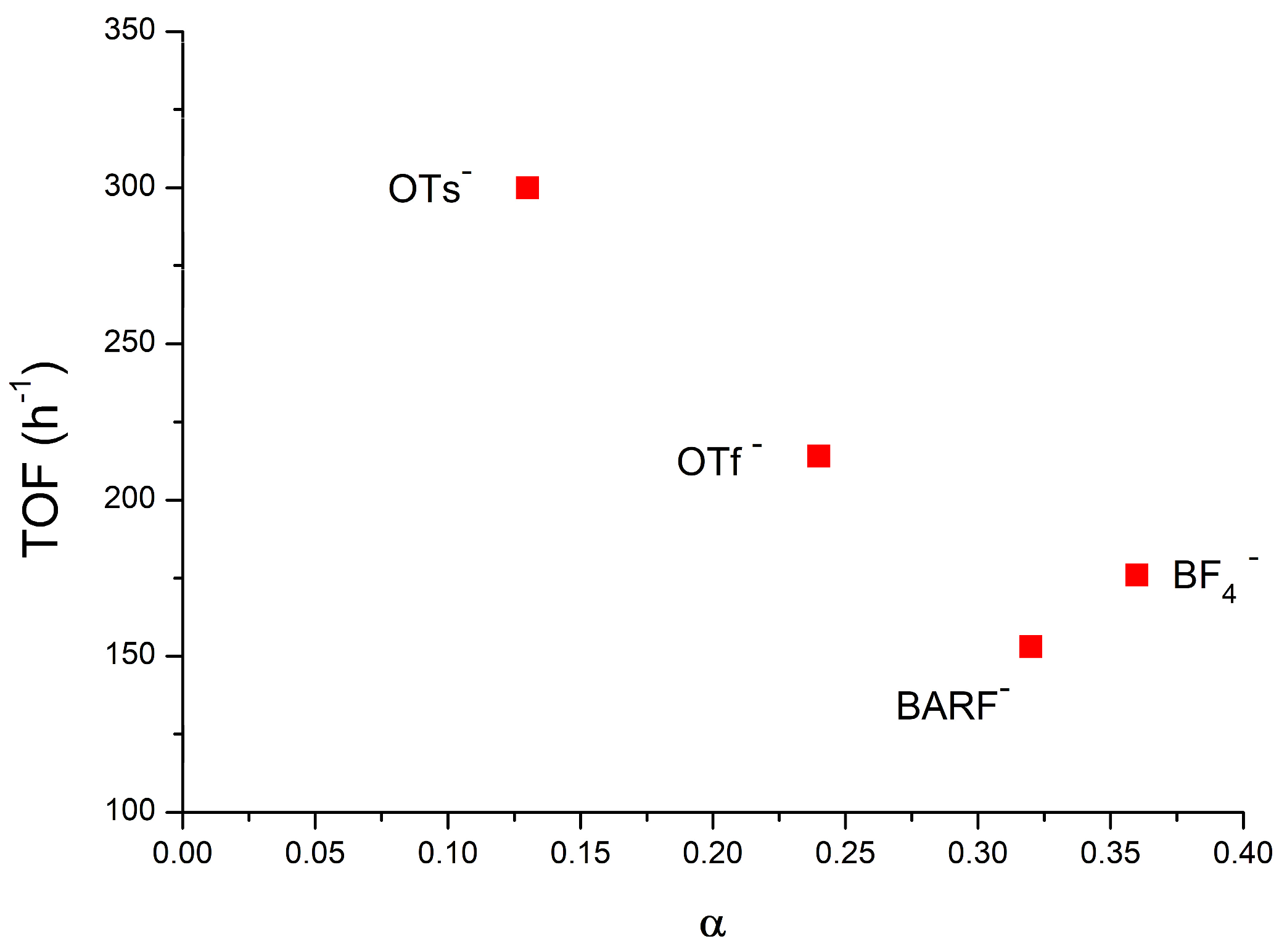
: [(NHC)Au(TME)]+ + X−} depends on the nature of the counterion (X−) when chloroform is the catalytic solvent: while the compounds containing OTs− and OTf− as the counterion gave a low α (which means a high number of ion pairs) of 0.13 and 0.24, respectively, the compounds containing BF4− and BArF4− showed higher α values of 0.36 and 0.32, respectively. These results experimentally confirm previous deductions based on catalytic and theoretical data: the lower the α value, the greater the catalytic activity because the anion that can activate methanol during a nucleophilic attack, although the lower propensity to activate methanol of BF4− and BArF4−, as suggested by the DFT calculations, cannot be completely overlooked. As for the effect of the solvent, α increases as the dielectric constant increases, as expected, and in particular, green solvents with high dielectric constants show a very high α (0.90, 0.84, 0.80, and 0.70 for propylene carbonate, γ-valerolactone, acetone, and methanol, respectively), thus confirming that the moderately high activity of NHC-Au-OTf in these solvents is due to the specific effect of polar functionalities (O-H, C=O, O-R) in activating methanol. Finally, the DOSY measurements conducted in p-Cymene show the formation of quadrupole species: under these conditions, the anion can better exercise its ‘template’ and ‘activating’ roles, giving the highest TOF.
