

Serine/Threonine Protein Kinases as Attractive Targets for Anti-Cancer Drugs—An Innovative Approach to Ligand Tuning Using Combined Quantum Chemical Calculations, Molecular Docking, Molecular Dynamic Simulations, and Network-like Similarity Graphs


Abstract
:
1. Introduction
1.1. Kinases—State of the Art
- (1)
- ATP-competitive inhibitors (targeting conserved orthosteric site): coumarins (elagic acid [25]); carboxyl acid derivatives ([5-oxo-5,6-dihydroindolo-(1,2a)quinazolin-7-yl]acetic acid (IQA) [26,27], 2,3,4,5-tetrabromocinnamic acid (TBCA) [28], and CX-5011 [29]); polyhalogenated benzimidazoles (4,5,6,7-tetraiodo-1H-benzimidazole (TBI), 4,5,6,7-tetrabromo-1H-benzimidazole (TBBz), and 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) [30]) and benzotriazoles (4,5,6,7-tetrabromobenzotriazole (TBB, TBBt) [31,32]); quinolones [33]; anthraquinones (Quinalizarin), xanthenones [34], and flavonoids (Quercetin) [35,36]; and hydroxycoumarines [37]), SRPIN803 derivatives [38,39], and bromoguaiacol derivatives of 1,2,4-triazole (GO289 [40]);
- (2)
- (3)
- (4)
- (5)
- Peptide-competitive inhibitors (substrate-competitive inhibitors): CIGB-300 (CIGB-325) [48].
