

Inhibition of Monoamine Oxidases by Pyridazinobenzylpiperidine Derivatives

 , and
, and
Abstract
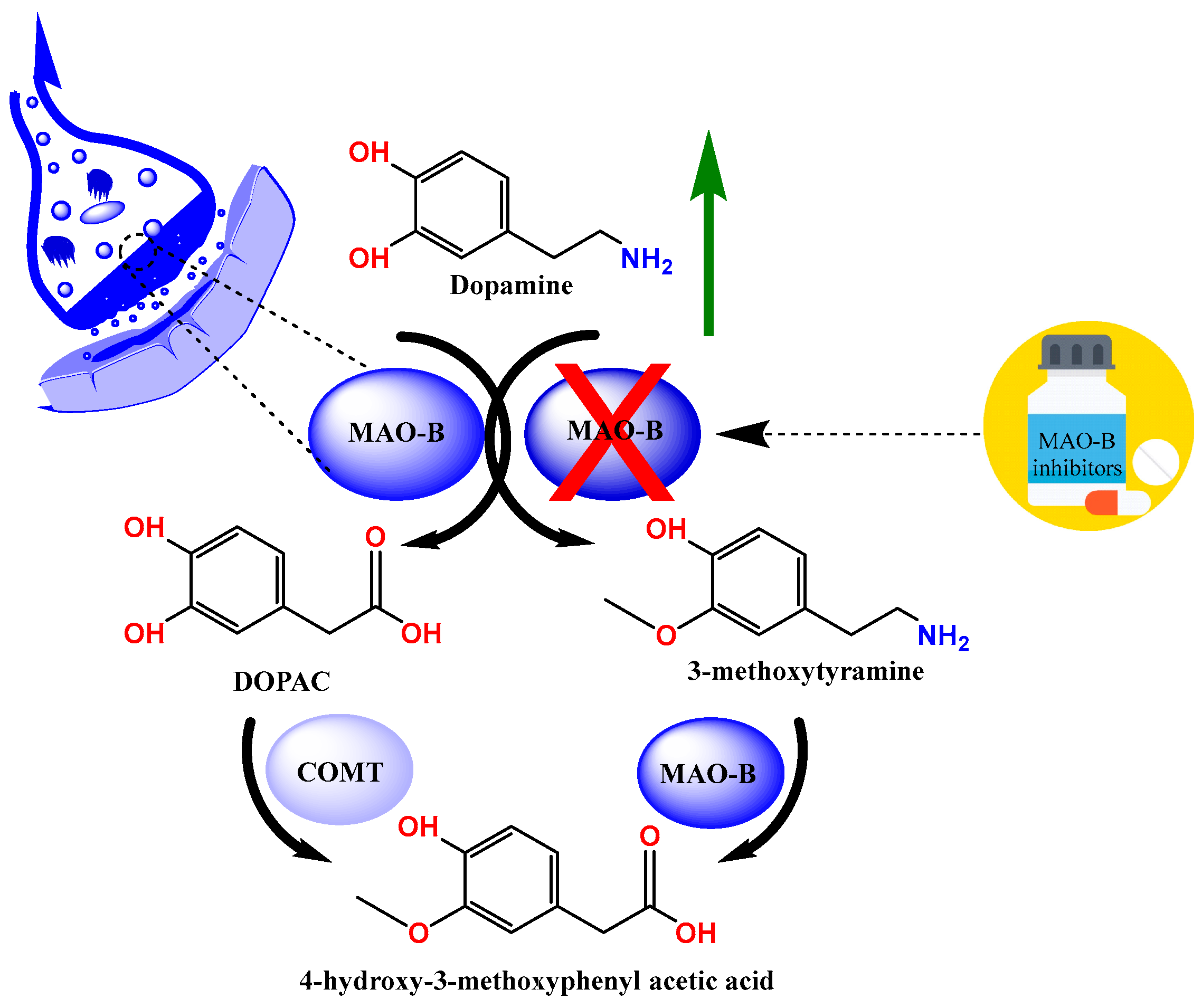
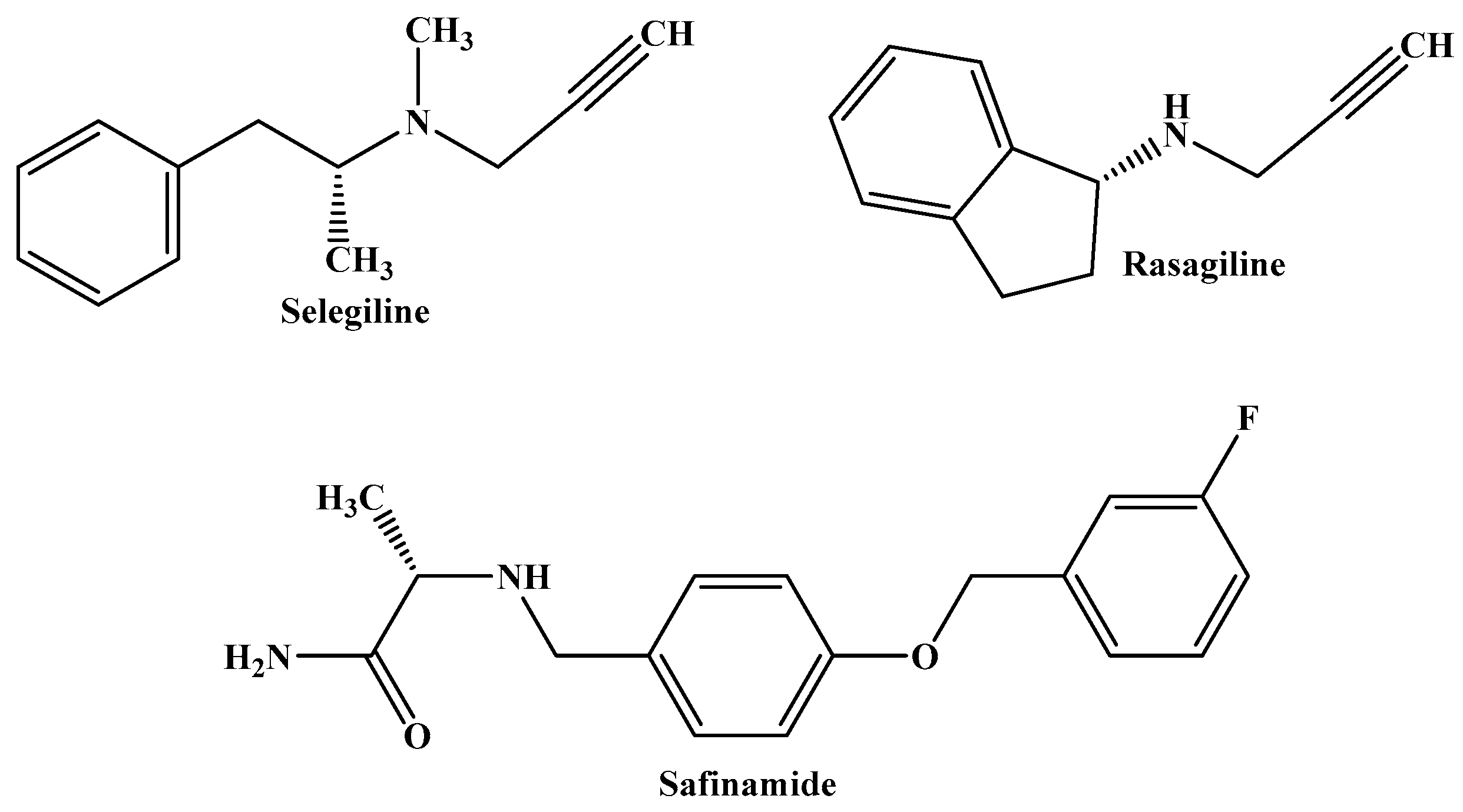
:1. Introduction
2. Results
2.1. Chemistry
2.2. Inhibition Studies
2.2.1. The Inhibitory Activities of MAO-A and MAO-B by the Compounds
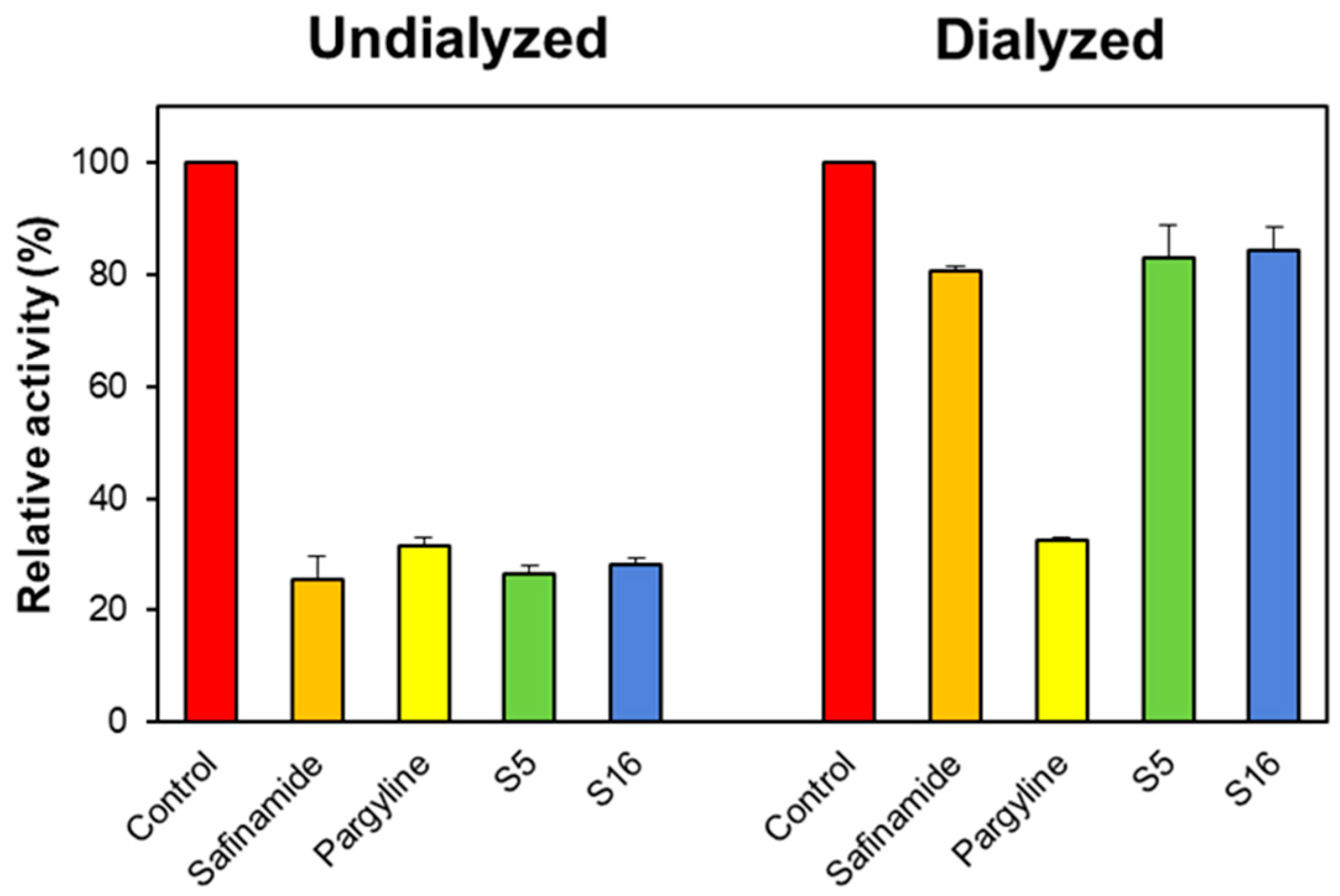
2.2.2. Reversibility Studies
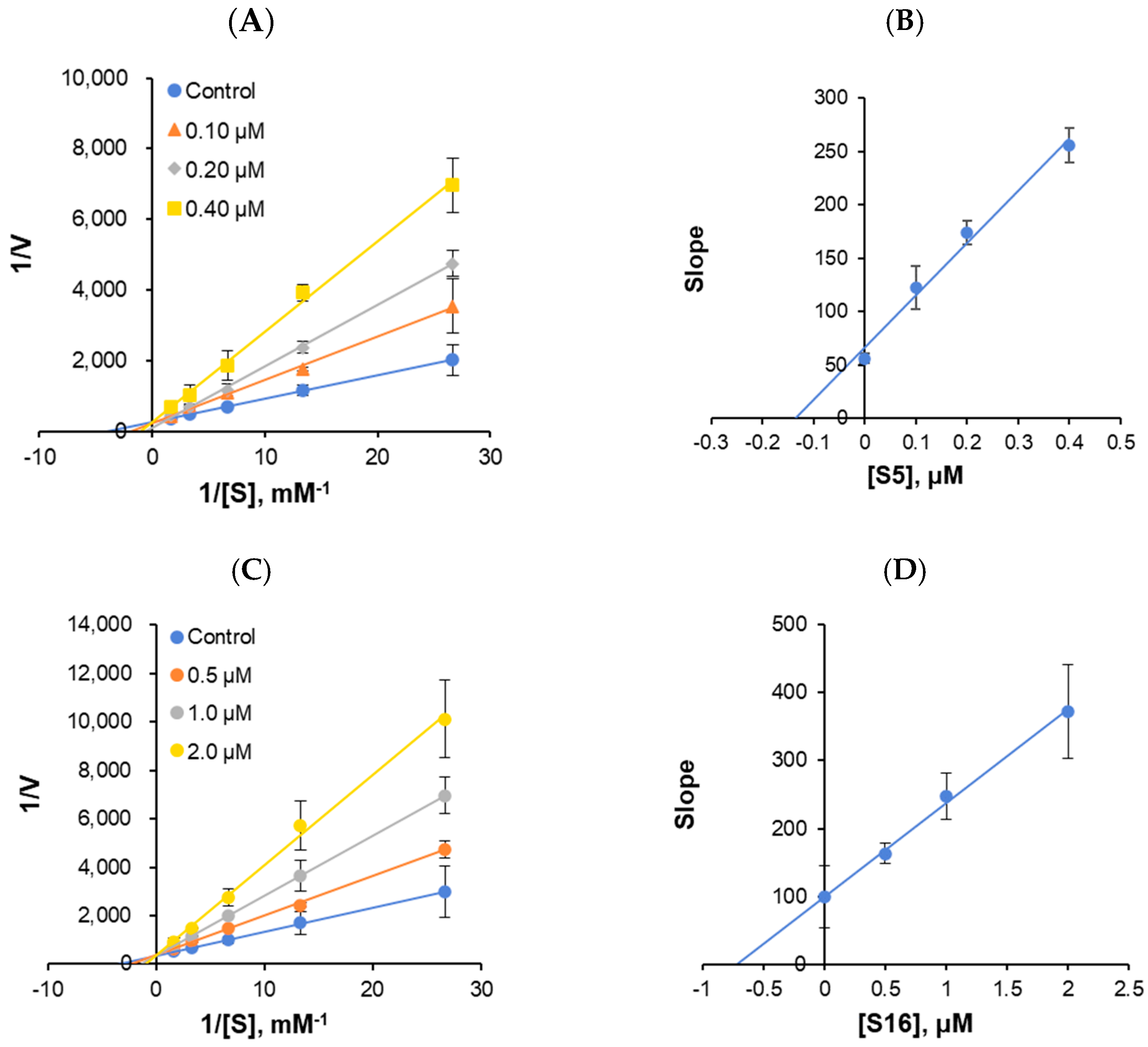
2.2.3. Enzyme and Inhibition Kinetics
2.3. Parallel Artificial Membrane Permeability Assay (PAMPA) for Blood–Brain Barrier (BBB) Permeation Study
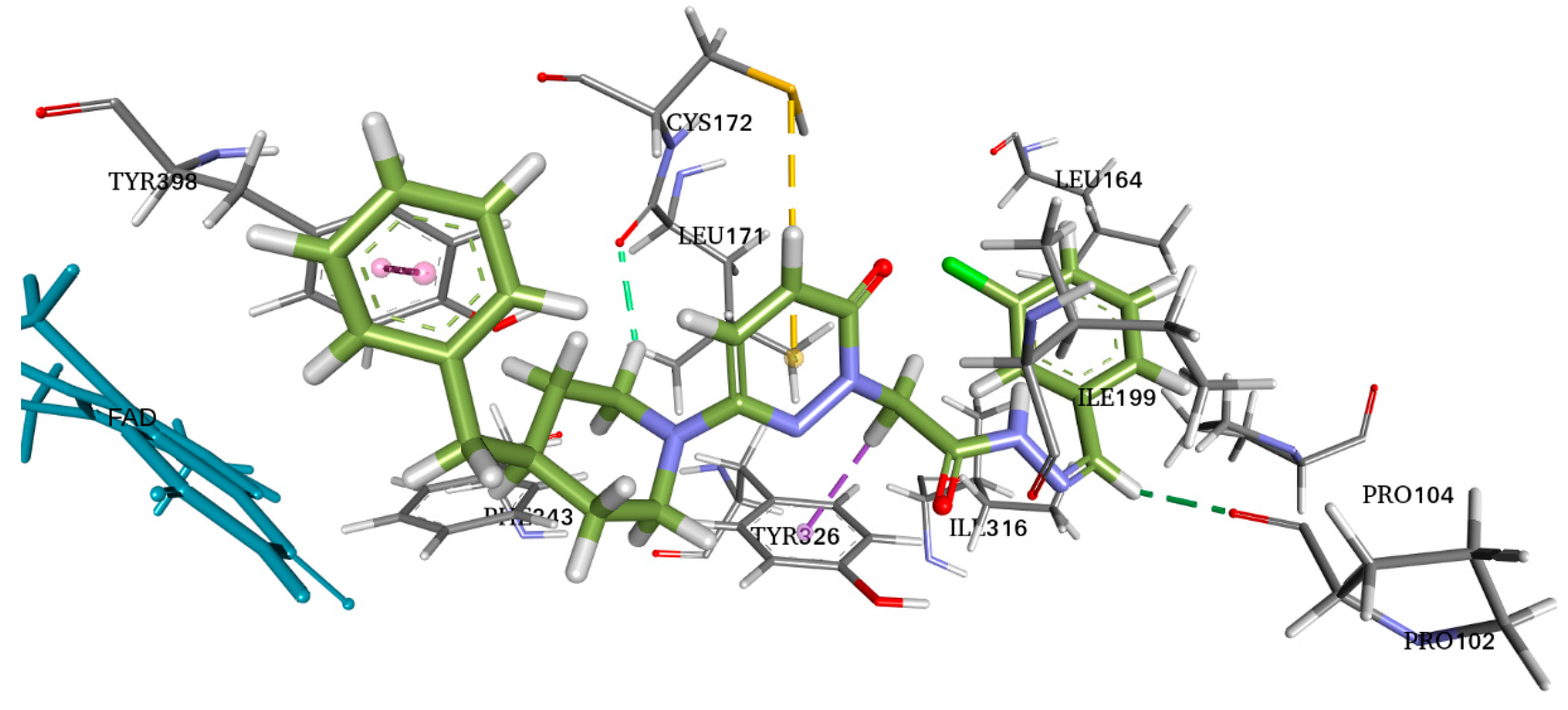
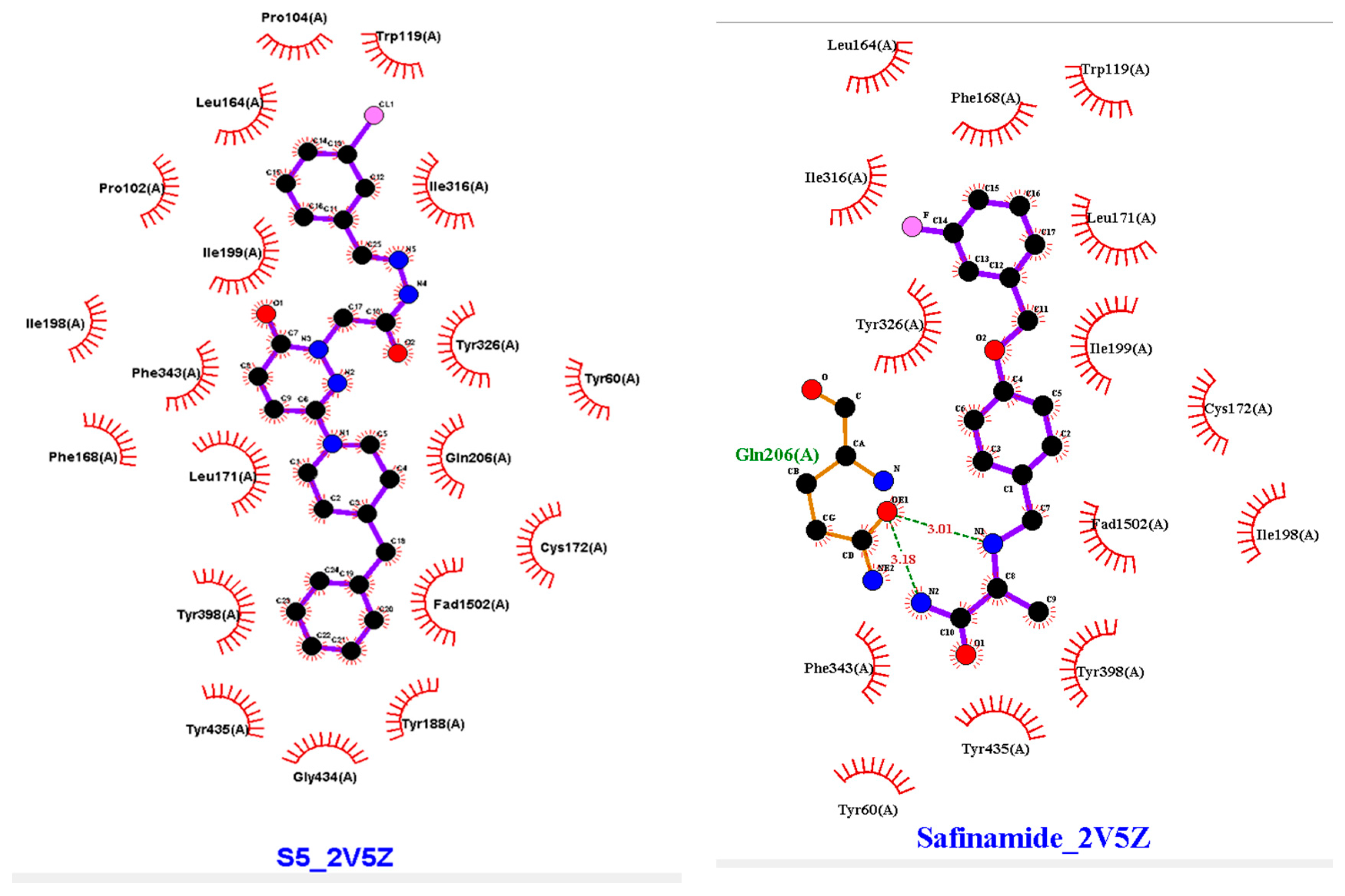
2.4. Molecular Docking
3. Discussion
3.1. Chemistry
3.2. The Inhibitory Activities of MAO-A and MAO-B by the Compounds
3.3. Molecular Docking
4. Materials and Methods
4.1. Chemistry
4.1.1. Experimental Procedure for Synthesis
4.1.2. Experimental Protocol for Producing the Chemicals Listed in the Title (S1–S24)
- N′-2-bromobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S3)
- N′-3-bromobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S4)
- N′-3-chlorobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S5)
- N′-3-fluorobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S7)
- N′-2-methylbenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S9)
- N′-3-methylbenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S10)
- N′-4-ethylbenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S15)
- N′-2-cyanobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S16)
- N′-3-cyanobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S17)
- N′-4-cyanobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S18)
- N′-2-methoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S19)
- N′-3,5-dimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S20)
- N′-2,4,6-trimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S21)
- N′-2,3-dimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S22)
- N′-3,4,5-trimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S23)
- N′-2,4-dimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S24)