[(NHC)Au(TME)]+ + X−} depends on the nature of the counterion (X−) when chloroform is the catalytic solvent: while the compounds containing OTs− and OTf− as the counterion gave a low α (which means a high number of ion pairs) of 0.13 and 0.24, respectively, the compounds containing BF4− and BArF4− showed higher α values of 0.36 and 0.32, respectively. These results experimentally confirm previous deductions based on catalytic and theoretical data: the lower the α value, the greater the catalytic activity because the anion that can activate methanol during a nucleophilic attack, although the lower propensity to activate methanol of BF4− and BArF4−, as suggested by the DFT calculations, cannot be completely overlooked. As for the effect of the solvent, α increases as the dielectric constant increases, as expected, and in particular, green solvents with high dielectric constants show a very high α (0.90, 0.84, 0.80, and 0.70 for propylene carbonate, γ-valerolactone, acetone, and methanol, respectively), thus confirming that the moderately high activity of NHC-Au-OTf in these solvents is due to the specific effect of polar functionalities (O-H, C=O, O-R) in activating methanol. Finally, the DOSY measurements conducted in p-Cymene show the formation of quadrupole species: under these conditions, the anion can better exercise its ‘template’ and ‘activating’ roles, giving the highest TOF.
1. Introduction
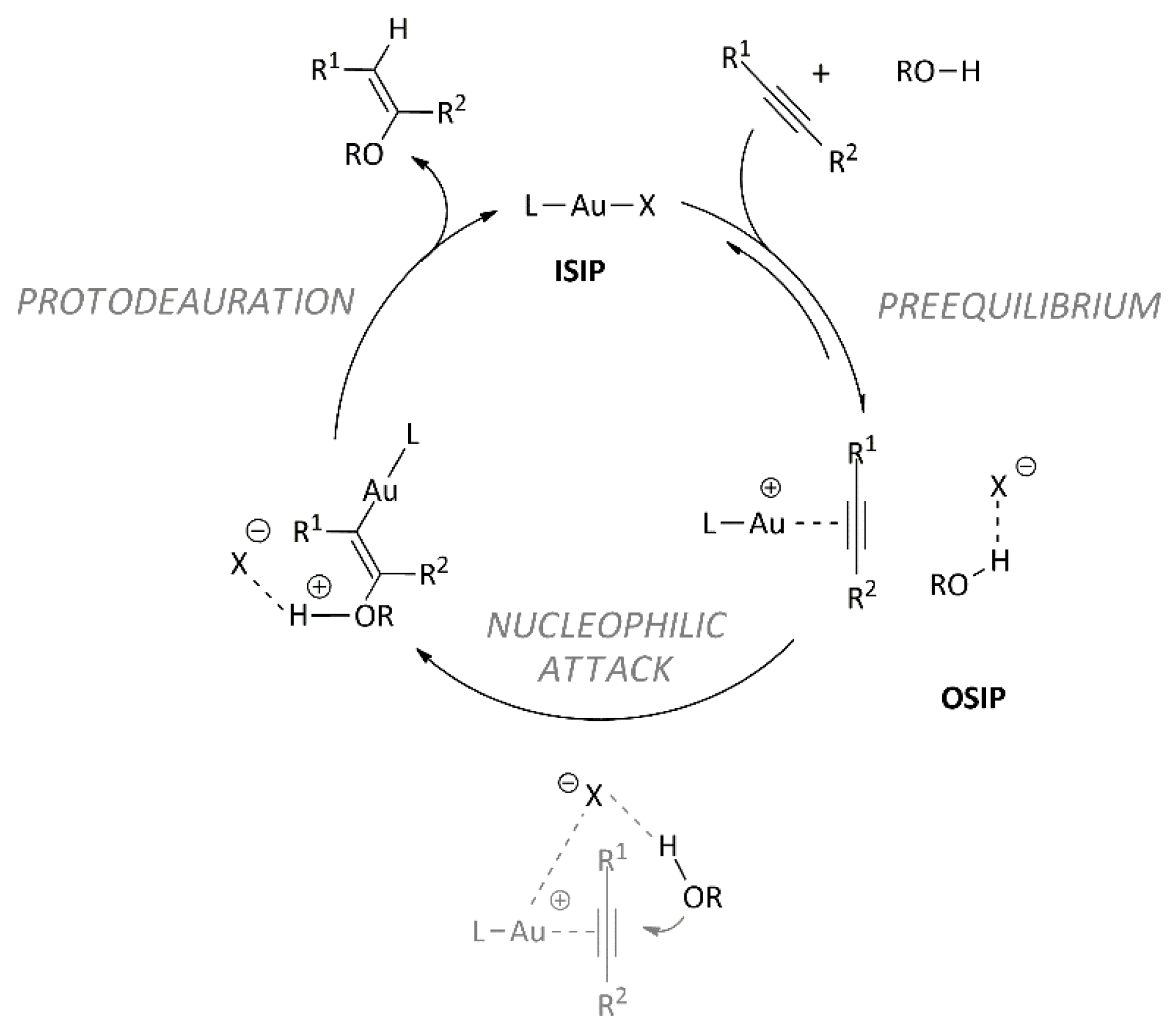
- The rate-determining step resulted in the nucleophilic attack of methanol on the coordinated alkyne (Figure 1), and the intermediate coordination ability, basicity, and hydrogen bond-accepting properties of OTs− and OTf− provide the best compromise for achieving an efficient catalyst (high TOF) [57].