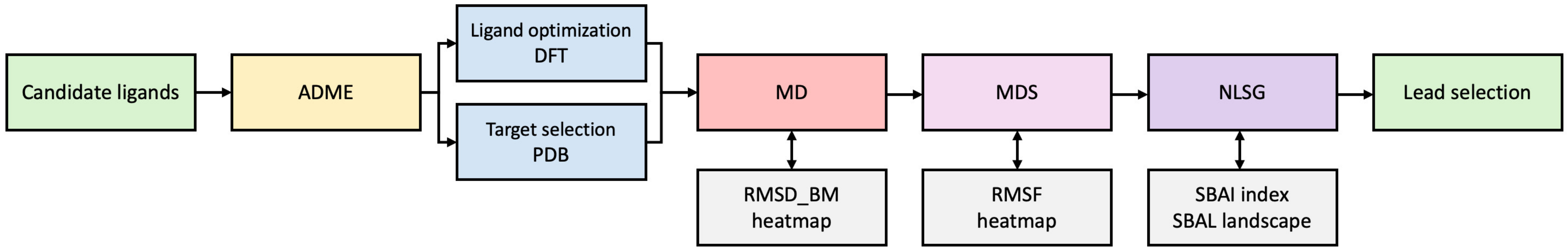
1.2. Research Motivation and Concept
- (1)
- The heatmaps approach, which has proven to be highly effective in
- -
- Identifying the strongest binding components as well as obstacles in the ligands;
- -
- The visualization of the flexibility/dynamics of protein residues.
- (2)
- The Structure Binding Affinity Landscape approach, based on the novel Structure-Binding Affinity Landscape Index (SBAI), that helped measure the degree to which binding affinity gained from molecular docking is gained or lost in response to a relatively small change in the ligand structure.
2. Results
2.1. Physicochemical Profile (ADME) and Key Pharmacokinetic Parameters of the Ligands
2.2. Target Binding Site and 3D Pharmacophore Analysis
2.3. Molecular Docking Results
2.3.1. Protein Kinase CK2α Target
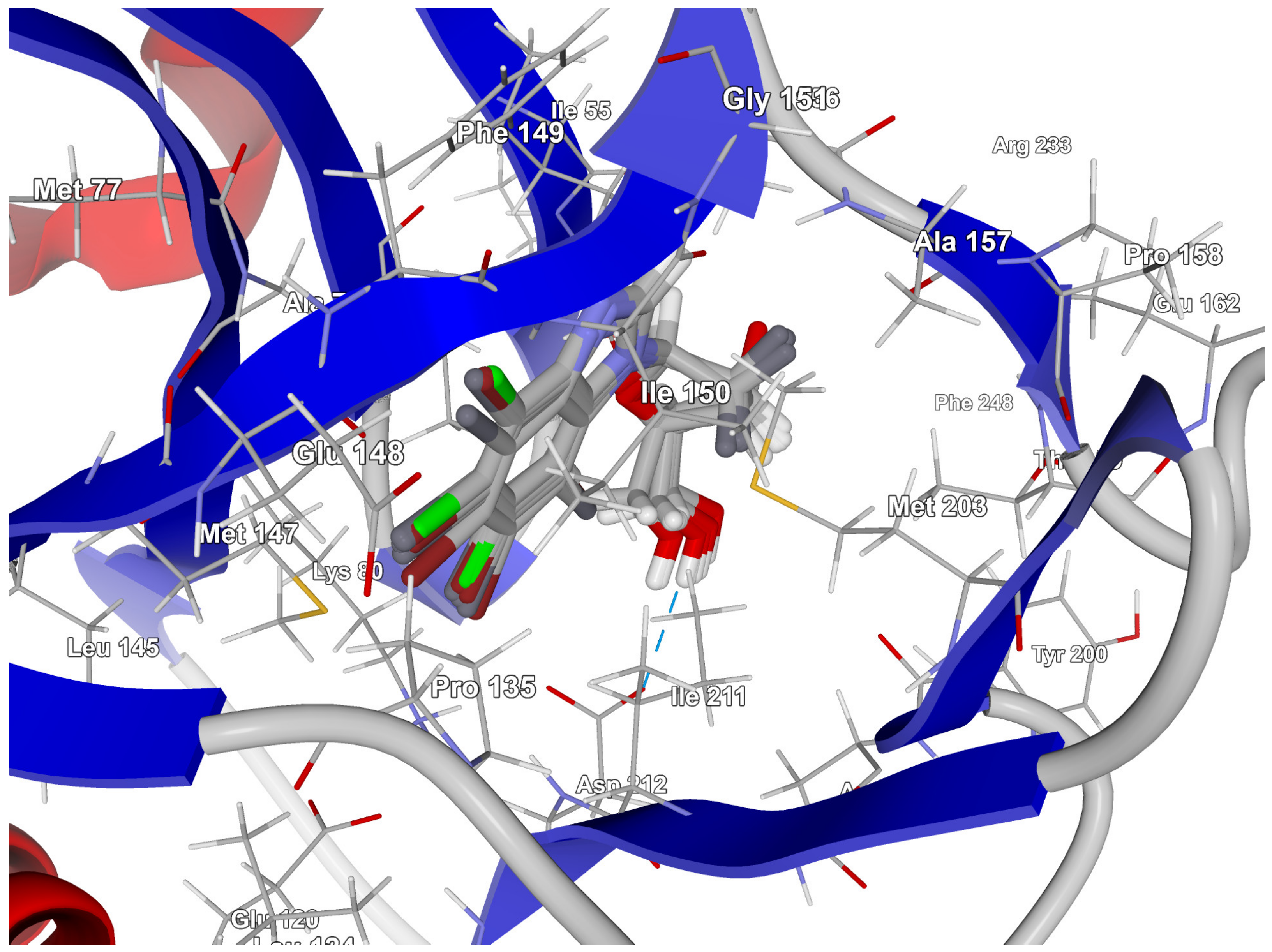
In-Depth Analysis of the Native Ligand Binding Mode
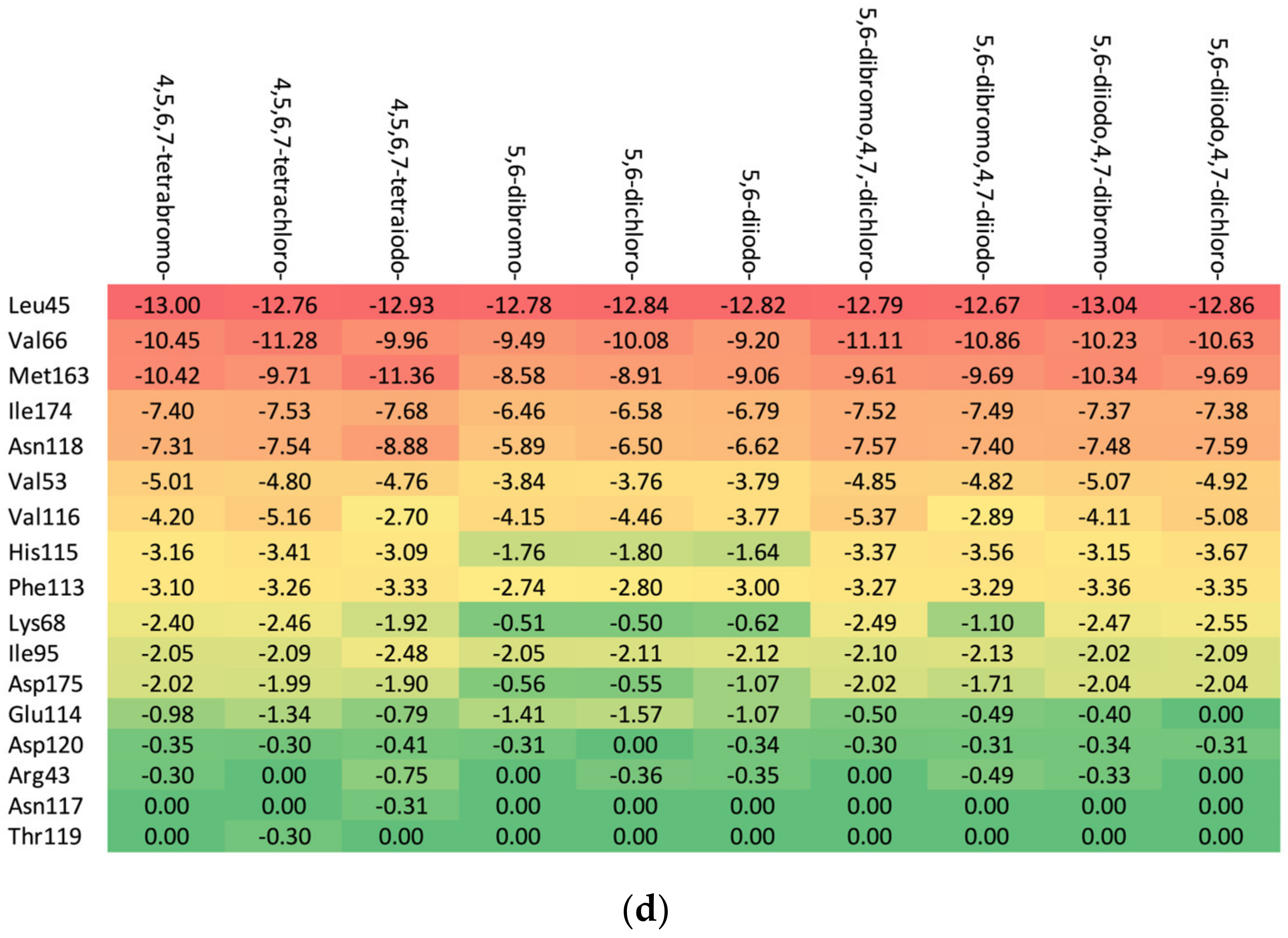
Residues Perspective
- (a)
- Ribose-: Met163 > Leu45 > Asn118 > Val66 > Ile174 > Val53.
- (b)
- 2′-deoxyribose-: Met163 > Leu45 > Val66 > Ile174 > Asn118 > Val53.
- (c)
- 2′-deoxy-2′,2′-difluoro-ribose-: Leu45 > Val66 > Met163 > Ile174 > Asn118 > Val53.