4.2. Enzyme Inhibition Studies
4.2.1. Enzyme Assays and Kinetics
4.2.2. An Inhibition Study of MAO-A and MAO-B by the Compounds
4.2.3. Reversibility Studies
4.3. Blood–Brain Barrier Permeability Study
4.4. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bohnen, N.I.; Yarnall, A.J.; Weil, R.S.; Moro, E.; Moehle, M.S.; Borghammer, P.; Bedard, M.A.; Albin, R.G. Cholinergic system changes in Parkinson’s disease: Emerging therapeutic approaches. Lancet Neurol. 2022, 21, 381–392. [Google Scholar] [CrossRef]
- Chew, Z.X.; Lim, C.L.; Ng, K.Y.; Chye, S.M.; Ling, A.P.; Koh, R.Y. The role of monoamine oxidase B inhibitors in the treatment of Parkinson’s disease-An update. CNS Neurol. Disord. Drug Targets 2023, 22, 329–352. [Google Scholar]
- Fujii, T.; Nagamori, S.; Wiriyasermkul, P.; Zheng, S.; Yago, A.; Shimizu, T.; Tabuchi, Y.; Okumura, T.; Fujii, T.; Takeshima, H.; et al. Parkinson’s disease-associated ATP13A2/PARK9 functions as a lysosomal H+, K+-ATPase. Nature Commun. 2023, 14, 2174. [Google Scholar] [CrossRef]
- Regensburger, M.; Ip, C.W.; Kohl, Z.; Schrader, C.; Urban, P.P.; Kassubek, J.; Jost, W.H. Clinical benefit of MAO-B and COMT inhibition in Parkinson’s disease: Practical considerations. J. Neural Transm. 2023, 130, 847–861. [Google Scholar] [CrossRef]
- Baweja, G.S.; Gupta, S.; Kumar, B.; Patel, P.; Asati, V. Recent updates on structural insights of MAO-B inhibitors: A review on target-based approach. Mol. Divers. 2023, 1–23. [Google Scholar] [CrossRef]
- Lv, Y.; Fan, M.; He, J.; Song, X.; Guo, J.; Gao, B.; Zhang, J.; Zhang, C.; Xie, Y. Discovery of Novel Benzimidazole Derivatives as Selective and Reversible Monoamine Oxidase B Inhibitors for Parkinson’s Disease Treatment. Eur. J. Med. Chem. 2024, 274, 116566. [Google Scholar] [CrossRef]
- Al-Saad, O.M.; Gabr, M.; Darwish, S.S.; Rullo, M.; Pisani, L.; Miniero, D.V.; Liuzzi, G.M.; Kany, A.M.; Hirch, A.K.N.; Abadi, A.H.; et al. Novel 6-hydroxybenzothiazol-2-carboxamides as potent and selective monoamine oxidase B inhibitors endowed with neuroprotective activity. Eur. J. Med. Chem. 2024, 269, 116266. [Google Scholar] [CrossRef]