- In the optimized geometry of the transition state, the anions OTs− and OTf− are located near the alkyne, interacting both with the metal center and with the methanol acting as a template, helping the methanol to assume its reactive position and activating the methanol through a hydrogen bond (enhancing the nucleophilicity of the alcohol). If the basicity of the anion is too low (BF4− and BARF−), the template effect is lost, and then the hydrogen bonding with the methanol does not take place [58].
- The polarity of the solvent is crucial in determining the catalytic activity of L-Au(I) complexes because it is related to the amount of ion pairs in the solution. Moreover, peculiar functional groups present in the solvent could promote the nucleophilic attack [54].
[(NHC)Au(TME)]+ + X−} and the amount of higher quadrupolar aggregates {2[(NHC)Au(TME)X] [(NHC)Au(TME)X]2} depends on both the nature of the counterion (X−) and solvent. These values relate to the performance of the (NHC)Au catalyst as a function of X− and the solvent and provide further insight into the effects of ion pairs in gold catalysis.2. Results and Discussion
3. Materials and Methods
3.1. Synthesis and Intramolecular Characterization
3.2. DOSY Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- Obradors, C.; Echavarren, A.M. Intriguing mechanistic labyrinths in gold(I) catalysis. Chem. Commun. 2014, 50, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Lackner, A.D.; Toste, F.D. Development of catalysts and ligands for enantioselective gold catalysis. Acc. Chem. Res. 2014, 47, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Dual gold catalysis. Acc. Chem. Res. 2014, 47, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Yeom, H.-S.; So, E.; Shin, S. Catalytic Access to α-Oxo Gold Carbenes by N–O Bond Oxidants. Acc. Chem. Res. 2014, 47, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. A Non-Diazo Approach to α-Oxo Gold Carbenes via Gold-Catalyzed Alkyne Oxidation. Acc. Chem. Res. 2014, 47, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Zhang, J. Gold-catalyzed cyclopropanation reactions using a carbenoid precursor toolbox. Chem. Soc. Rev. 2015, 44, 677–698. [Google Scholar] [CrossRef] [PubMed]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [PubMed]
- Pflästerer, D.; Hashmi, A.S.K. Gold catalysis in total synthesis—Recent achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef] [PubMed]
- Alyabyev, S.B.; Beletskaya, I.P. Gold as a catalyst. Part I. Nucleophilic addition to the triple bond. Russ. Chem. Rev. 2017, 86, 689–749. [Google Scholar] [CrossRef]
- Alyabyev, S.B.; Beletskaya, I.P. Gold as a catalyst. Part II. Alkynes in the reactions of carbon–carbon bond formation. Russ. Chem. Rev. 2018, 87, 948–1047. [Google Scholar] [CrossRef]
- Nijamudheen, A.; Datta, A. Gold-Catalyzed Cross-Coupling Reactions: An Overview of Design Strategies, Mechanistic Studies, and Applications. Chem.-Eur. J. 2020, 26, 1442–1487. [Google Scholar] [CrossRef]
- Collado, A.; Nelson, D.J.; Nolan, S.P. Optimizing Catalyst and Reaction Conditions in Gold(I) Catalysis–Ligand Development. Chem. Rev. 2021, 121, 8559–8612. [Google Scholar]
- Chintawar, C.C.; Yadav, A.K.; Kumar, A.; Sancheti, S.P.; Patil, N.T. Divergent Gold Catalysis: Unlocking Molecular Diversity through Catalyst Control. Chem. Rev. 2021, 121, 8478–8558. [Google Scholar] [CrossRef]
- Wang, W.; Hammond, G.B.; Xu, B. Ligand Effects and Ligand Design in Homogeneous Gold(I) Catalysis. J. Am. Chem. Soc. 2012, 134, 5697–5705. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Bandini, M. Counterion Effects in Homogeneous Gold Catalysis. ACS Catal. 2015, 5, 1638–1652. [Google Scholar] [CrossRef]
- Xia, Y.; Dudnik, A.S.; Gevorgyan, V.; Li, Y.J. Mechanistic insights into the gold-catalyzed cycloisomerization of bromoallenyl ketones: Ligand-controlled regioselectivity. Am. Chem. Soc. 2008, 130, 6940–6941. [Google Scholar] [CrossRef]
