
Stable Radical Isoporphyrin Copolymer Prepared with Di(phenylphosphane)
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
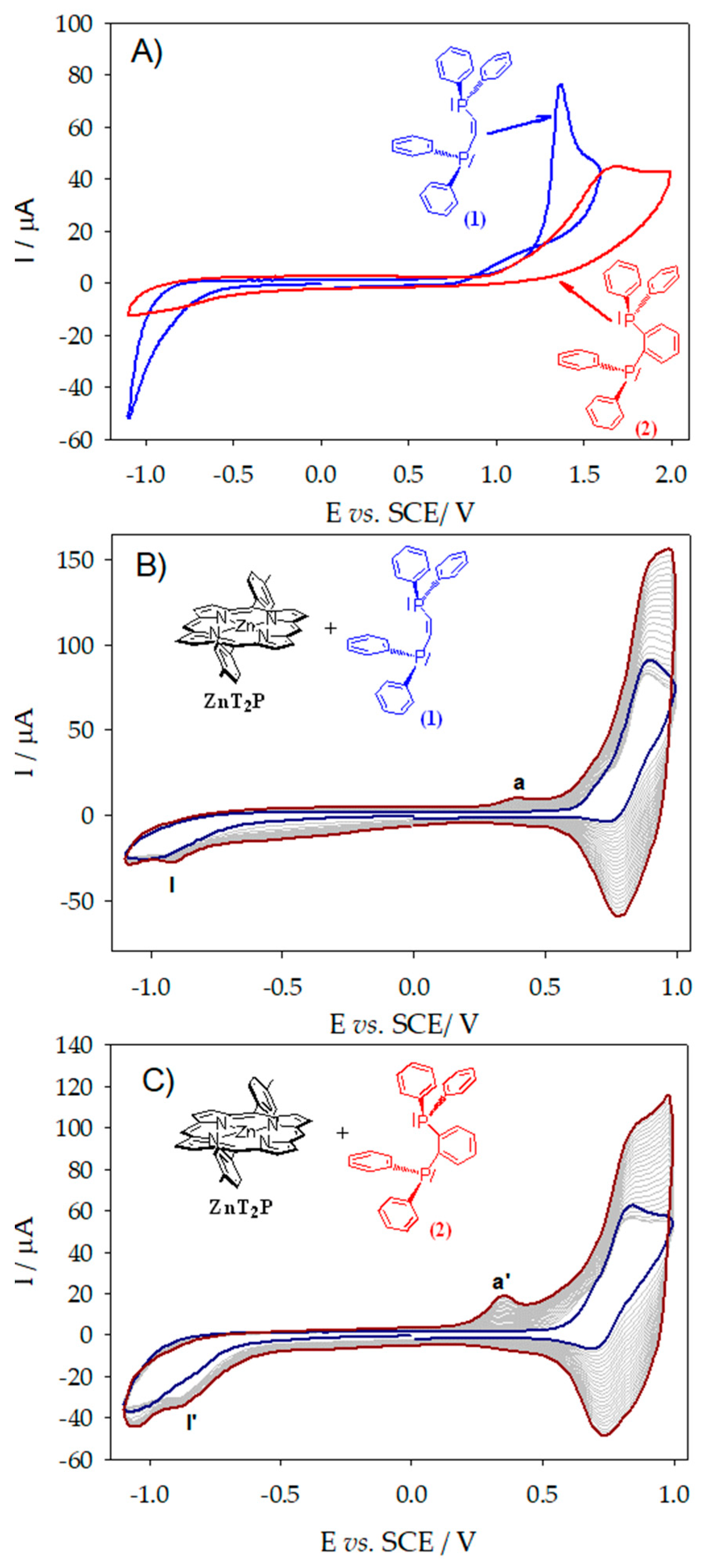
2.1. Electropolymerization of Isoporphyrin Copolymers
2.2. Electrochemical Quartz Cristal Microbalance (EQCM)
2.3. X-ray Photoelectron Spectroscopy (XPS)
2.4. Electron Paramagnetic Resonance (EPR)
2.5. UV–Visible–NIR Spectroscopy
2.6. Film Morphology (Atomic Force Microscopy, AFM)
2.7. Electrochemistry of the Copolymer
2.8. Electrochemical Impedance Spectroscopy (EIS) and Photoelectrochemical Properties
3. Materials and Methods
3.1. Reagents
3.2. Electrochemistry
3.3. Electropolymerization
3.4. Electrochemical Quartz Cristal Microbalance (EQCM)
3.5. X-ray Photoelectron Spectroscopy
3.6. Atomic Force Microscopy (AFM)
3.7. EPR Measurements
3.8. Photoelectrochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, B.; Smith, K.M. Novel meso-substitution reactions of zinc (II) octaethylporphyrin. Tetrahedron Lett. 1977, 18, 3079–3082. [Google Scholar] [CrossRef]
- Smith, K.M.; Barnett, G.H.; Evans, B.; Martynenko, Z. Novel meso-substitution reactions of metalloporphyrins. J. Am. Chem. Soc. 1979, 101, 5953–5961. [Google Scholar] [CrossRef]
- Padilla, A.G.; Wu, S.-M.; Shine, H.J. Reaction of zinc tetraphenylporphyrin cation radical perchlorate with pyridine. J. Chem. Soc. Chem. Commun. 1976, 7, 236–237. [Google Scholar] [CrossRef]
- Shine, H.J.; Padilla, A.G.; Wu, S.-M. Ion radicals. 45. Reactions of zinc tetraphenylporphyrin cation radical perchlorate with nucleophiles. J. Org. Chem. 1979, 44, 4069–4075. [Google Scholar] [CrossRef]
- Malek, A.; Latos-Grazynski, L.; Bartczak, T.J.; Zadlo, A. Reactions of the iron(III) tetraphenylporphyrin.pi. cation radical with triphenylphosphine and the nitrite anion. Formation of β-substituted iron(III) porphyrins. Inorg. Chem. 1991, 30, 3222–3230. [Google Scholar] [CrossRef]
- Giraudeau, A.; El Kahef, L. β-Substitution de la méso-terraphénylporphyrine de zinc par voie électrochimique. Can. J. Chem. 1991, 69, 1161–1165. [Google Scholar] [CrossRef]
- Dimé, A.K.D.; Cattey, H.; Lucas, D.; Devillers, C.H. Electrosynthesis and X-ray crystallographic Structure of ZnII meso-triaryltriphenylphosphonium porphyrin and structural comparison with MgII meso-triphenylphosphonium porphine. Eur. J. Inorg. Chem. 2018, 2018, 4834–4841. [Google Scholar] [CrossRef]