- Kondeva-Burdina, M.; Mateev, E.; Angelov, B.; Tzankova, V.; Georgieva, M. In Silico Evaluation and In Vitro Determination of Neuroprotective and MAO-B Inhibitory Effects of Pyrrole-Based Hydrazones: A Therapeutic Approach to Parkinson’s Disease. Molecules 2022, 27, 8485. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Oh, J.M.; Abdelgawad, M.A.; Abourehab, M.A.; Tengli, A.K.; Singh, A.K.; Ahmad, I.; Patel, H.; Mathew, B.; Kim, H. Development of isopropyl-tailed chalcones as a new class of selective MAO-B inhibitors for the treatment of Parkinson’s disorder. ACS Omega 2023, 8, 6908–6917. [Google Scholar] [CrossRef] [PubMed]
- Çeçen, M.; Oh, J.M.; Özdemir, Z.; Büyüktuncel, S.E.; Uysal, M.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Design, synthesis, and biological evaluation of pyridazinones containing the (2-fluorophenyl) piperazine moiety as selective MAO-B inhibitors. Molecules 2020, 25, 5371. [Google Scholar] [CrossRef] [PubMed]
- Alagöz, M.A.; Oh, J.M.; Zenni, Y.N.; Özdemir, Z.; Abdelgawad, M.A.; Naguib, I.A.; Ghoneim, M.M.; Gambacorta, N.; Nicolotti, O.; Kim, H.; et al. Development of a novel class of pyridazinone derivatives as selective MAO-B inhibitors. Molecules 2022, 27, 3801. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X.; Gao, C.; Liu, Y.; Ma, Z.; Zang, J.; Wang, H.; Liu, L.; Liu, Y.; Sun, H.; et al. Molecular Mechanism and Structure-activity Relationship of the Inhibition Effect between Monoamine Oxidase and Selegiline Analogues. Curr. Comput.-Aid. Drug 2024, 20, 474–485. [Google Scholar] [CrossRef]
- Hirano, M.; Samukawa, M.; Isono, C.; Kusunoki, S.; Nagai, Y. The effect of rasagiline on swallowing function in Parkinson’s disease. Heliyon 2024, 10, e23407. [Google Scholar] [CrossRef]
- Blair, H.A.; Dhillon, S. Safinamide: A review in Parkinson’s disease. CNS Drugs 2017, 31, 169–176. [Google Scholar] [CrossRef]
- Özdemir, Z.; Gökçe, M.; Karakurt, A. Synthesis and analgesic, anti-inflammatory and antimicrobial evaluation of 6-substituted-3 (2H)-pyridazinone-2-acetyl-2-(substituted benzal) hydrazone derivatives. FABAD J. Pharm. Sci. 2012, 37, 111–122. [Google Scholar]