Ligand Perspective
2.3.2. Proto-Oncogene Serine/Threonine-Protein Kinase PIM-1 Target
2.3.3. Atypical Protein Kinase RIO1 Target
2.4. Protein Flexibility and Molecular Dynamics Simulations
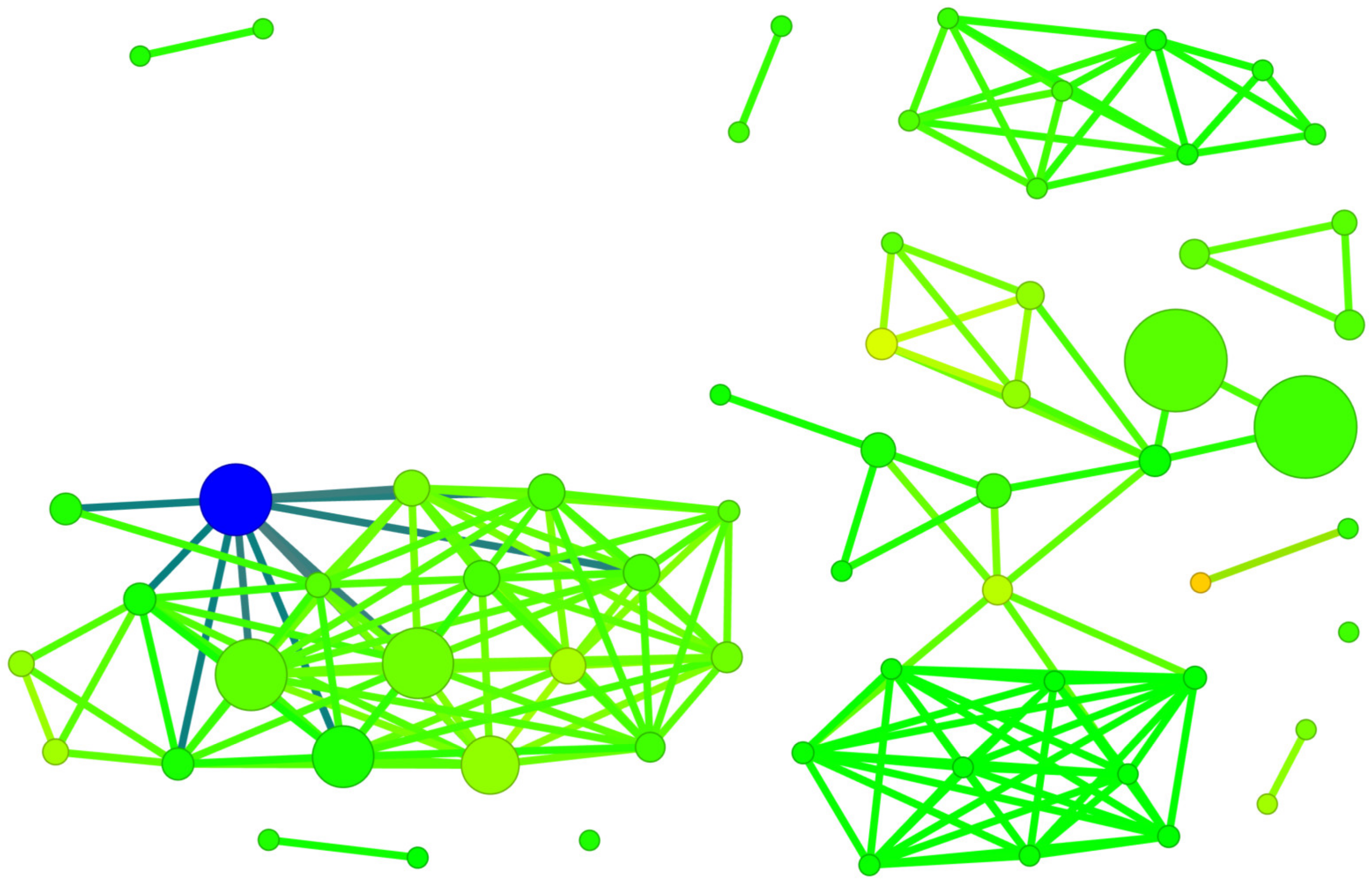
2.5. Structure-Binding Affinity Analysis Using Network-like Similarity Graphs
2.5.1. Structure-Activity Landscape Index
2.5.2. Structure-Binding Affinity Index and Structure-Binding Affinity Landscape
3. Materials and Methods
3.1. ADME and Membrane Permeability Prediction
3.2. Density Functional Theory
3.3. Molecular Docking
3.4. Molecular Dynamic Simulations
3.5. Evaluation of the Binding Modes
3.5.1. Root-Mean-Square Deviation of the Binding Mode
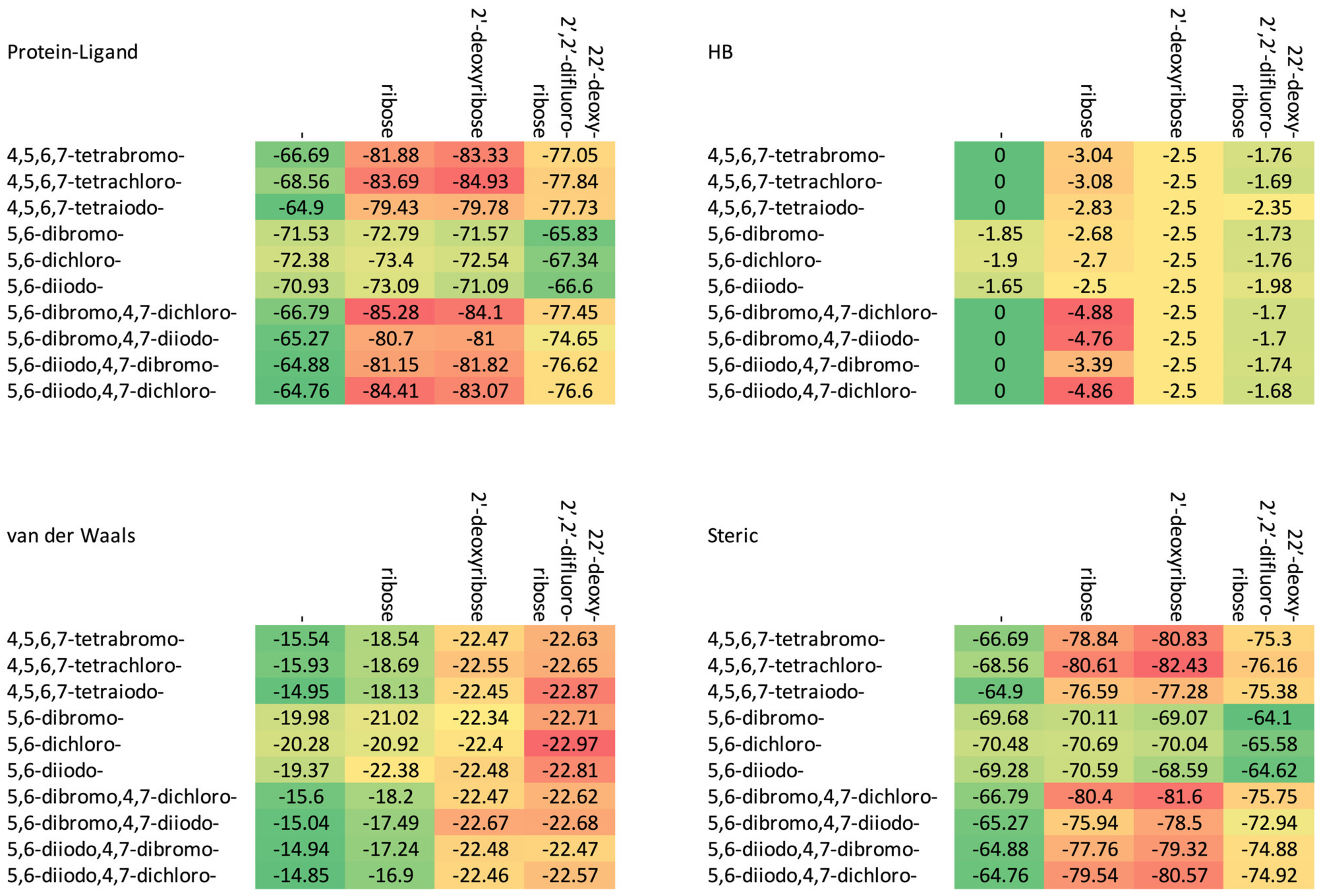
3.5.2. Heatmaps
- -
- -
- The protein–ligand binding energy;
- -
- The normalized B-factors;
- -
- The root-mean-square fluctuation (RMSF) of a structure.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amaral, P.; Carbonell-Sala, S.; De La Vega, F.M.; Faial, T.; Frankish, A.; Gingeras, T.; Guigo, R.; Harrow, J.L.; Hatzigeorgiou, A.G.; Johnson, R.; et al. The Status of the Human Gene Catalogue. Nature 2023, 622, 41–47. [Google Scholar] [CrossRef]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten Things You Should Know about Protein Kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR Core Data Resource in 2021: New Developments and Updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef]
- Reinecke, M.; Heinzlmeir, S.; Wilhelm, M.; Médard, G.; Klaeger, S.; Kuster, B. Kinobeads: A Chemical Proteomic Approach for Kinase Inhibitor Selectivity Profiling and Target Discovery. In Methods and Principles in Medicinal Chemistry; Plowright, A.T., Ed.; Wiley: New York, NY, USA, 2019; pp. 97–130. ISBN 978-3-527-34529-8. [Google Scholar]
- Litchfield, D.W. Protein Kinase CK2: Structure, Regulation and Role in Cellular Decisions of Life and Death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef]
- Stephenson, E.H.; Higgins, J.M.G. Pharmacological Approaches to Understanding Protein Kinase Signaling Networks. Front. Pharmacol. 2023, 14, 1310135. [Google Scholar] [CrossRef]
- Duncan, J.S.; Litchfield, D.W. Too Much of a Good Thing: The Role of Protein Kinase CK2 in Tumorigenesis and Prospects for Therapeutic Inhibition of CK2. Biochim. Biophys. Acta. 2008, 1784, 33–47. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein Kinase CK2 in Health and Disease: CK2: A Key Player in Cancer Biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef]
- Chua, M.; Ortega, C.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Duncan, J.S.; Turowec, J.P.; Vilk, G.; Li, S.S.C.; Gloor, G.B.; Litchfield, D.W. Regulation of Cell Proliferation and Survival: Convergence of Protein Kinases and Caspases. Biochim. Biophys. Acta 2010, 1804, 505–510. [Google Scholar] [CrossRef]
- Ruzzene, M.; Pinna, L.A. Addiction to Protein Kinase CK2: A Common Denominator of Diverse Cancer Cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein Kinase CK2—A Key Suppressor of Apoptosis. Adv. Enzym. Regul. 2008, 48, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Ramji, D.P. Protein Kinase CK2, an Important Regulator of the Inflammatory Response? J. Mol. Med. 2008, 86, 887–897. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.A.; Benveniste, E.N. Protein Kinase CK2: An Emerging Regulator of Immunity. Trends Immunol. 2018, 39, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.M.S.; Lima, C.S.; Silva-Neto, M.A.C.; Da Poian, A.T. Mayaro Virus Infection Cycle Relies on Casein Kinase 2 Activity. Biochem. Biophys. Res. Comm. 2002, 296, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Brunati, A.M.; Saggioro, D.; Chieco-Bianci, L.; Pinna, L.A. Altered Protein Kinase Activities of Lymphoid Cells Transformed by Abelson and Moloney Leukemia Viruses. FEBS Lett. 1986, 206, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Matthews, D.A.; Wadd, S.; Scott, J.E.; Kean, J.; Graham, S.; Russell, W.C.; Clements, J.B. Interaction between Herpes Simplex Virus Type 1 IE63 Protein and Cellular Protein P32. J. Virol. 2000, 74, 11322–11328. [Google Scholar] [CrossRef] [PubMed]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Gietz, R.D.; Graham, K.C.; Litchfield, D.W. Interactions between the Subunits of Casein Kinase II. J. Biol. Chem. 1995, 270, 13017–13021. [Google Scholar] [CrossRef]