- Ruhlmann, L.; Giraudeau, A. One-pot electrochemical generation of a porphyrin dimer with bis(diphenylphosphonium)acetylene bridge. Chem. Commun. 1996, 17, 2007–2008. [Google Scholar] [CrossRef]
- Ruhlmann, L.; Giraudeau, A. A first series of diphosphonium electrochemically bridged porphyrins. Eur. J. Inorg. Chem. 2001, 2001, 659–668. [Google Scholar] [CrossRef]
- Ruhlmann, L.; Gross, M.; Giraudeau, A. Bisporphyrins with bischlorin features obtained by direct anodic coupling of porphyrins, Chem. Eur. J. 2003, 9, 5085–5096. [Google Scholar] [CrossRef]
- Holmes-Smith, A.S.; Zheng, X.; Uttamlal, M. Characterization of an electropolymerized Pt(II) diamino phenyl porphyrin polymer suitable for oxygen sensing. Meas. Sci. Technol. 2006, 17, 3328–3334. [Google Scholar] [CrossRef]
- Walter, M.G.; Wamser, C.C. Synthesis and Characterization of Electropolymerized Nanostructured Aminophenylporphyrin Films. J. Phys. Chem. C 2010, 114, 7563–7574. [Google Scholar] [CrossRef]
- Arce, R.; del Río, R.; Ruiz-León, D.; Velez, J.; Isaacs, M.; del Valle, M.A.; Aguirre, M.J. Evidence for the Formation of a Copolymer by Simultaneous Electropolymerization of p-Tetraaminophenyl Porphyrin Cobalt (II)) and o-Phenylenediamine on Glassy Carbon. Int. J. Electrochem. Sci. 2012, 7, 11596–11608. [Google Scholar] [CrossRef]
- Pérez-Morales, M.; de Miguel, G.; Muñoz, E.; Martín-Romero, M.T.; Camacho, L. Reversible binding of molecular dioxygen to CoTSPP electrodeposited films from aqueous basic media. Electrochem. Commun. 2006, 8, 638–642. [Google Scholar] [CrossRef]
- Trevin, S.; Bedioui, F.; Devynck, J. New electropolymerized nickel porphyrin films. Application to the detection of nitric oxide in aqueous solution. J. Electroanal. Chem. 1996, 408, 261–265. [Google Scholar] [CrossRef]
- Boudiaf, M.; Liang, Y.; Lamare, R.; Weiss, J.; Bonnefont, A.; Ruhlmann, L. Stable isoporphyrin copolymer: Electrochemical mechanism and behavior and photovoltaic properties. Electrochim. Acta 2019, 309, 432–449. [Google Scholar] [CrossRef]
- Liang, Y.; Sokolov, M.N.; Mikhaylov, M.A.; Ibrahim, H.; Goldmann, M.; Choua, S.; Le Breton, N.; Boudon, C.; Badets, V.; Bonnefont, A.; et al. A 3D electropolymerized thin film based on an isoporphyrin and on a pyridine end-decorated molybdenum(II) halide cluster: Photoelectrochemical and impedance properties. Electrochim. Acta 2021, 388, 138493. [Google Scholar] [CrossRef]