- Özdemir, Z.; Alagöz, M.A.; Uslu, H.; Karakurt, A.; Erikci, A.; Ucar, G.; Uysal, M. Synthesis, molecular modelling and biological activity of some pyridazinone derivatives as selective human monoamine oxidase-B inhibitors. Pharmacol. Rep. 2020, 72, 692–704. [Google Scholar] [CrossRef]
- Hamed, M.Y.; Aly, A.F.; Abdullah, N.H.; Ismail, M.F. Synthesis, characterization and antifungal evaluation of novel pyridazin-3 (2 H)-one derivatives. Polycycl. Aromat. Comp. 2023, 43, 2356–2375. [Google Scholar] [CrossRef]
- Partap, S.; Akhtar, M.J.; Yar, M.S.; Hassan, M.Z.; Siddiqui, A.A. Pyridazinone hybrids: Design, synthesis and evaluation as potential anticonvulsant agents. Bioorg. Chem. 2018, 77, 74–83. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gebarowski, T.; Świątek, P. Design, synthesis, biological evaluation and in silico studies of novel pyrrolo [3,4-d] pyridazinone derivatives with promising anti-inflammatory and antioxidant activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Imran, M.; Nayeem, N. Synthesis and antihypertensive activity of some novel pyridazinones. Orient. J. Chem. 2016, 32, 267–274. [Google Scholar] [CrossRef]
- Yamada, T.; Shimamura, H.; Tsukamoto, Y.; Yamaguchi, A.; Ohki, M. Pyridazinones. 3. Synthesis, antisecretory, and antiulcer activities of 2-cyanoguanidine derivatives. J. Med. Chem. 1983, 26, 1144–1149. [Google Scholar] [CrossRef]
- Mikus, J.; Świątek, P.; Przybyła, P.; Krzyżak, E.; Marciniak, A.; Kotynia, A.; Redzicka, A.; Wiatrak, B.; Jawień, P.; Gębarowski, T.; et al. Synthesis, Biological, Spectroscopic and Computational Investigations of Novel N-Acylhydrazone Derivatives of Pyrrolo [3,4-d] pyridazinone as Dual COX/LOX Inhibitors. Molecules 2023, 28, 5479. [Google Scholar] [CrossRef]
- He, Z.X.; Gong, Y.P.; Zhang, X.; Ma, L.Y.; Zhao, W. Pyridazine as a privileged structure: An updated review on anticancer activity of pyridazine containing bioactive molecules. Eur. J. Med. Chem. 2021, 209, 112946. [Google Scholar] [CrossRef]