- Niefind, K. Crystal Structure of the Catalytic Subunit of Protein Kinase CK2 from Zea Mays at 2.1A resolution. EMBO J. 1998, 17, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Clausen, M.H.; Nielsen, T.E. Allosteric Small-Molecule Kinase Inhibitors. Pharmacol. Therapeut. 2015, 156, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Dong, J.; Song, K.; Wang, T.; Huang, W.; Zhang, J.; Yang, X.; Shen, Y.; Zhang, J. A Novel Allosteric Site in Casein Kinase 2α Discovered Using Combining Bioinformatics and Biochemistry Methods. Acta Pharmacol. Sin. 2017, 38, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Bonvini, P.; Zorzi, E.; Poletto, G.; Pagano, M.A.; Sarno, S.; Donella-Deana, A.; Zagotto, G.; Rosolen, A.; Pinna, L.A.; et al. Identification of ellagic acid as potent inhibitor of protein kinase CK2: A successful example of a virtual screening application. J. Med. Chem. 2006, 49, 2363–2366. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Ruzzene, M.; Frascella, P.; Pagano, M.A.; Meggio, F.; Zambon, A.; Mazzorana, M.; Maira, G.D.; Lucchini, V.; Pinna, L.A. Development and Exploitation of CK2 Inhibitors. Mol. Cell Biochem. 2005, 274, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Zien, P.; Duncan, J.S.; Skierski, J.; Bretner, M.; Litchfield, D.W.; Shugar, D. Tetrabromobenzotriazole (TBBt) and Tetrabromobenzimidazole (TBBz) as Selective Inhibitors of Protein Kinase CK2: Evaluation of Their Effects on Cells and Different Molecular Forms of Human CK2. Biochim. Bioph. Acta 2005, 1754, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Poletto, G.; Di Maira, G.; Cozza, G.; Ruzzene, M.; Sarno, S.; Bain, J.; Elliott, M.; Moro, S.; Zagotto, G.; et al. Tetrabromocinnamic Acid (TBCA) and Related Compounds Represent a New Class of Specific Protein Kinase CK2 Inhibitors. ChemBioChem 2007, 8, 129–139. [Google Scholar] [CrossRef]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented Selectivity and Structural Determinants of a New Class of Protein Kinase CK2 Inhibitors in Clinical Trials for the Treatment of Cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Bain, J.; Kazimierczuk, Z.; Sarno, S.; Ruzzene, M.; Di Maira, G.; Elliott, M.; Orzeszko, A.; Cozza, G.; Meggio, F.; et al. The Selectivity of Inhibitors of Protein Kinase CK2: An Update. Biochem. J. 2008, 415, 353–365. [Google Scholar] [CrossRef]
- Sarno, S.; Reddy, H.; Meggio, F.; Ruzzene, M.; Davies, S.P.; Donella-Deana, A.; Shugar, D.; Pinna, L.A. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP Site-directed Inhibitor of Protein Kinase CK2 (‘Casein Kinase-2′). FEBS Lett. 2001, 496, 44–48. [Google Scholar] [CrossRef]
- Vangrevelinghe, E.; Zimmermann, K.; Schoepfer, J.; Portmann, R.; Fabbro, D.; Furet, P. Discovery of a Potent and Selective Protein Kinase CK2 Inhibitor by High-Throughput Docking. J. Med. Chem. 2003, 46, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Mazzorana, M.; Pinna, L.A.; Battistutta, R. A Structural Insight into CK2 Inhibition. Mol. Cell Biochem. 2008, 316, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, L.; Fresco, P.; Pinto, E.; Sousa, E.; Pinto, M.; Gonçalves, J. Inhibition of Protein Kinase C by Synthetic Xanthone Derivatives. Bioorg. Med. Chem. 2003, 11, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pagano, M.A.; Moro, S.; Zagotto, G.; Ruzzene, M.; Sarno, S.; Cozza, G.; Bain, J.; Elliott, M.; Deana, A.D.; et al. Inhibition of Protein Kinase CK2 by Condensed Polyphenolic Derivatives. An in Vitro and in Vivo Study. Biochemistry 2004, 43, 12931–12936. [Google Scholar] [CrossRef] [PubMed]
- Lolli, G.; Cozza, G.; Mazzorana, M.; Tibaldi, E.; Cesaro, L.; Donella-Deana, A.; Meggio, F.; Venerando, A.; Franchin, C.; Sarno, S.; et al. Inhibition of Protein Kinase CK2 by Flavonoids and Tyrphostins. A Structural Insight. Biochemistry 2012, 51, 6097–6107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhong, R. Docking and 3D-QSAR Studies of 7-Hydroxycoumarin Derivatives as CK2 Inhibitors. Eur. J. Med. Chem. 2010, 45, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Dalle Vedove, A.; Zonta, F.; Zanforlin, E.; Demitri, N.; Ribaudo, G.; Cazzanelli, G.; Ongaro, A.; Sarno, S.; Zagotto, G.; Battistutta, R.; et al. A Novel Class of Selective CK2 Inhibitors Targeting Its Open Hinge Conformation. Eur. J. Med. Chem. 2020, 195, 112267. [Google Scholar] [CrossRef]
- Morooka, S.; Hoshina, M.; Kii, I.; Okabe, T.; Kojima, H.; Inoue, N.; Okuno, Y.; Denawa, M.; Yoshida, S.; Fukuhara, J.; et al. Identification of a Dual Inhibitor of SRPK1 and CK2 That Attenuates Pathological Angiogenesis of Macular Degeneration in Mice. Mol. Pharmacol. 2015, 88, 316–325. [Google Scholar] [CrossRef]