- Jia, M.; Cera, G.; Perrotta, D.; Monari, M.; Bandini, M. Taming Gold (I)–Counterion Interplay in the De-aromatization of Indoles with Allenamides. Chem. Eur. J. 2014, 20, 9875–9878. [Google Scholar] [CrossRef] [PubMed]
- Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A.S.K. An Industrial Perspective on Counter Anions in Gold Catalysis: Underestimated with Respect to “Ligand Effects”. Adv. Synth. Catal. 2018, 360, 2493–2502. [Google Scholar] [CrossRef]
- Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A.S.K. An Industrial Perspective on Counter Anions in Gold Catalysis: On Alternative Counter Anions. Adv. Synth. Catal. 2018, 360, 3949–3959. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, S.; Xu, B. Stable Yet Reactive Cationic Gold Catalysts with Carbon Based Counterions. RSC Adv. 2016, 6, 77830–77833. [Google Scholar] [CrossRef]
- Nieto-Oberhuber, C.; López, S.; Muñoz, M.P.; Cárdenas, D.J.; Buñuel, E.; Nevado, C.; Echavarren, A.M. Divergent Mechanisms forthe Skeletal Rearrangement and [2 + 2] Cycloaddition of Enynes Catalyzed by Gold. Angew. Chem. Int. Ed. 2005, 44, 6146–6148. [Google Scholar] [CrossRef] [PubMed]
- Mézailles, N.; Ricard, L.; Gagosz, F. Phosphine Gold(I) Bis-(trifluoromethanesulfonyl)imidate Complexes as New Highly Efficient and Air-Stable Catalysts for the Cycloisomerization of Enynes. Org. Lett. 2005, 7, 4133–4136. [Google Scholar] [CrossRef] [PubMed]
- Veenboer, R.M.P.; Collado, A.; Dupuy, S.; Lebl, T.; Falivene, L.; Cavallo, L.; Cordes, D.B.; Slawin, A.M.Z.; Cazin, C.S.J.; Nolan, S.P. Inner-Sphere versus Outer-Sphere Coordination of BF4− in a NHC-Gold(I) Complex. Organometallics 2017, 36, 2861–2869. [Google Scholar] [CrossRef]
- Kovács, G.; Ujaque, G.; Lledós, A. The Reaction Mechanism of the Hydroamination of Alkenes Catalyzed by Gold(I)-Phosphine: The Role of the Counterion and the N-Nucleophile Substituents in the Proton-Transfer Step. J. Am. Chem. Soc. 2008, 130, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Han, J.; Okoromoba, O.E.; Shimizu, N.; Amii, H.; Tormena, C.F.; Hammond, G.B.; Xu, B. Predicting Counterion Effects Using a Gold Affinity Index and a Hydrogen Bonding Basicity Index. Org. Lett. 2017, 19, 5848–5851. [Google Scholar] [CrossRef]
- Roithovà, J.; Jankovà, Š.; Jašìkovà, L.; Vàňa, J.; Hybelbauerovà, S. Gold–gold cooperation in the addition of methanol to alkynes. Angew. Chem. Int. Ed. 2012, 51, 8378–8382. [Google Scholar] [CrossRef] [PubMed]
- Campeau, D.; León Rayo, D.F.; Mansour, A.; Muratov, K.; Gagosz, F. Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes, and Alkenes. Chem. Rev. 2021, 121, 8756–8867. [Google Scholar]
- Kumar, M.; Jasinski, J.; Hammond, G.B.; Xu, B. Alkyne/Alkene/Allene-Induced Disproportionation of Cationic Gold(I) Catalyst. Chem.-Eur. J. 2014, 20, 3113–3119. [Google Scholar] [CrossRef]
- Li, D.; Zang, W.; Bird, M.J.; Hyland, C.J.T.; Shi, M. Gold-Catalyzed Conversion of Highly Strained Compounds. Chem. Rev. 2021, 121, 8685–8755. [Google Scholar] [CrossRef]
- Mato, M.; Franchino, A.; García-Morales, C.; Echavarren, A.M. Gold-Catalyzed Synthesis of Small Rings. Chem. Rev. 2021, 121, 8613–8684. [Google Scholar] [CrossRef]
- Brooner, R.E.M.; Brown, T.J.; Chee, M.A.; Widenhoefer, R.A. Effect of Substitution, Ring Size, and Counterion on the Intermediates Generated in the Gold-Catalyzed Intramolecular Hydroalkoxylation of Allenes. Organometallics 2016, 35, 2014–2021. [Google Scholar] [CrossRef]
- Wang, T.; Hashmi, A.S.K. 1,2-Migrations onto Gold Carbene Centers. Chem. Rev. 2021, 121, 8948–8978. [Google Scholar] [CrossRef] [PubMed]