- Giovannoni, M.P.; Ciciani, G.; Cilibrizzi, A.; Crocetti, L.; Daniele, S.; Mannelli, L.D.C.; Ghelardini, C.; Giacomelli, C.; Guerrini, G.; Martini, C.; et al. Further studies on pyrazolo[1′,5′:1,6] pyrimido[4,5-d] pyridazin-4(3H)-ones as potent and selective human A1 adenosine receptor antagonists. Eur. J. Med. Chem. 2015, 89, 32–41. [Google Scholar] [CrossRef]
- Cao, X.; Chen, Y.; Zhang, Y.; Qiu, Y.; Yu, M.; Xu, X.; Liu, X.; Liu, B.-F.; Zhang, G. Synthesis and biological evaluation of new 6-hydroxypyridazinone benzisoxazoles: Potential multi-receptor-targeting atypical antipsychotics. Eur. J. Med. Chem. 2016, 124, 713–728. [Google Scholar] [CrossRef]
- Abd-Rabo, Z.S.; George, R.F.; Zaafar, D.K.; Gawish, A.Y.; Serry, A.M. Design, synthesis, and biological evaluation of some new 2-phenyl-3,6-pyridazinedione derivatives as PDE-5 inhibitors. Bioorg. Chem. 2024, 145, 107213. [Google Scholar] [CrossRef]
- Xing, X.; Chang, L.C.; Kong, Q.; Colton, C.K.; Lai, L.; Glicksman, M.A.; Lin, C.L.G.; Cuny, G.D. Structure–activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg. Med. Chem. Lett. 2011, 21, 5774–5777. [Google Scholar] [CrossRef]
- Asif, M.; Almehmadi, M.M.; Alsaiari, A.A. Diverse pharmacological potential of pyridazine analogs against various diseases. Med. Chem. 2024, 20, 245–267. [Google Scholar]
- Rosa, F.A.; Gonçalves, D.S.; Pianoski, K.E.; da Silva, M.J.; Ames, F.Q.; Aguiar, R.P.; Volpato, H.; Lazarin-Bidóia, D.; Nakamura, C.V.; Bersani-Amado, C.A. Discovery of a new pyrido [2,3-d] pyridazine-2, 8-dione derivative as a potential anti-inflammatory agent through COX-1/COX-2 dual inhibition. RSC Med. Chem. 2024, 15, 1038–1045. [Google Scholar] [CrossRef]
- Allam, H.A.; Kamel, A.A.; El-Daly, M.; George, R.F. Synthesis and vasodilator activity of some pyridazin-3(2H)-one based compounds. Future Med. Chem. 2020, 12, 37–50. [Google Scholar] [CrossRef]
- Besada, P.; Viña, D.; Costas, T.; Costas-Lago, M.C.; Vila, N.; Torres-Terán, I.; Sturlese, M.; Moro, S.; Terán, C. Pyridazinones containing dithiocarbamoyl moieties as a new class of selective MAO-B inhibitors. Bioorg. Chem. 2021, 115, 105203. [Google Scholar] [CrossRef]