- Oshima, T.; Niwa, Y.; Kuwata, K.; Srivastava, A.; Hyoda, T.; Tsuchiya, Y.; Kumagai, M.; Tsuyuguchi, M.; Tamaru, T.; Sugiyama, A.; et al. Cell-Based Screen Identifies a New Potent and Highly Selective CK2 Inhibitor for Modulation of Circadian Rhythms and Cancer Cell Growth. Sci. Adv. 2019, 5, eaau9060. [Google Scholar] [CrossRef]
- Laudet, B.; Barette, C.; Dulery, V.; Renaudet, O.; Dumy, P.; Metz, A.; Prudent, R.; Deshiere, A.; Dideberg, O.; Filhol, O.; et al. Structure-Based Design of Small Peptide Inhibitors of Protein Kinase CK2 Subunit Interaction. Biochemical J. 2007, 408, 363–373. [Google Scholar] [CrossRef]
- Prudent, R.; Moucadel, V.; Laudet, B.; Barette, C.; Lafanechère, L.; Hasenknopf, B.; Li, J.; Bareyt, S.; Lacôte, E.; Thorimbert, S.; et al. Identification of Polyoxometalates as Nanomolar Noncompetitive Inhibitors of Protein Kinase CK2. Chem. Biol. 2008, 15, 3–692. [Google Scholar] [CrossRef] [PubMed]
- Viht, K.; Saaver, S.; Vahter, J.; Enkvist, E.; Lavogina, D.; Sinijärv, H.; Raidaru, G.; Guerra, B.; Issinger, O.-G.; Uri, A. Acetoxymethyl Ester of Tetrabromobenzimidazole–Peptoid Conjugate for Inhibition of Protein Kinase CK2 in Living Cells. Bioconjug. Chem. 2015, 26, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Zanin, S.; Sarno, S.; Costa, E.; Girardi, C.; Ribaudo, G.; Salvi, M.; Zagotto, G.; Ruzzene, M.; Pinna, L.A. Design, Validation and Efficacy of Bisubstrate Inhibitors Specifically Affecting Ecto-CK2 Kinase Activity. Biochem. J. 2015, 471, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-Chlorophenylamino)Benzo[c][2,6]Naphthyridine-8-Carboxylic Acid (CX-4945), the First Clinical Stage Inhibitor of Protein Kinase CK2 for the Treatment of Cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Sarno, S.; Ruzzene, M.; Girardi, C.; Orzeszko, A.; Kazimierczuk, Z.; Zagotto, G.; Bonaiuto, E.; Di Paolo, M.L.; Pinna, L.A. Exploiting the Repertoire of CK2 Inhibitors to Target DYRK and PIM Kinases. Biochim. Biophys. Acta 2013, 1834, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Girardi, C.; Ranchio, A.; Lolli, G.; Sarno, S.; Orzeszko, A.; Kazimierczuk, Z.; Battistutta, R.; Ruzzene, M.; Pinna, L.A. Cell-Permeable Dual Inhibitors of Protein Kinases CK2 and PIM-1: Structural Features and Pharmacological Potential. Cell. Mol. Life Sci. 2014, 71, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Baladrón, I.; Valenzuela, C.; Perera, Y. CIGB-300: A Peptide-Based Drug That Impairs the Protein Kinase CK2-Mediated Phosphorylation. Semin. Oncol. 2018, 45, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Papinutto, E.; Franchin, C.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Orzeszko, A.; Zanotti, G.; Battistutta, R.; et al. ATP Site-Directed Inhibitors of Protein Kinase CK2: An Update. CTMC 2011, 11, 1340–1351. [Google Scholar] [CrossRef]
- Baumli, S.; Endicott, J.A.; Johnson, L.N. Halogen Bonds Form the Basis for Selective P-TEFb Inhibition by DRB. Chem. Biol. 2010, 17, 931–936. [Google Scholar] [CrossRef]
- Kuo, Y.-H.; Lai, T.-C.; Chang, C.-H.; Hsieh, H.-C.; Yang, F.-M.; Hu, M.-C. 5,6-Dichloro-1-β-D-Ribofuranosylbenzimidazole (DRB) Induces Apoptosis in Breast Cancer Cells through Inhibiting of Mcl-1 Expression. Sci. Rep. 2023, 13, 12621. [Google Scholar] [CrossRef]
- Koronkiewicz, M.; Chilmonczyk, Z.; Kazimerczuk, Z.; Orzeszko, A. Deoxynucleosides with Benzimidazoles as Aglycone Moiety Are Potent Anticancer Agents. Eur. J. Pharmacol. 2018, 820, 146–155. [Google Scholar] [CrossRef]
- Kubiński, K.; Masłyk, M.; Orzeszko, A. Benzimidazole Inhibitors of Protein Kinase CK2 Potently Inhibit the Activity of Atypical Protein Kinase Rio1. Mol. Cell Biochem. 2017, 426, 195–203. [Google Scholar] [CrossRef]
- Battistutta, R.; Mazzorana, M.; Cendron, L.; Borto, A.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Moro, S.; Pinna, L.A. The ATP-Binding Site of Protein Kinase CK2 Holds a Positive Electrostatic Area and Conserved Water Molecules. ChemBioChem 2007, 8, 1804–1809. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An Overview of Halogen Bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Fourmigué, M. Halogen Bonding: Recent Advances. Curr. Opin. Solid State Mat. Sci. 2009, 13, 36–45. [Google Scholar] [CrossRef]
- Johnson, M.A.; Maggiora, G.M. (Eds.) Concepts and Applications of Molecular Similarity; Wiley& Sons: New York, NY, USA, 1991; p. 393. ISBN 978-0-471-62175-1. [Google Scholar]