- Inisan, C.; Saillard, J.-Y.; Guilard, R.; Tabard, A.; Mest, Y. Electrooxidation of porphyrin free bases: Fate of the π-cation radical. New J. Chem. 1998, 22, 823–830. [Google Scholar] [CrossRef]
- Zheng, W.; Shan, N.; Yu, L.; Wang, X. UV–visible, fluorescence and EPR properties of porphyrins and metalloporphyrins. Dye. Pigment. 2008, 77, 153–157. [Google Scholar] [CrossRef]
- Lan, M.; Zhao, H.; Yuan, H.; Jiang, C.; Zuo, S.; Jiang, Y. Absorption and EPR spectra of some porphyrins and metalloporphyrins. Dye. Pigment. 2007, 74, 357–362. [Google Scholar] [CrossRef]
- Fajer, J. Structural effects in chemistry and biology. J. Porphyr. Phthalocyanines 2000, 4, 382–390. [Google Scholar] [CrossRef]
- Shelnutt, J.A.; Song, X.; Ma, J.G.; Jia, S.L.; Jentzen, W.; Medforth, C.J. Nonplanar porphyrins and their significance in proteins. Chem. Soc. Rev. 1998, 27, 31–42. [Google Scholar] [CrossRef]
- Barkigia, K.M.; Berber, M.D.; Fajer, J.; Medforth, C.J.; Renner, M.W.; Smith, K.M. Nonplanar porphyrins. X-ray structures of (2,3,7,8,12,13,17,18-octaethyl- and -octamethyl-5,10,15,20-tetraphenylporphinato) zinc(II). J. Am. Chem. Soc. 1990, 112, 8851–8857. [Google Scholar] [CrossRef]
- Sparks, L.D.; Medforth, C.J.; Park, M.S.; Chamberlain, J.R.; Ondrias, M.R.; Senge, M.O.; Smith, K.M.; Shelnutt, J.A. Metal dependence of the nonplanar distortion of octaalkyltetraphenylporphyrins. J. Am. Chem. Soc. 1993, 115, 581–592. [Google Scholar] [CrossRef]
- Senge, M.O.; Renner, M.W.; Kalisch, W.W.; Fajer, J. Molecular structure of (5,10,15,20-tetrabutyl-2,3,7,8,12,13,17,18-octaethylporphyrinato)nickel(II)d correlation of nonplanarity with frontier orbital shifts. J. Chem. Soc. Dalton Trans. 2000, 3, 381–385. [Google Scholar] [CrossRef]
- D’Souza, F.; Zandler, M.E.; Tagliatesta, P.; Ou, Z.; Shao, J.; Van Caemelbecke, C.E.; Kadish, K.M. Electronic, spectral, and electrochemical properties of (TPPBrx)Zn where TPPBrx is the dianion of b-brominated-pyrrole tetraphenylporphyrin and x varies from 0 to 8. Inorg. Chem. 1998, 37, 4567–4572. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Barrett, A.G.M.; Hoffman, B.M. Super-charged Porphyrazines: Synthesis and physical properties of octacationic tetraazaporphyrins. Inorg. Chem. 1999, 38, 6143–6151. [Google Scholar] [CrossRef] [PubMed]
- Bernard, C.; Gisselbrecht, J.-P.; Gross, M.; Vogel, E.; Lausmann, M. Redox properties of porphycenes and metalloporphycenes. A comparison with porphyrins. Inorg. Chem. 1994, 33, 2393–2401. [Google Scholar] [CrossRef]