- Mazej, T.; Knez, D.; Meden, A.; Gobec, S.; Sova, M. 4-phenethyl-1-propargylpiperidine-derived dual inhibitors of butyrylcholinesterase and monoamine oxidase B. Molecules 2021, 26, 4118. [Google Scholar] [CrossRef]
- Costas-Lago, M.C.; Besada, P.; Rodríguez-Enríquez, F.; Viña, D.; Vilar, S.; Uriarte, E.; Borges, F.; Terán, C. Synthesis and structure-activity relationship study of novel 3-heteroarylcoumarins based on pyridazine scaffold as selective MAO-B inhibitors. Eur. J. Med. Chem. 2017, 139, 1–11. [Google Scholar] [CrossRef]
- Baek, S.C.; Park, M.H.; Ryu, H.W.; Lee, J.P.; Kang, M.-G.; Park, D.; Park, C.M.; Oh, S.-R.; Kim, H. Rhamnocitrin isolated from Prunus padus var. seoulensis: A potent and selective reversible inhibitor of human monoamine oxidase A. Bioorg. Chem. 2019, 83, 317–325. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Oh, J.M.; Kim, H.J.; Mun, S.-G.; Al-Karmalawy, A.A.; Alnajjar, R.; Choi, Y.-J.; Kim, J.-J.; Nam, G.; Kim, H.; et al. Novel coumarin benzamides as potent and reversible monoamine oxidase-B inhibitors: Design, synthesis, and neuroprotective effects. Bioorg. Chem. 2024, 142, 106939. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Jejurikar, B.L.; Rohane, S.H. Drug designing in discovery studio. Asian J. Res. Chem. 2021, 14, 135–138. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
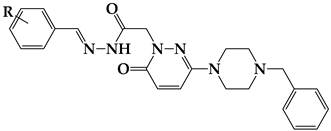 | |||||
|---|---|---|---|---|---|
| Compound | R | Yield (%) | M.P. (°C) | M.W. | Molecular Formula |
| S1 | H | 30.45 | 210–211 | 429.23 | C25H27N5O2 |
| S2 | 4-Br | 58.08 | 189–190 | 507.13 | C25H26BrN5O2 |
| S3 | 2-Br | 66.39 | 188–189 | 507.13 | C25H26BrN5O2 |
| S4 | 3-Br | 69.67 | 190–191 | 507.13 | C25H26BrN5O2 |
| S5 | 3-Cl | 87.29 | 137–138 | 464.14 | C25H26ClN5O2 |
| S6 | 4-Cl | 69.67 | 240–241 | 464.14 | C25H26ClN5O2 |
| S7 | 3-F | 92.33 | 197–198 | 448.21 | C25H26FN5O2 |
| S8 | 4-F | 85.48 | 198–199 | 448.21 | C25H26FN5O2 |
| S9 | 2-CH3 | 43.56 | 222–223 | 444.24 | C26H29N5O2 |
