Base-Free Oxidation of HMF to FDCA over Ru/Cu-Co-O·MgO under Aqueous Conditions

Abstract
:1. Introduction
2. Results and Discussion
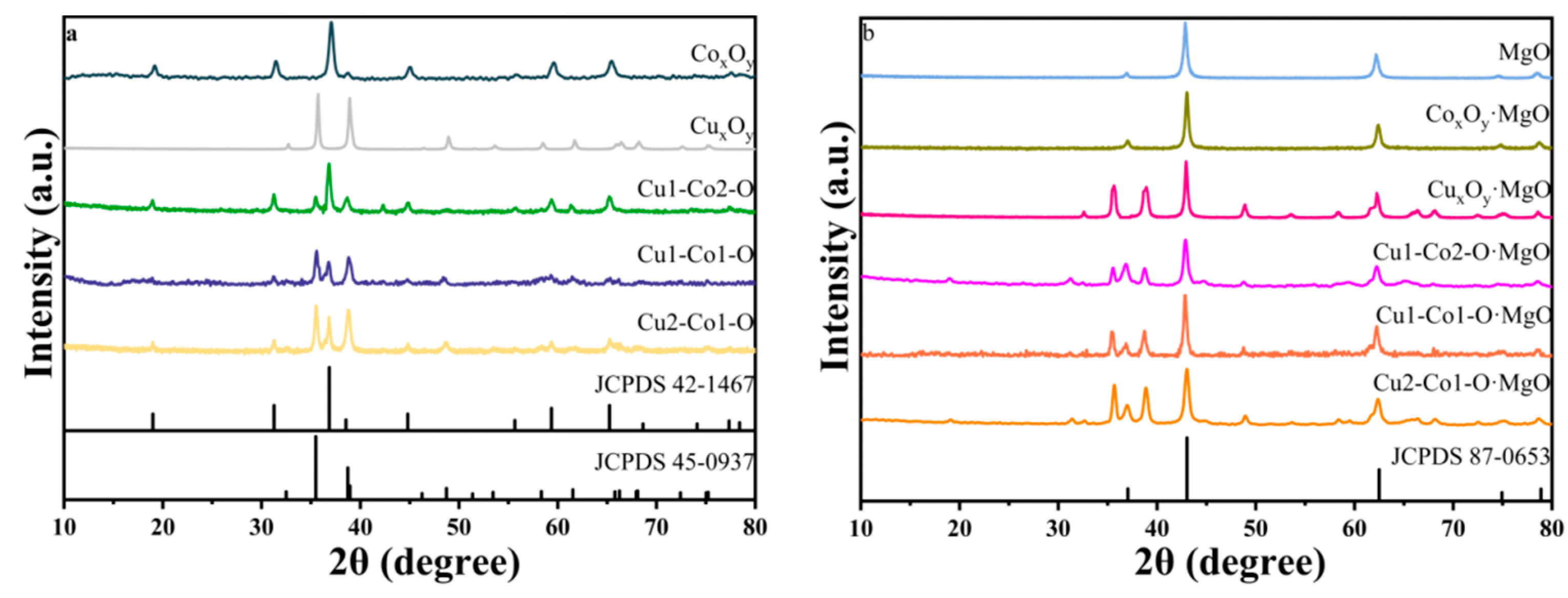
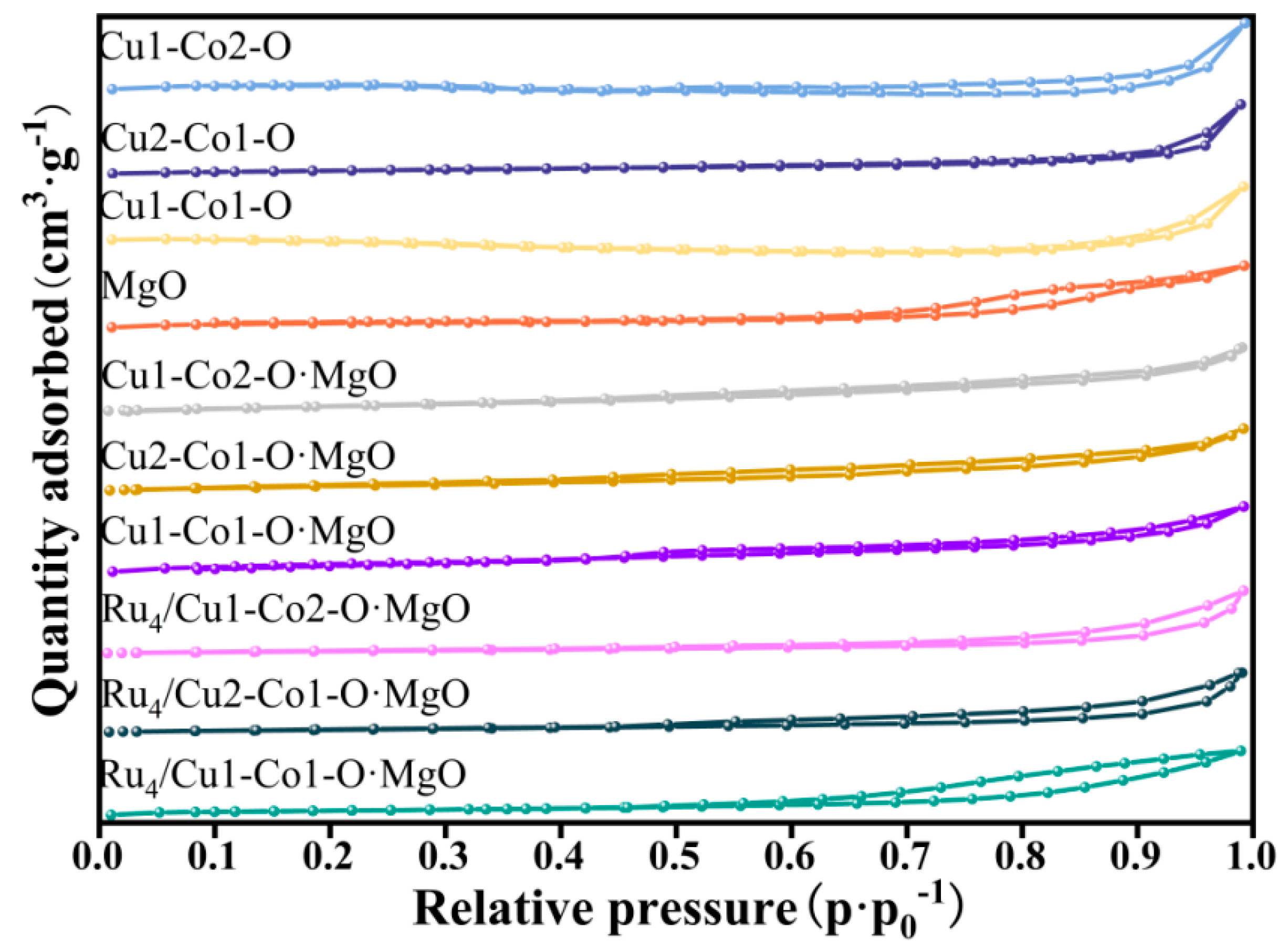
2.1. Structure of Catalysts
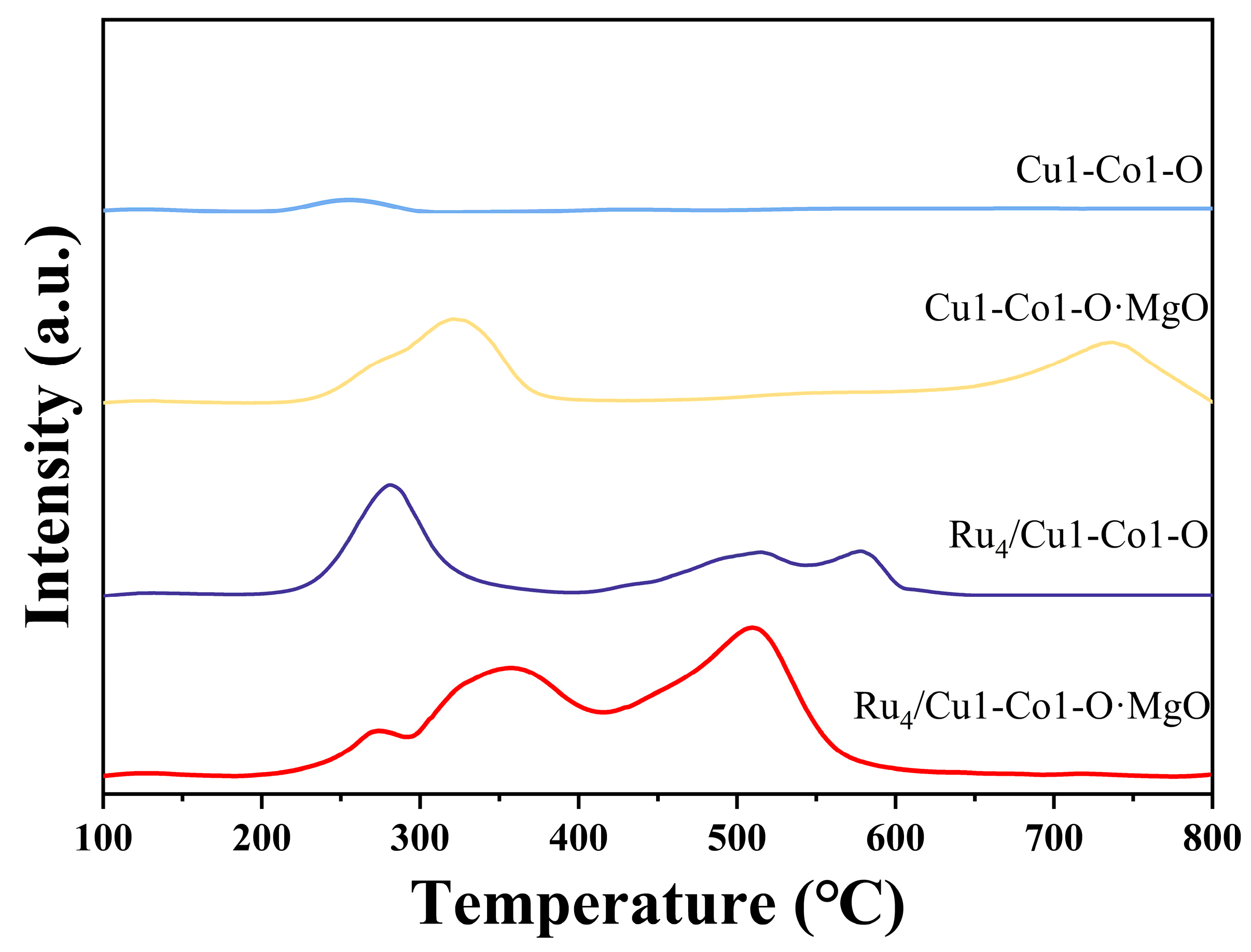
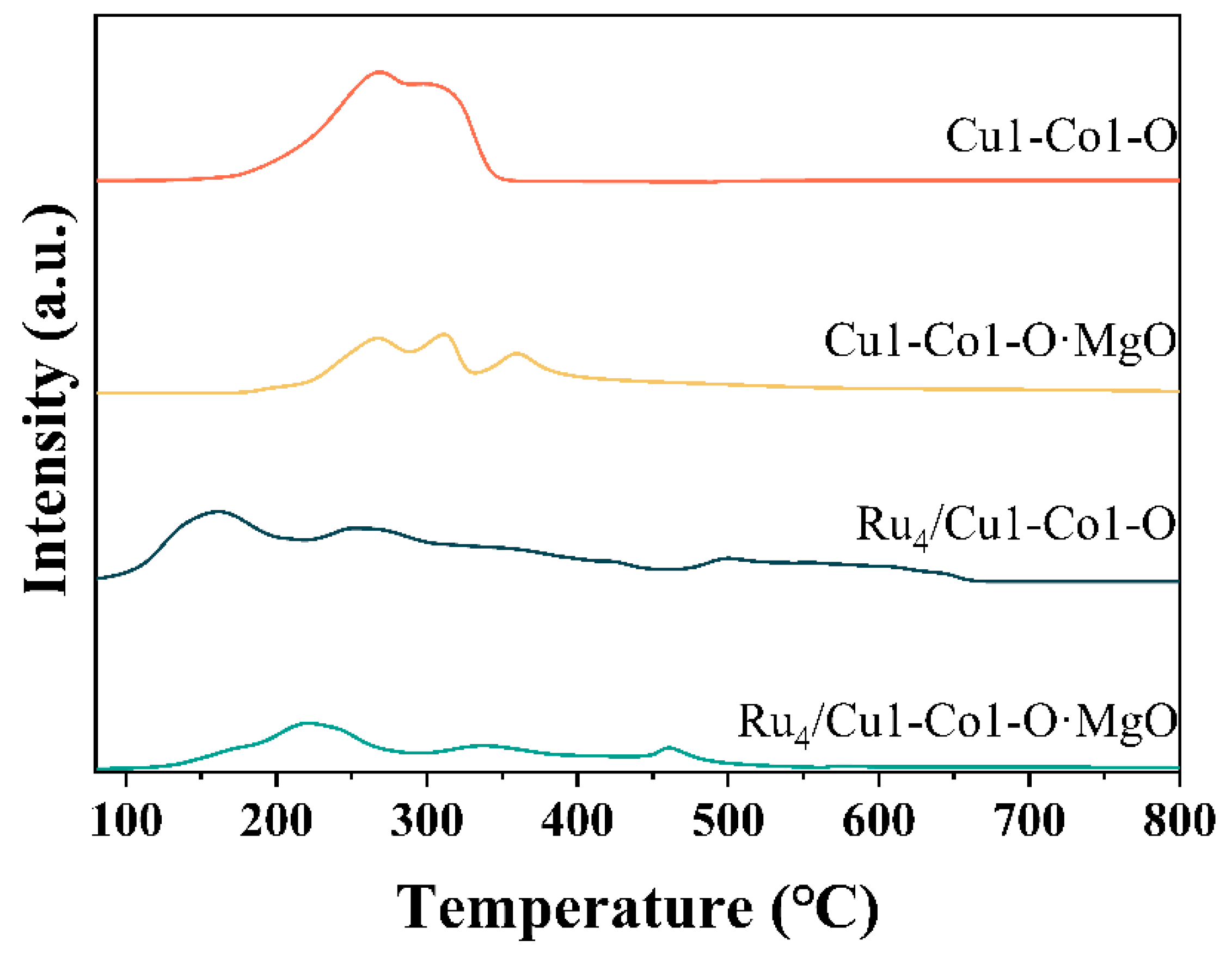
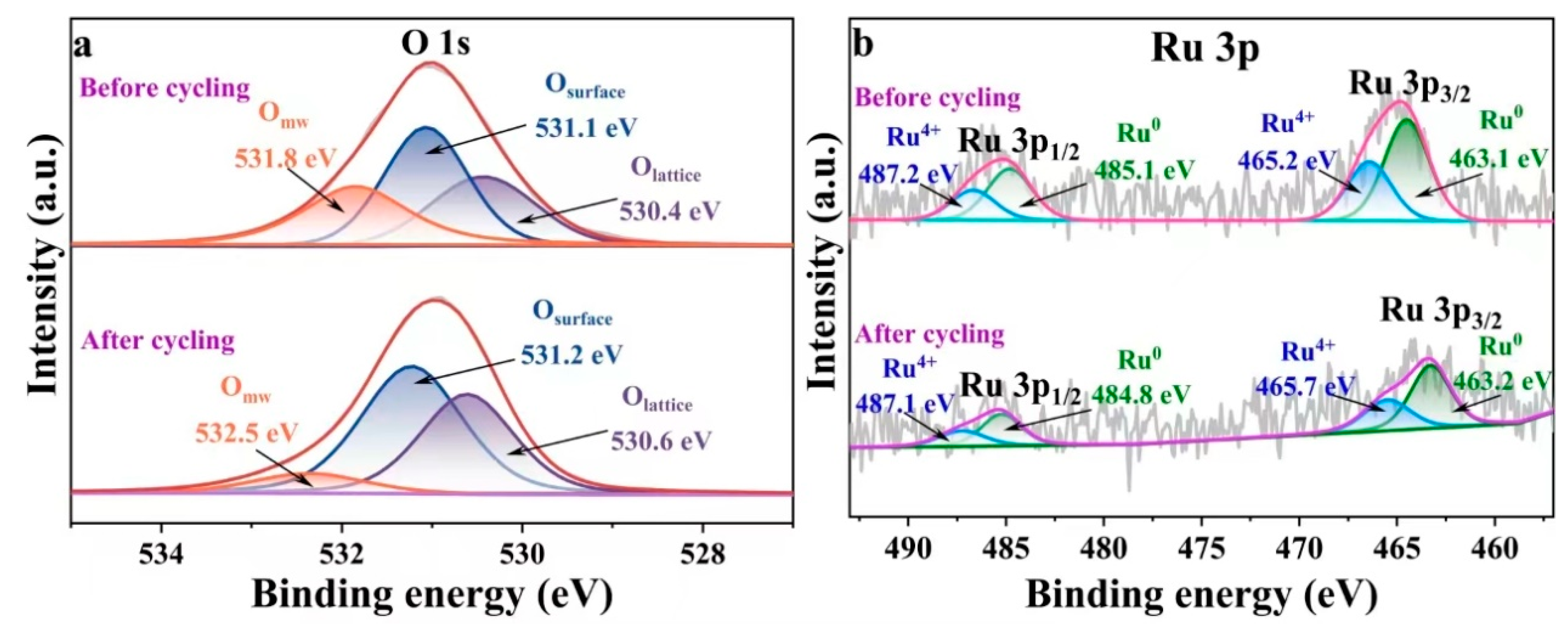
2.2. Evaluation of the Surface Chemistry of Catalysts
2.3. Morphological Analysis of the Catalyst
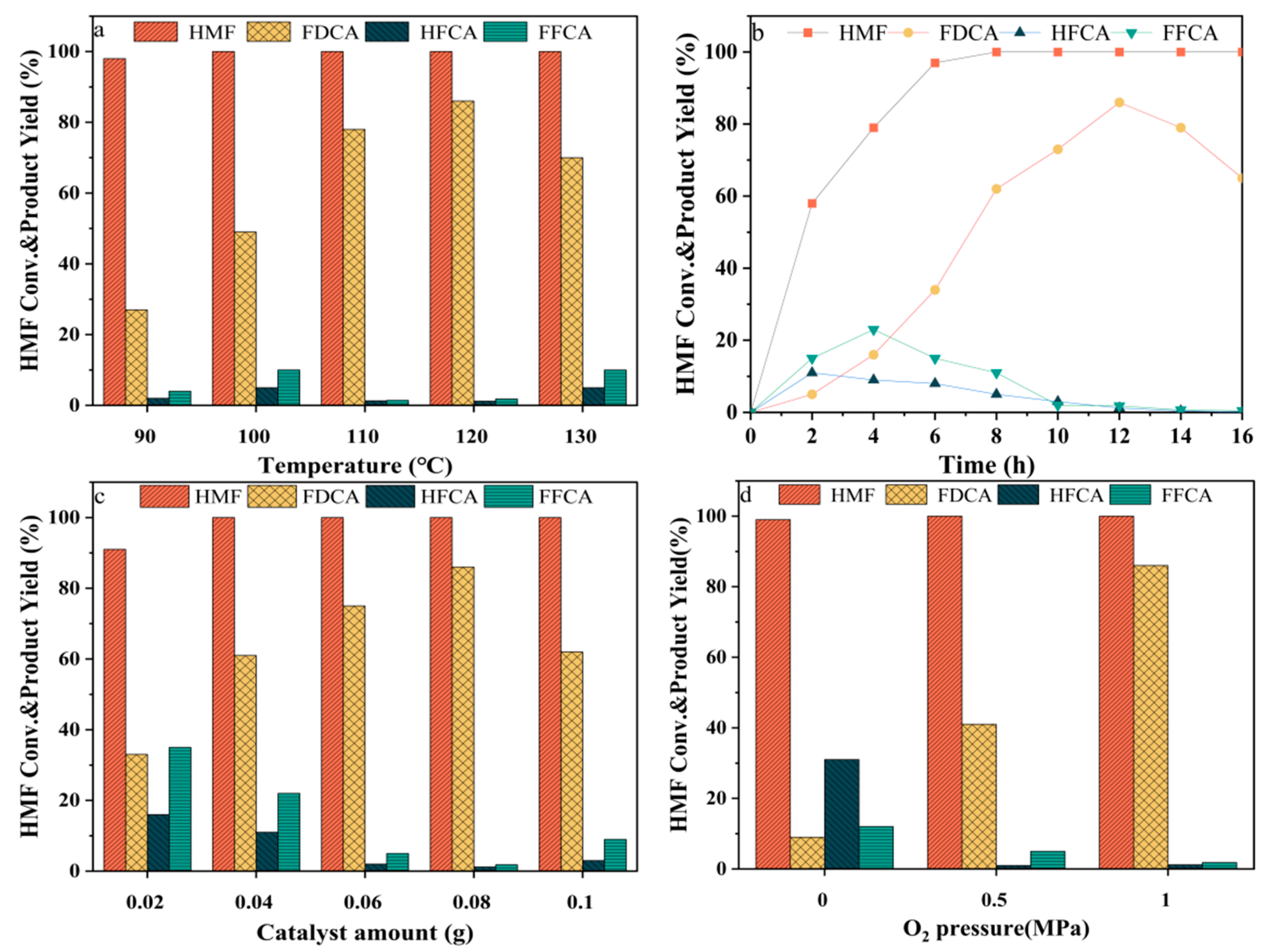