- Bistoni, G.; Belanzoni, P.; Belpassi, L.; Tarantelli, F. π activation of alkynes in homogeneous and heterogeneous gold catalysis. J. Phys. Chem. A 2016, 120, 5239–5247. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.A.; Belpassi, L.; Tarantelli, F.; Zuccaccia, D.; Harvey, J.N.; Belanzoni, P. Dioxygen insertion into the gold(I)-hydride bond: Spin orbit coupling effects in the spotlight for oxidative addition. Chem. Sci. 2016, 7, 7034–7039. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.A.; Belpassi, L.; Tarantelli, F.; Harvey, J.N.; Belanzoni, P. The ligand effect on the oxidative addition of dioxygen to gold(I)-hydride complexes. Dalton Trans. 2017, 46, 11679–11690. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, L.; Ciancaleoni, G.; Tarantelli, F.; Zuccaccia, D.; Belanzoni, P. Unraveling the Anion/Ligand Interplay in the Reaction Mechanism of Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2017, 36, 2364–2376. [Google Scholar] [CrossRef]
- Sorbelli, D.; Nunes dos Santos Comprido, L.; Knizia, G.; Hashmi, A.S.K.; Belpassi, L.; Belanzoni, P.; Klein, J.E.M.N. Cationic gold(I) diarylallenylidene complexes: Bonding features and ligand effects. ChemPhysChem 2019, 20, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Sorbelli, D.; Belanzoni, P.; Belpassi, L. Tuning the gold(I)-carbon σ bond in gold-alkynyl complexes through structural modifications of the NHC ancillary ligand: Effect on spectroscopic observables and reactivity. Eur. J. Inorg. Chem. 2021, 2021, 2401–2416. [Google Scholar] [CrossRef]
- Segato, J.; Baratta, W.; Belanzoni, P.; Belpassi, L.; Del Zotto, A.; Zuccaccia, D. Experimental and Theoretical Investigation of the Cycloisomerization of N-propargylcarboxamide Catalyzed by NHC−Au−X in Green Solvents. Inor. Chem. Acta. 2021, 552, 120372. [Google Scholar] [CrossRef]
- Sabatelli, F.; Segato, J.; Belpassi, L.; Del Zotto, A.; Zuccaccia, D.; Belanzoni, P. Monitoring of the Pre-Equilibrium Step in the Alkyne Hydration Reaction Catalyzed by Au(III) Complexes: A Computational Study Based on Experimental Evidences. Molecules 2021, 26, 2445. [Google Scholar] [CrossRef]
- Segato, J.; Del Zotto, A.; Belpassi, L.; Belanzoni, P.; Zuccaccia, D. Hydration of alkynes catalyzed by [Au(X)(L)(ppy)]X in the green solvent γ-valerolactone under acid-free conditions: The importance of the pre-equilibrium step. Catal. Sci. Technol. 2020, 10, 7757–7767. [Google Scholar] [CrossRef]
- Hamilton, G.L.; Kang, E.J.; Mba, M.; Dean Toste, F.D. A powerful chiral counterion strategy for asymmetric transition metal catalysis. Science 2007, 317, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Zuccaccia, D.; Belpassi, L.; Tarantelli, F.; Macchioni, A. Ion Pairing in Cationic Olefin−Gold(I) Complexes. J. Am. Chem. Soc. 2009, 131, 3170–3171. [Google Scholar] [CrossRef] [PubMed]
- Salvi, N.; Belpassi, L.; Zuccaccia, D.; Tarantelli, F.; Macchioni, A. Ion pairing in NHC gold(I) olefin complexes: A combined experimental/theoretical study. J. Organomet. Chem. 2010, 695, 2679–2686. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Belpassi, L.; Tarantelli, F.; Zuccaccia, D.; Macchioni, A. A combined NMR/DFT study on the ion pair structure of [(PR12R2)Au(η2-3-hexyne)]BF4 complexes. Dalton Trans. 2013, 42, 4122–4131. [Google Scholar] [CrossRef] [PubMed]
- Segato, J.; Aneggi, A.; Baratta, W.; Campagnolo, F.; Belpassi, L.; Belanzoni, P.; Zuccaccia, D. Experimental and Theoretical Investigation of Ion Pairing in Gold(III) Catalysts. Organometallics 2023, 42, 2973–2982. [Google Scholar] [CrossRef]
- Biasiolo, L.; Ciancaleoni, G.; Belpassi, L.; Bistoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D. Relationship between the anion/cation relative orientation and the catalytic activity of nitrogen acyclic carbene–gold catalysts. Catal. Sci. Technol. 2015, 5, 1558–1567. [Google Scholar] [CrossRef]
- The turning point towards a better understanding of how gold (I) can catalyze the addition of an alcohol molecule to an alkyne is dated 1998. This important work was developed by Teles and co-workers, who analyzed the catalytic properties of different L-Au-X complexes in terms of activity (TOF) and stability (TON) in relation to the nature of the neutral ligand L and the anion X−. [Teles, J.H.; Brode, S.; Chabanas, M.; Angew. Chem. Int. Ed. 1998, 37, 1415–1418.]