- Kasha, M. Relation between exciton bands and conduction bands in molecular lamellar systems. Rev. Mod. Phys. 1959, 31, 162–169. [Google Scholar] [CrossRef]
- Sessler, J.L.; Johnson, M.R.; Creager, S.E.; Fettinger, J.C.; Ibers, J.A. Synthesis and characterization of quinone-substituted octaalkyl porphyrin monomers and dimers. J. Am. Chem. Soc. 1990, 112, 9310–9329. [Google Scholar] [CrossRef]
- Abhilash, G.J.; Bhuyan, J.; Singh, P.; Maji, S.; Pal, K.; Sarkar, S. NO2-Mediated meso-hydroxylation of iron(III) porphyrin. Inorg. Chem. 2009, 48, 1790–1792. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Soret Band/nm | Q and NIR Bands/nm | π–π* Band/nm |
|---|---|---|---|
| ZnT2P 1 | 415 (405.4) | 546 (18.9), 584 (4.9) | |
| cis-1,2-bis(diphenyl-phosphino)ethene 1 | 214 (44.9), 257 (19.7) | ||
| 1,2-bis(diphenyl-phosphino)benzene 2 | 214 (58.7), 278 (17.9) | ||
| poly-ZnT2isoP1 3 | 432, 505 | 587, 633, 810 | |
| poly-ZnT2isoP2 4 | 432, 497 | 574, 632, 805 |
| Compounds | Ring Oxidation | Reduction of the -P+(Ph)2- | Ring Reduction | Oxidation of Phosphane -P+●(Ph)2−/ -P(Ph2)- | ||||
|---|---|---|---|---|---|---|---|---|
| peak a or a’ | peak I or I’ | |||||||
| ZnT2P 1 | 1.08 (150) | 0.79 (90) | −1.41 (160) | −1.84 (170) | ||||
| cis-1,2-bis(diphenyl phosphino)ethene) | 1.37 irr | |||||||
| 1,2-bis(diphenyl phosphino)benzene | 1.68 irr | |||||||
| poly-ZnT2isoP1 | 1.39 irr | 1.15 irr | +0.86 (250) | +0.42 irr | −0.91 irr | |||
| poly-ZnT2isoP2● | 1.40 irr | 1.17 irr | +0.88 (196) | +0.38 irr | −0.87 irr | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Bonnefont, A.; Badets, V.; Boudon, C.; Goldmann, M.; Diot, G.; Choua, S.; Le Breton, N.; Ruhlmann, L. Stable Radical Isoporphyrin Copolymer Prepared with Di(phenylphosphane). Molecules 2024, 29, 3056. https://doi.org/10.3390/molecules29133056
Liang Y, Bonnefont A, Badets V, Boudon C, Goldmann M, Diot G, Choua S, Le Breton N, Ruhlmann L. Stable Radical Isoporphyrin Copolymer Prepared with Di(phenylphosphane). Molecules. 2024; 29(13):3056. https://doi.org/10.3390/molecules29133056
Chicago/Turabian StyleLiang, Yiming, Antoine Bonnefont, Vasilica Badets, Corinne Boudon, Michel Goldmann, Guillaume Diot, Sylvie Choua, Nolwenn Le Breton, and Laurent Ruhlmann. 2024. "Stable Radical Isoporphyrin Copolymer Prepared with Di(phenylphosphane)" Molecules 29, no. 13: 3056. https://doi.org/10.3390/molecules29133056