| S10 | 3-CH3 | 86.98 | 106–107 | 444.24 | C26H29N5O2 |
| S11 | 3-OCH3 | 95.26 | 168–169 | 459.24 | C26H29N5O3 |
| S12 | 4-OCH3 | 78.13 | 178–179 | 459.24 | C26H29N5O3 |
| S13 | 4-N(CH3)2 | 72.18 | 108–109 | 472.26 | C27H32N6O2 |
| S14 | 4-NO2 | 59.51 | 199–200 | 474.21 | C25H26N6O4 |
| S15 | 4-C2H5 | 78.62 | 225–226 | 458.26 | C27H31N5O2 |
| S16 | 2-CN | 66.85 | 189–190 | 455.22 | C26H26N6O2 |
| S17 | 3-CN | 86.49 | 128–129 | 455.22 | C26H26N6O2 |
| S18 | 4-CN | 69.47 | 129–130 | 455.22 | C26H26N6O2 |
| S19 | 2-OCH3 | 92.37 | 206–207 | 459.24 | C26H29N5O3 |
| S20 | 3,5-diOCH3 | 97.37 | 208–209 | 490.25 | C27H31N5O4 |
| S21 | 2,4,6-triOCH3 | 54.81 | 160–161 | 520.26 | C28H33N5O5 |
| S22 | 2,3-diOCH3 | 76.95 | 212–213 | 490.25 | C27H31N5O4 |
| S23 | 3,4,5-triOCH3 | 86.16 | 146–147 | 520.26 | C28H33N5O5 |
| S24 | 2,4-diOCH3 | 61.26 | 174–175 | 490.25 | C27H31N5O4 |
| Compound | Residual Activity at 10 µM (%) | IC50 (µM) | SI b | ||
|---|---|---|---|---|---|
| MAO-A | MAO-B | MAO-A | MAO-B | ||
| S1 | 63.41 ± 2.24 | 36.47 ± 8.32 | 16.469 ± 0.047 | 7.046 ± 0.086 | 2.34 |
| S2 | 54.76 ± 3.10 | 56.47 ± 4.99 | 10.949 ± 1.194 | 10.744 ± 0.565 | 1.02 |
| S3 | 34.35 ± 1.93 | 50.59 ± 0.01 | 4.128 ± 1.124 | 4.753 ± 0.155 | 0.87 |
| S4 | 56.91 ± 9.78 | 33.53 ± 2.50 | 26.966 ± 0.797 | 6.171 ± 0.155 | 4.37 |
| S5 | 25.58 ± 3.85 | −24.57 ± 8.51 | 3.857 ± 0.770 | 0.203 ± 0.004 | 19.04 |
| S6 | 42.22 ± 3.22 | 26.71 ± 3.83 | 7.136 ± 1.066 | 4.955 ± 0.353 | 1.44 |
| S7 | 12.81 ± 7.24 | 1.03 ± 4.63 | 4.739 ± 0.624 | 1.711 ± 0.009 | 2.77 |
| S8 | 22.36 ± 3.07 | 31.85 ± 2.62 | 7.994 ± 1.281 | 5.371 ± 0.123 | 1.49 |
| S9 | 23.19 ± 1.20 | 32.89 ± 5.19 | 4.508 ± 0.589 | 7.237 ± 0.994 | 0.62 |
| S10 | 16.14 ± 4.26 | 9.31 ± 2.04 | 4.526 ± 0.424 | 3.549 ± 0.001 | 1.28 |
| S11 | 16.61 ± 9.23 | 14.52 ± 1.14 | 5.064 ± 0.006 | 1.709 ± 0.437 | 2.96 |
| S12 | 7.71 ± 8.25 | 38.46 ± 0.36 | 4.642 ± 0.001 | 6.463 ± 0.035 | 0.72 |
| S13 | 36.08 ± 1.01 | 37.56 ± 2.50 | 5.634 ± 1.182 | 6.480 ± 0.559 | 0.87 |
| S14 | 67.08 ± 5.19 | 39.77 ± 4.14 | 24.170 ± 0.283 | 3.624 ± 0.540 | 6.67 |