- Biasiolo, L.; Trinchillo, M.; Belanzoni, P.; Belpassi, L.; Busico, V.; Ciancaleoni, G.; D’Amora, A.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D. Unexpected Anion Effect in the Alkoxylation of Alkynes Catalyzed by N-Heterocyclic Carbene (NHC) Cationic Gold Complexes. Chem. Eur. J. 2014, 20, 14594–14598. [Google Scholar] [CrossRef]
- Savka, R.; Plenio, H. Metal Complexes of Very Bulky N,N′-Diarylimidazolylidene N-Heterocyclic Carbene (NHC) Ligands with 2,4,6-Cycloalkyl Substituents. Eur. J. Inorg. Chem. 2014, 36, 6246–6253. [Google Scholar] [CrossRef]
- Gaggioli, C.A.; Ciancaleoni, G.; Zuccaccia, D.; Bistoni, G.; Belpassi, L.; Tarantelli, F.; Belanzoni, P. Strong Electron-Donating Ligands Accelerate the Protodeauration Step in Gold(I)-Catalyzed Reactions: A Quantitative Understanding of the Ligand Effect. Organometallics 2016, 35, 2275–2285. [Google Scholar] [CrossRef]
- Zuccaccia, D.; Belanzoni, P.; Belpassi, L.; Ciancaleoni, G.; Del Zotto, A. Role of ion pairing in the mechanisms of Au(I)-catalysed reactions: Theory and experiment. In RSC Catalysis Series; Royal Society of Chemistry: London, UK, 2019; Chapter 26; pp. 564–578. [Google Scholar] [CrossRef]
- Gatto, M.; Del Zotto, A.; Segato, J.; Zuccaccia, D. Hydration of Alkynes Catalyzed by L–Au–X under Solvent-and Acid-Free Conditions: New Insights into an Efficient, General, and Green Methodology. Organometallics 2018, 37, 4685–4691. [Google Scholar] [CrossRef]
- Gatto, M.; Baratta, W.; Belanzoni, P.; Belpassi, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. Hydration and alkoxylation of alkynes catalyzed by NHC-Au-OTf. Green Chem. 2018, 20, 2125–2134. [Google Scholar] [CrossRef]
- Sorbelli, D.; Segato, J.; Del Zotto, A.; Belpassi, L.; Zuccaccia, D.; Belanzoni, P. The mechanism of the gold(I)-catalyzed Meyer–Schuster rearrangement of 1-phenyl-2-propyn-1-ol via 4-endo-dig cyclization. Dalton Trans. 2021, 50, 5154–5160. [Google Scholar] [CrossRef] [PubMed]
- Zuccaccia, D.; Del Zotto, A.; Baratta, W. The pivotal role of the counterion in gold catalyzed hydration and alkoxylation of alkynes. Coord. Chem. Rev. 2019, 396, 103–116. [Google Scholar] [CrossRef]
- Trinchillo, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Busico, V.; D’Amora, A.; D’Amore, L.; Del Zotto, A.; Tarantelli, F.; Tuzi, A.; et al. Extensive Experimental and Computational Study of Counterion Effect in the Reaction Mechanism of NHC-Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2016, 35, 641–654. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Belpassi, L.; Zuccaccia, D.; Tarantelli, F.; Belanzoni, P. Counterion Effect in the Reaction Mechanism of NHC Gold(I)-Catalyzed Alkoxylation of Alkynes: Computational Insight into Experiment. ACS Catal. 2015, 5, 803–814. [Google Scholar] [CrossRef]
- Bellachioma, G.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. NMR investigation of non-covalent aggregation of coordination compounds ranging from dimers and ion pairs up to nano-aggregates. Coord. Chem. Rev. 2008, 252, 2224–2238. [Google Scholar] [CrossRef]
- Zaccaria, F.; Sian, L.; Zuccaccia, C.; Macchioni, A. Ion pairing in transition metal catalyzed olefin polymerization. Adv. Organomet. Chem. 2020, 73, 1–78. [Google Scholar] [CrossRef]
- Balzano, F.; Cuzzola, A.; Diversi, P.; Ghiotto, F.; Uccello-Barretta, G. Cationic Gold(I) Phosphanyl Thiolates: Aurophilic Interactions in the Solid State and in Solution. Eur. J. Inorg. Chem. 2007, 2007, 5556–5562. [Google Scholar] [CrossRef]
- Hamdoun, G.; Bour, C.; Gandon, V.; Dumez, J.N. Empirical Estimation of the Molecular Weight of Gold Complexes in Solution by Pulsed-Field Gradient NMR. Organometallics 2018, 37, 4692–4698. [Google Scholar] [CrossRef]
- Landrini, M.; De Paolis, E.; Macchioni, A.; Tensi, L.; Hrobárik, P.; Rocchigiani, L. Ion Pairing in Cationic Au(I)(μ-H)2WCp2 Bimetallic Dihydrides. Eur. J. Inorg. Chem. 2022, 29, 2022. [Google Scholar] [CrossRef]
- Biasiolo, L.; Belpassi, L.; Ciancaleoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D. Diffusion NMR measurements on cationic linear gold(I) complexes. Polyhedron 2015, 92, 52–59. [Google Scholar] [CrossRef]
- Lau, V.M.; Gorin, C.F.; Kanan, M.W. Electrostatic control of regioselectivity via ion pairing in a Au(I)-catalyzed rearrangement. Chem. Sci. 2014, 5, 4975. [Google Scholar] [CrossRef]