2.4. Optimisation of Catalyst Preparation Conditions
2.5. Evaluation of Catalyst Activity
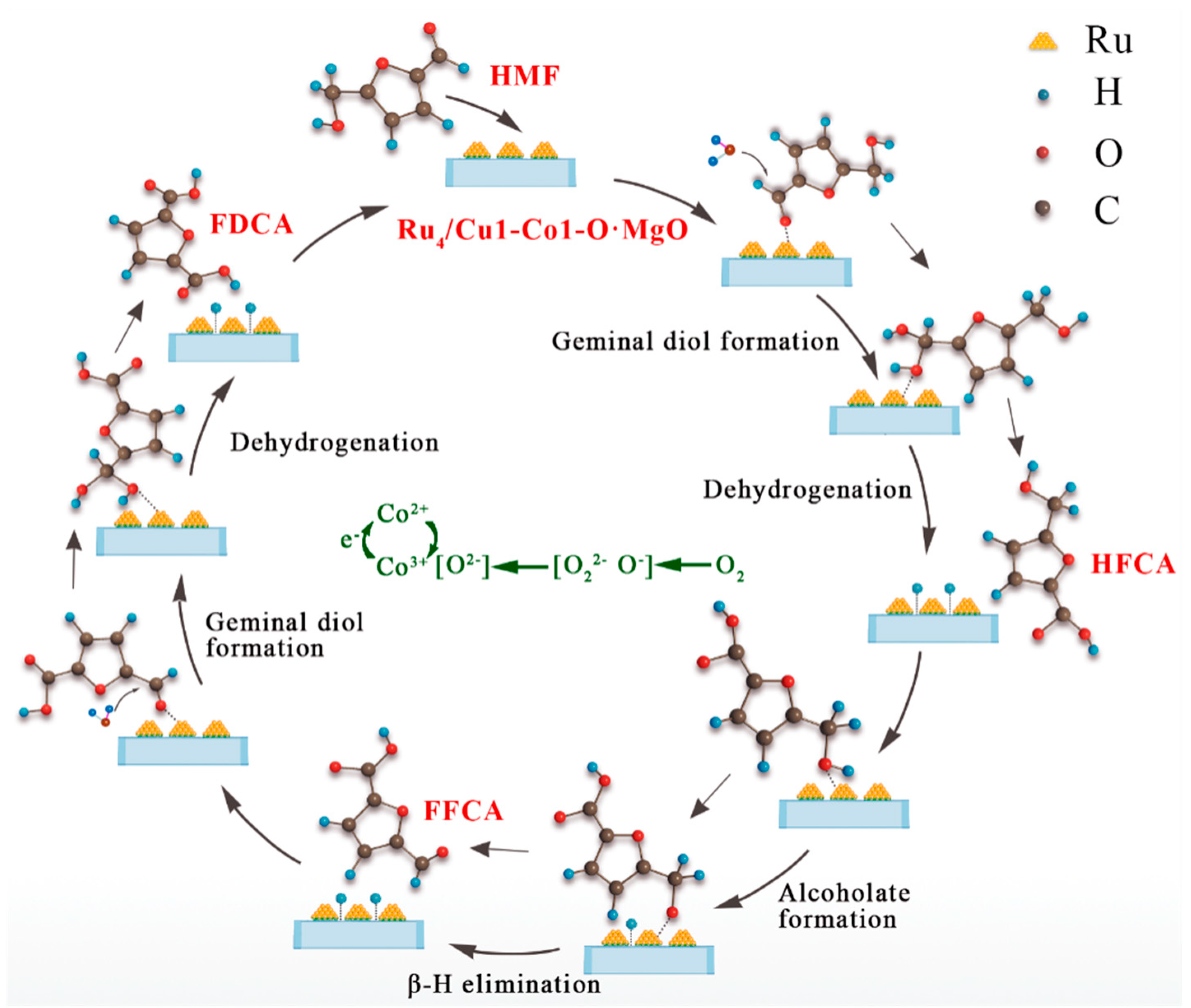
2.6. Potential Reaction Mechanism of the Catalyst
2.7. Separation and Purification of FDCA
3. Experimental Section
3.1. Preparation of the Catalyst
3.2. Catalytic Reaction Equipment and Product Detection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aranha, D.J.; Gogate, P.R. A review on green and efficient synthesis of 5-hydroxymethylfurfural (HMF) and 2,5-furandicarboxylic acid (FDCA) from sustainable biomass. Ind. Eng. Chem. Res. 2023, 62, 3053–3078. [Google Scholar] [CrossRef]
- Cui, Y.; Deng, C.; Fan, L.; Qiu, Y.; Zhao, L. Progress in the biosynthesis of bio-based PET and PEF polyester monomers. Green Chem. 2023, 25, 5836–5857. [Google Scholar] [CrossRef]
- Menegazzo, F.; Fantinel, T.; Signoretto, M.; Pinna, F.; Manzoli, M. On the process for furfural and HMF oxidative esterification over Au/ZrO2. J. Catal. 2014, 319, 61–70. [Google Scholar] [CrossRef]
- Han, X.; Geng, L.; Guo, Y.; Jia, R.; Liu, X.; Zhang, Y.; Wang, Y. Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Pt/C-O-Mg catalyst. Green Chem. 2016, 18, 1597–1604. [Google Scholar] [CrossRef]
- Rass, H.A.; Essayem, N.; Besson, M. Selective aerobic oxidation of 5-HMF into 2,5-furandicarboxylic acid with Pt catalysts supported on TiO2-and ZrO2-based supports. ChemSusChem 2015, 8, 1206–1217. [Google Scholar] [CrossRef]
- Siyo, B.; Schneider, M.; Radnik, J.; Pohl, M.M.; Langer, P.; Steinfeldt, N. Influence of support on the aerobic