- Latosińska, J.N.; Latosińska, M.; Seliger, J.; Žagar, V.; Apih, T. Butterfly Effect in Cytarabine: Combined NMR-NQR Experiment, Solid-State Computational Modeling, Quantitative Structure-Property Relationships and Molecular Docking Study. Pharmaceuticals 2024, 17, 445. [Google Scholar] [CrossRef]
- Latosińska, J.N.; Latosińska, M.; Maurin, J.K.; Orzeszko, A.; Kazimierczuk, Z. Quantum-Chemical Insight into Structure–Reactivity Relationship in 4,5,6,7-Tetrahalogeno-1H-benzimidazoles: A Combined X-ray, DSC, DFT/QTAIM, Hirshfeld Surface-Based, and Molecular Docking Approach. J. Phys. Chem. A 2014, 118, 2089–2106. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Inte grated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Zeng, W.; Siramshetty, V.B.; Williams, J.; Kabir, M.; Hagen, N.; Padilha, E.C.; Wang, A.Q.; Mathé, E.A.; Xu, X.; et al. Development and validation of PAMPA-BBB QSAR model to predict brain penetration potential of novel drug candidates. Front. Pharmacol. 2023, 14, 1291246. [Google Scholar] [CrossRef]
- Brear, P.; De Fusco, C.; Atkinson, E.L.; Iegre, J.; Francis-Newton, N.J.; Venkitaraman, A.R.; Hyvönen, M.; Spring, D.R. A Fragment-Based Approach Leading to the Discovery of Inhibitors of CK2α with a Novel Mechanism of Action. RSC Med. Chem. 2022, 13, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D. PDB Protein Data Bank. Available online: https://www.rcsb.org/structure/5kgd (accessed on 29 March 2024).
- Kiburu, I.N.; LaRonde-LeBlanc, N. Interaction of Rio1 Kinase with Toyocamycin Reveals a Conformational Switch That Controls Oligomeric State and Catalytic Activity. PLoS ONE 2012, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Cerca, S.; Kiburu, I.; Thomson, E.; LaRonde, N.; Hurt, E. Dominant Rio1 Kinase/ATPase Catalytic Mutant Induces Trapping of Late Pre-40S Biogenesis Factors in 80S-like Ribosomes. Nucleic Acids Res. 2014, 42, 8635–8647. [Google Scholar] [CrossRef]
- Wang, S.; Xie, J.; Pei, J.; Lai, L. CavityPlus 2022 Update: An Integrated Platform for Comprehensive Protein Cavity Detection and Property Analyses with User-Friendly Tools and Cavity Databases. J. Mol. Biol. 2023, 435, 168141. [Google Scholar] [CrossRef]
- Pagano, M.A.; Andrzejewska, M.; Ruzzene, M.; Sarno, S.; Cesaro, L.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Optimization of Protein Kinase CK2 Inhibitors Derived from 4,5,6,7-Tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. [Google Scholar] [CrossRef]
- Battistutta, R.; De Moliner, E.; Sarno, S.; Zanotti, G.; Pinna, L.A. Structural features underlying selective inhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Prot. Sci. 2001, 10, 2200–2206. [Google Scholar] [CrossRef]
- Battistutta, R.; Mazzorana, M.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Pinna, L.A. Inspecting the structure-activity relationship of protein kinase CK2 inhibitors derived from tetrabromo-benzimidazole. Chem. Biol. 2005, 12, 1211–1219. [Google Scholar] [CrossRef]
- Wawer, M.; Peltason, L.; Weskamp, N.; Teckentrup, A.; Bajorath, J. Structure–Activity Relationship Anatomy by Network-like Similarity Graphs and Local Structure–Activity Relationship Indices. J. Med. Chem. 2008, 51, 6075–6084. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.; Van Drie, J.H. Structure–Activity Landscape Index: Identifying and Quantifying Activity Cliffs. J. Chem. Inf. Model. 2008, 48, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Guner, O. (Ed.) Pharmacophore Perception, Development, and Use in Drug Design; International University Line: La Jolla, CA, USA, 1999; p. 560. [Google Scholar]
- Latosińska, M.; Latosińska, J.N. The Chameleon Strategy—A Recipe for Effective Ligands Screening for Viral Targets Based on Four Novel Structure-Binding Strength Indices. Viruses 2024, 16, 1073. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Latosińska, M.; Latosińska, J.N. Favipiravir Analogues as Inhibitors of SARS-CoV-2 RNA-Dependent RNA Polymerase, Combined Quantum Chemical Modeling, Quantitative Structure-Property Relationship, and Molecular Docking Study. Molecules 2024, 29, 441. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Yang, J.; Chen, C. GEMDOCK: A Generic Evolutionary Method for Molecular Docking. Proteins 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Gehlhaar, D.K.; Bouzida, D.; Rejto, P.A. Fully Automated and Rapid Flexible Docking of Inhibitors Covalently Bound to Serine Proteases. In Evolutionary Programming VII; Porto, V.W., Saravanan, N., Waagen, D., Eiben, A.E., Eds.; Springer Science: Berlin/Heidelberg, Germany, 1998; Volume 1447, pp. 449–461. [Google Scholar]