| S15 | 30.12 ± 8.10 | 54.44 ± 4.05 | 3.691 ± 0.655 | 5.336 ± 0.667 | 0.69 |
| S16 | 33.16 ± 9.25 | −12.90 ± 1.14 | 6.149 ± 0.305 | 0.979 ± 0.206 | 6.28 |
| S17 | 9.98 ± 0.57 | 0.00 ± 0.01 | 6.283 ± 0.306 | 3.099 ± 0.088 | 2.03 |
| S18 | 35.94 ± 2.54 | 48.37 ± 0.08 | 7.192 ± 0.201 | 3.513 ± 0.811 | 2.05 |
| S19 | 7.83 ± 2.05 | 20.76 ± 1.76 | 4.662 ± 0.307 | 1.000 ± 0.326 | 4.66 |
| S20 | 68.02 ± 4.21 | 11.71 ± 2.41 | 18.999 ± 2.714 | 3.631 ± 0.142 | 5.23 |
| S21 | 32.79 ± 2.18 | 48.45 ± 6.43 | 7.471 ± 0.344 | 6.123 ± 0.662 | 1.22 |
| S22 | −8.50 ± 2.12 | 39.00 ± 2.83 | 5.938 ± 0.264 | 7.871 ± 0.208 | 0.75 |
| S23 | 78.79 ± 0.30 | 53.11 ± 2.67 | 28.373 ± 2.953 | 10.949 ± 0.405 | 2.59 |
| S24 | 20.70 ± 1.34 | 54.66 ± 3.76 | 4.128 ± 0.107 | 10.660 ± 2.476 | 0.39 |
| Toloxatone | 11.72 ± 0.39 | 81.89 ± 2.68 | 1.245 ± 0.233 | >40 | >32.13 |
| Clorgyline | −0.30 ± 0.43 | 25.84 ± 0.23 | 0.013 ± 0.008 | 1.853 ± 0.112 | 142.54 |
| Safinamide | 69.04 ± 1.36 | −15.61 ± 0.86 | >40 | 0.021 ± 0.001 | >1904 |
| Pargyline | 19.88 ± 6.82 | −11.08 ± 1.53 | 2.403 ± 0.358 | 0.14 ± 0.01 | 17.16 |
| Compound | Experimental Pe (×10−6 cm·s−1) | Prediction |
|---|---|---|
| S5 | 5.75 ± 0.13 | CNS+ |
| S16 | 3.78 ± 0.20 | CNS+ |
| Selegiline | 5.69 ± 0.04 | CNS+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.M.; Zenni, Y.N.; Özdemir, Z.; Kumar, S.; Kılıç, S.; Akdağ, M.; Özçelik, A.B.; Kim, H.; Mathew, B. Inhibition of Monoamine Oxidases by Pyridazinobenzylpiperidine Derivatives. Molecules 2024, 29, 3097. https://doi.org/10.3390/molecules29133097
Oh JM, Zenni YN, Özdemir Z, Kumar S, Kılıç S, Akdağ M, Özçelik AB, Kim H, Mathew B. Inhibition of Monoamine Oxidases by Pyridazinobenzylpiperidine Derivatives. Molecules. 2024; 29(13):3097. https://doi.org/10.3390/molecules29133097
Chicago/Turabian StyleOh, Jong Min, Yaren Nur Zenni, Zeynep Özdemir, Sunil Kumar, Semanur Kılıç, Mevlüt Akdağ, Azime Berna Özçelik, Hoon Kim, and Bijo Mathew. 2024. "Inhibition of Monoamine Oxidases by Pyridazinobenzylpiperidine Derivatives" Molecules 29, no. 13: 3097. https://doi.org/10.3390/molecules29133097