- Zuccaccia, D.; Clot, E.; Macchioni, A. Aggregation in solution of neutral half-sandwich Ru(II) precatalysts for transfer hydrogenation. New J. Chem. 2005, 29, 430. [Google Scholar] [CrossRef]
- Brown, T.J.; Dickens, M.G.; Widenhoefer, R.A. Syntheses, X-ray Crystal Structures, and Solution Behavior of Monomeric, Cationic, Two-Coordinate Gold(I) π-Alkene Complexes. J. Am. Chem. Soc. 2009, 131, 6350–6351. [Google Scholar] [CrossRef]
- Gierer, A.; Wirtz, K.Z. Naturforsch., Teil A 1953, 8, 522. Available online: http://znaturforsch.com/aa/v08a/c08a.htm (accessed on 9 May 2024).
- Spernol, A.; Wirtz, K.Z. Naturforsch., Teil A 1953, 8, 532. Available online: http://znaturforsch.com/aa/v08a/c08a.htm (accessed on 9 May 2024).
- Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Determining accurate molecular sizes in solution through NMR diffusion spectroscopy. Chem. Soc. Rev. 2008, 37, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Clot, E.; Macchioni, A. Self-Aggregation Tendency of All Species Involved in the Catalytic Cycle of Bifunctional Transfer Hydrogenation. Organometallics 2009, 28, 960. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. Combining Diffusion NMR and Conductometric Measurements to Evaluate the Hydrodynamic Volume of Ions and Ion Pairs. Organometallics 2007, 26, 3624. [Google Scholar] [CrossRef]
- The hydrodynamic volume measured in catalytic conditions is about 23% greater than those obtained in chloroform, but the low concentration (1.3 mM) of 1Cl and the presence of both methanol and TME in the mixture suggest that no aggregation occurs and that is the ‘real’ volume in catalytic conditions. It can be assumed that the volume of the fragment Au–Cl is almost negligible compared to the volume of the ligand NHC.
- Rocchigiani, L.; Bellachioma, G.; Ciancaleoni, G.; Crocchianti, S.; Laganà, A.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. Anion-Dependent Tendency of Di-Long-Chain Quaternary Ammonium Salts to Form Ion Quadruples and Higher Aggregates in Benzene. ChemPhysChem 2010, 11, 3243. [Google Scholar] [CrossRef]
- Lu, Z.; Li, T.; Mudshinge, S.R.; Xu, B.; Hammond, G.B. Optimization of Catalysts and Conditions in Gold(I) Catalysis-Counterion and Additive Effects. Chem. Rev. 2021, 121, 8452–8477. [Google Scholar] [CrossRef] [PubMed]
- Sian, L.; Macchioni, A.; Zuccaccia, C. Understanding the Role of Metallocenium Ion-Pair Aggregates on the Rate of Olefin Insertion into the Metal-Carbon Bond. ACS Catal. 2020, 10, 1591–1606. [Google Scholar] [CrossRef]
- Wang, W.; Kumar, M.; Hammond, G.B.; Xu, B. Enhanced Reactivity in Homogeneous Gold Catalysis through Hydrogen Bonding. Org. Lett. 2014, 16, 636. [Google Scholar] [CrossRef] [PubMed]
- Zhdanko, A.; Maier, M.M. The Mechanism of Gold(I)-Catalyzed Hydroalkoxylation of Alkynes: An Extensive Experimental Study. Chem.-Eur. J. 2014, 20, 1918. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Keresztes, I.; Hopson, R.; Williard, P.G. Characterization of Reactive Intermediates by Multinuclear Diffusion-Ordered NMR Spectroscopy (DOSY). Acc. Chem. Res. 2009, 42, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.S. Diffusion Ordered Nuclear Magnetic Resonance Spectroscopy: Principles and Applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Sinnaeve, D. The Stejskal–Tanner Equation Generalized for Any Gradient Shape—An Overview of Most Pulse Sequences Measuring Free Diffusion. Concepts Magn. Reson. Part A 2012, 40A, 39–65. [Google Scholar] [CrossRef]
- Jerschow, A.; Müller, N. Suppression of Convection Artifacts in Stimulated-Echo Diffusion Experiments. Double-Stimulated-Echo Experiments. J. Magn. Reson. 1997, 125, 372–375. [Google Scholar] [CrossRef]
- Jerschow, A.; Müller, N. Convection Compensation in Gradient Enhanced Nuclear Magnetic Resonance Spectroscopy. J. Magn. Reson. 1998, 132, 13–18. [Google Scholar] [CrossRef]
- Tyrrell, H.J.V.; Harris, K.R. Diffusion in Liquids: A Theoretical and Experimental Study; Butterworths monographs in chemistry; Butterworths: London, UK, 1984; Available online: https://shop.elsevier.com/books/liquids-and-liquid-mixtures/perlmutter/978-0-408-24193-9 (accessed on 9 May 2024).