- Vangone, A.; Schaarschmidt, J.; Koukos, P.; Geng, C.; Citro, N.; Trellet, M.E.; Xue, L.; Bonvin, A.M.J.J. Large-scale prediction of binding affinity in protein-small ligand complexes: The PRODIGY-LIG web server. Bioinformatics 2019, 35, 1585–1587. [Google Scholar] [CrossRef]
- Kurkcuoglu, Z.; Koukos, P.I.; Citro, N.; Trellet, M.E.; Rodrigues, J.P.G.L.M.; Moreira, I.S.; Roel-Touris, J.; Melquiond, A.S.J.; Geng, C.; Schaarschmidt, J.; et al. Performance of HADDOCK and a simple contact-based protein-ligand binding affinity predictor in the D3R Grand Challenge 2. J. Comp. Aid. Mol. Des. 2018, 32, 175–185. [Google Scholar] [CrossRef]
- Diedrich, K.; Krause, B.; Berg, O.; Rarey, M. PoseEdit: Enhanced ligand binding mode communication by interactive 2D diagrams. J. Comput. Aided Mol. Des. 2023, 37, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Structure | Predicted Max pKd Ligandability | Predicted Average pKd | DrugScore (The Degree of Druggability) | Druggability | Surface Area [Å2] | Volume [Å3] | Pharmacophore |
|---|---|---|---|---|---|---|---|---|
| CK2α | 4KWP | 11.59 | 6.90 | 785.00 | Strong | 835.50 | 1309.25 | Hydrophobic centers 7 H-Bond acceptor center 1 |
| 10.30 | 6.15 | −31.00 | Medium | 718.50 | 944.88 | - | ||
| 8.55 | 5.55 | −1022.00 | Weak | 288.75 | 414.62 | - | ||
| 8AEC | 9.40 | 5.84 | 682.00 | Strong | 554.75 | 543.62 | Hydrophobic centers 9 | |
| 7.48 | 5.18 | −1204.00 | Weak | 222.00 | 291.75 | - | ||
| PIM-1 | 4DTK | 11.42 | 6.53 | 1150.00 | Strong | 683.75 | 871.62 | H-Bond donor center 6 H-Bond acceptor center 3 Hydrophobic center 2 |
| 11.06 | 6.41 | 182.00 | Medium | 166.00 | 106.38 | - | ||
| 5KGD | 10.77 | 6.13 | 1155.00 | Strong | 639.50 | 785.88 | H-Bond acceptor center 1 Hydrophobic center 9 | |
| 9.85 | 6.00 | −779.00 | Weak | 271.00 | 408.88 | - | ||
| 8.34 | 5.48 | −682.00 | Weak | 377.50 | 450.00 | - | ||
| RIO1 | 3RE4 | 9.93 | 6.02 | 514.00 | Medium | 610.50 | 918.25 | H-Bond donor center 6 H-Bond acceptor center 5 Hydrophobic center 2 |
| 6.51 | 4.85 | −1315.00 | Weak | - | - | - | ||
| Optimal | >6 | >6 | Strong: ≥600; 600 > Medium > −180 | Strong/Medium | - | - | - |
| Glycone | Ligand | Eprotein–ligand | Etotal | Steric | van der Waals | Hydrogen Bond | Binding Affinity |
|---|---|---|---|---|---|---|---|
| - | 4,5,6,7-tetrabromo- | −66.69 | −67.37 | −66.69 | −15.54 | 0 | −8.67 |
| 4,5,6,7-tetrachloro- | −68.56 | −66.16 | −68.56 | −15.93 | 0 | −8.67 | |
| 4,5,6,7-tetraiodo- | −64.90 | −66.47 | −64.90 | −14.95 | 0 | −8.67 | |
| 5,6-dibromo- | −71.53 | −69.37 | −69.68 | −19.98 | −1.85 | −7.16 | |
| 5,6-dichloro- | −72.38 | −69.70 | −70.48 | −20.28 | −1.90 | −7.17 | |
| 5,6-diiodo- | −70.93 | −68.88 | −69.28 | −19.37 | −1.65 | −7.14 | |
| 5,6-dibromo,4,7-dichloro- | −66.79 | −66.43 | −66.79 | −15.60 | 0 | −8.61 | |
| 5,6-dibromo,4,7-diiodo- | −65.27 | −67.01 | −65.27 | −15.04 | 0 | −8.61 | |
| 5,6-diiodo,4,7-dibromo- | −64.88 | −66.72 | −64.88 | −14.94 | 0 | −8.60 | |
| 5,6-diiodo,4,7-dichloro- | −64.76 | −65.50 | −64.76 | −14.85 | 0 | −8.61 | |
| ribose | 4,5,6,7-tetrabromo- | −81.88 | −82.32 | −78.84 | −18.54 | −3.04 | −7.56 |
| 4,5,6,7-tetrachloro- | −83.69 | −80.81 | −80.61 | −18.69 | −3.08 | −7.56 | |
| 4,5,6,7-tetraiodo- | −79.43 | −81.02 | −76.59 | −18.13 | −2.83 | −7.54 | |
| 5,6-dibromo- | −72.79 | −69.69 | −70.11 | −21.02 | −2.68 | −5.83 | |
| 5,6-dichloro- | −73.40 | −69.78 | −70.69 | −20.92 | −2.70 | −5.83 | |
| 5,6-diiodo- | −73.09 | −70.32 | −70.59 | −22.38 | −2.50 | −5.81 | |
| 5,6-dibromo,4,7-dichloro- | −85.28 | −83.80 | −80.40 | −18.20 | −4.88 | −7.72 | |
| 5,6-dibromo,4,7-diiodo- | −80.70 | −82.01 | −75.94 | −17.49 | −4.76 | −7.71 | |
| 5,6-diiodo,4,7-dibromo- | −81.15 | −82.32 | −77.76 | −17.24 | −3.39 | −7.55 | |
| 5,6-diiodo,4,7-dichloro- | −84.41 | −84.04 | −79.54 | −16.90 | −4.86 | −7.72 | |
| 2′-deoxyribose | 4,5,6,7-tetrabromo- | −83.33 | −81.57 | −80.83 | −22.47 | −2.50 | −8.06 |
| 4,5,6,7-tetrachloro- | −84.93 | −86.28 | −82.43 | −22.55 | −2.50 | −7.94 | |
| 4,5,6,7-tetraiodo- | −79.78 | −85.01 | −77.28 | −22.45 | −2.50 | −7.94 | |
| 5,6-dibromo- | −71.57 | −71.27 | −69.07 | −22.34 | −2.50 | −6.29 | |
| 5,6-dichloro- | −72.54 | −71.98 | −70.04 | −22.40 | −2.50 | −6.29 | |
| 5,6-diiodo- | −71.09 | −70.96 | −68.59 | −22.48 | −2.50 | −6.29 | |