- Mills, R. Self-Diffusion in Normal and Heavy Water in the Range 1–45 °C. J. Phys. Chem. 1973, 77, 685–688. [Google Scholar] [CrossRef]
- Chen, H.C.; Chen, S.H. Diffusion of Crown Ethers in Alcohols. J. Phys. Chem. 1984, 88, 5118–5121. [Google Scholar] [CrossRef]
- Zuccaccia, D.; Macchioni, A. An Accurate Methodology to Identify the Level of Aggregation in Solution by PGSE NMR Measurements: The Case of Half-Sandwich Diamino Ruthenium(II) Salts. Organometallics 2005, 24, 3476–3486. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | Dt+ | Dt− | rH+ | VH+ | rH− | VH− | N+ | N− | α |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1Cl (937) | 6.99 | 6.1 | 937 | 1.00 | |||||
| 2 | 1(TME)OTs (1245) | 5.93 | 5.24 | 6.6 | 1188 | 6.4 | 1098 | 0.95 | 0.88 | 0.13 |
| 3 | 1(TME)OTf (1152) | 6.40 | 7.38 | 6.2 | 998 | 6.0 | 900 | 0.87 | 0.78 | 0.24 |
| 4 | 1(TME)BF4 (1089) | 6.01 | 7.65 | 6.9 | 1376 | 5.6 | 720 | 1.26 | 0.66 | 0.36 |
| 5 | 1(TME) BARF (1927) | 6.84 | 5.58 | 6.7 | 1260 | 7.3 | 1596 | 0.65 | 0.82 | 0.32 |
| Entries | Solvent | Dt+ | Dt− | rH+ | VH+ | rH− | VH− | N+ | N− | α |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | p-cymene | 3.60 | 5.26 | 8.4 | 2509 | 7.1 | 1499 | 2.18 | 1.3 | 0 |
| 2 | chloroform * | 6.40 | 7.38 | 6.2 | 998 | 6.0 | 900 | 0.87 | 0.78 | 0.24 |
| 3 | methanol | 6.57 | 10.5 | 6.5 | 1166 | 4.2 | 306 | 1.01 | 0.26 | 0.70 |
| 4 | acetone | 14.6 | 10.2 | 6.1 | 946 | 3.9 | 250 | 0.82 | 0.22 | 0.80 |
| 5 | γ-valerolactone | 3.52 | 7.94 | 6.3 | 1067 | 4.0 | 270 | 0.93 | 0.23 | 0.84 |
| 6 | propylene carbonate | 3.56 | 9.47 | 6.3 | 1023 | 3.7 | 217 | 0.89 | 0.18 | 0.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campagnolo, F.; Aneggi, E.; Baratta, W.; Munir, T.; Zuccaccia, D. Diffusion Nuclear Magnetic Resonance Measurements on Cationic Gold (I) Complexes in Catalytic Conditions: Counterion and Solvent Effects. Molecules 2024, 29, 3018. https://doi.org/10.3390/molecules29133018
Campagnolo F, Aneggi E, Baratta W, Munir T, Zuccaccia D. Diffusion Nuclear Magnetic Resonance Measurements on Cationic Gold (I) Complexes in Catalytic Conditions: Counterion and Solvent Effects. Molecules. 2024; 29(13):3018. https://doi.org/10.3390/molecules29133018
Chicago/Turabian StyleCampagnolo, Filippo, Eleonora Aneggi, Walter Baratta, Talha Munir, and Daniele Zuccaccia. 2024. "Diffusion Nuclear Magnetic Resonance Measurements on Cationic Gold (I) Complexes in Catalytic Conditions: Counterion and Solvent Effects" Molecules 29, no. 13: 3018. https://doi.org/10.3390/molecules29133018
APA StyleCampagnolo, F., Aneggi, E., Baratta, W., Munir, T., & Zuccaccia, D. (2024). Diffusion Nuclear Magnetic Resonance Measurements on Cationic Gold (I) Complexes in Catalytic Conditions: Counterion and Solvent Effects. Molecules, 29(13), 3018. https://doi.org/10.3390/molecules29133018