| 5,6-dibromo,4,7-dichloro- | −84.10 | −87.25 | −81.60 | −22.47 | −2.50 | −7.94 | |
| 5,6-dibromo,4,7-diiodo- | −81.00 | −86.09 | −78.50 | −22.67 | −2.50 | −7.94 | |
| 5,6-diiodo,4,7-dibromo- | −81.82 | −86.77 | −79.32 | −22.48 | −2.50 | −7.94 | |
| 5,6-diiodo,4,7-dichloro- | −83.07 | −87.08 | −80.57 | −22.46 | −2.50 | −7.94 | |
| 2′-deoxy- 2′,2′-difluoro-ribose | 4,5,6,7-tetrabromo- | −77.05 | −61.06 | −75.30 | −22.63 | −1.76 | −9.53 |
| 4,5,6,7-tetrachloro- | −77.84 | −60.06 | −76.16 | −22.65 | −1.69 | −9.52 | |
| 4,5,6,7-tetraiodo- | −77.73 | −63.48 | −75.38 | −22.87 | −2.35 | −9.59 | |
| 5,6-dibromo- | −65.83 | −44.21 | −64.10 | −22.71 | −1.73 | −7.89 | |
| 5,6-dichloro- | −67.34 | −44.52 | −65.58 | −22.97 | −1.76 | −7.89 | |
| 5,6-diiodo- | −66.60 | −44.22 | −64.62 | −22.81 | −1.98 | −7.92 | |
| 5,6-dibromo,4,7-dichloro- | −77.45 | −61.53 | −75.75 | −22.62 | −1.70 | −9.52 | |
| 5,6-dibromo,4,7-diiodo- | −74.65 | −60.67 | −72.94 | −22.68 | −1.70 | −9.52 | |
| 5,6-diiodo,4,7-dibromo- | −76.62 | −62.32 | −74.88 | −22.47 | −1.74 | −9.53 | |
| 5,6-diiodo,4,7-dichloro- | −76.60 | −61.45 | −74.92 | −22.57 | −1.68 | −9.52 |
| Ligand | Eprotein–ligand | Etotal | Steric | van der Waals | Hydrogen Bonds | Binding Affinity |
|---|---|---|---|---|---|---|
| 4,5,6,7-tetrabromo- | −91.79 | −77.06 | −87.79 | −15.63 | −4.00 | −9.80 |
| 4,5,6,7-tetrachloro- | −96.61 | −79.36 | −92.83 | −19.63 | −3.79 | −9.78 |
| 4,5,6,7-tetraiodo- | −83.33 | −70.88 | −79.35 | −21.12 | −3.98 | −9.79 |
| 5,6-dibromo- | −94.13 | −81.73 | −89.13 | −18.71 | −5.00 | −8.29 |
| 5,6-dichloro- | −96.82 | −83.81 | −91.82 | −18.87 | −5.00 | −8.29 |
| 5,6-diiodo- | −90.29 | −78.04 | −85.29 | −18.55 | −5.00 | −8.29 |
| 5,6-dibromo,4,7-dichloro- | −91.90 | −76.44 | −87.97 | −20.62 | −3.93 | −9.79 |
| 5,6-dibromo,4,7-diiodo- | −88.96 | −76.35 | −84.99 | −21.96 | −3.97 | −9.79 |
| 5,6-diiodo,4,7-dibromo- | −85.14 | −71.34 | −81.18 | −20.38 | −3.96 | −9.79 |
| 5,6-diiodo,4,7-dichloro- | −86.87 | −72.25 | −82.89 | −20.36 | −3.97 | −9.80 |
| Ligand | Eprotein–ligand | Etotal | Steric | van der Waals | Hydrogen Bond | Binding Affinity |
|---|---|---|---|---|---|---|
| 4,5,6,7-tetrabromo- | −81.75 | −60.95 | −80.65 | −24.66 | −1.09 | −9.45 |
| 4,5,6,7-tetrachloro- | −86.01 | −64.41 | −85.09 | −23.19 | −0.93 | −9.44 |
| 4,5,6,7-tetraiodo- | −75.61 | −57.06 | −75.61 | −25.47 | 0 | −9.32 |
| 5,6-dibromo- | −81.87 | −71.31 | −79.97 | −22.28 | −1.90 | −7.91 |
| 5,6-dichloro- | −84.21 | −73.30 | −82.23 | −21.75 | −1.99 | −7.92 |
| 5,6-diiodo- | −79.76 | −69.26 | −77.74 | −21.75 | −2.03 | −7.93 |
| 5,6-dibromo,4,7-dichloro- | −83.77 | −64.10 | −82.95 | −22.97 | −0.82 | −9.43 |
| 5,6-dibromo,4,7-diiodo- | −80.14 | −61.61 | −80.14 | −27.26 | 0 | −9.32 |
| 5,6-diiodo,4,7-dibromo- | −79.29 | −60.33 | −78.86 | −24.06 | −0.43 | −9.38 |
| 5,6-diiodo,4,7-dichloro- | −81.66 | −62.87 | −80.77 | −22.98 | −0.90 | −9.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latosińska, M.; Latosińska, J.N. Serine/Threonine Protein Kinases as Attractive Targets for Anti-Cancer Drugs—An Innovative Approach to Ligand Tuning Using Combined Quantum Chemical Calculations, Molecular Docking, Molecular Dynamic Simulations, and Network-like Similarity Graphs. Molecules 2024, 29, 3199. https://doi.org/10.3390/molecules29133199
Latosińska M, Latosińska JN. Serine/Threonine Protein Kinases as Attractive Targets for Anti-Cancer Drugs—An Innovative Approach to Ligand Tuning Using Combined Quantum Chemical Calculations, Molecular Docking, Molecular Dynamic Simulations, and Network-like Similarity Graphs. Molecules. 2024; 29(13):3199. https://doi.org/10.3390/molecules29133199
Chicago/Turabian StyleLatosińska, Magdalena, and Jolanta Natalia Latosińska. 2024. "Serine/Threonine Protein Kinases as Attractive Targets for Anti-Cancer Drugs—An Innovative Approach to Ligand Tuning Using Combined Quantum Chemical Calculations, Molecular Docking, Molecular Dynamic Simulations, and Network-like Similarity Graphs" Molecules 29, no. 13: 3199. https://doi.org/10.3390/molecules29133199
APA StyleLatosińska, M., & Latosińska, J. N. (2024). Serine/Threonine Protein Kinases as Attractive Targets for Anti-Cancer Drugs—An Innovative Approach to Ligand Tuning Using Combined Quantum Chemical Calculations, Molecular Docking, Molecular Dynamic Simulations, and Network-like Similarity Graphs. Molecules, 29(13), 3199. https://doi.org/10.3390/molecules29133199