oxidation of HMF into FDCA over preformed Pd nanoparticle based materials. Appl. Catal. A-Gen. 2014, 478, 107–116. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Y.; Chu, G.; Wang, S.; Wang, C.; Zhang, Y.; Zhang, L.; Mei, J. Surface chemistry regulation on particle-support interaction of ruthenium and Cr-Fe oxides for selective oxidation of 5-hydroxymethylfurfural. Chem. Eng. J. 2023, 474, 145670. [Google Scholar] [CrossRef]
- Antonyraj, C.A.; Huynh, N.T.T.; Lee, K.W.; Kim, Y.J.; Shin, S.; Shin, J.S.; Cho, J.K. Base-free oxidation of 5-hydroxymethyl-2-furfural to 2,5-furan dicarboxylic acid over basic metal oxide-supported ruthenium catalysts under aqueous conditions. Chem. Sci. 2018, 130, 156. [Google Scholar] [CrossRef]
- Gao, T.; Yin, Y.; Fang, W.; Cao, Q. Highly dispersed ruthenium nanoparticles on hydroxyapatite as selective and reusable catalyst for aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid under base-free conditions. Mol. Catal. 2018, 450, 55–64. [Google Scholar] [CrossRef]
- Gorbanev, Y.Y.; Kegnæs, S.; Riisager, A. Selective aerobic oxidation of 5-hydroxymethylfurfural in water over solid ruthenium hydroxide catalysts with magnesium-based supports. Catal. Lett. 2011, 141, 1752–1760. [Google Scholar] [CrossRef]
- Gao, T.; Zhang, H.; Hu, C.; Jing, F.; Fang, W. Base-free aerobic oxidation of 5-hydroxymethylfurfural on a Ru(0) center in cooperation with a Co(II)/Co(III) redox pair over the one-pot synthesized Ru-Co composites. Ind. Eng. Chem. Res. 2020, 59, 17200–17209. [Google Scholar] [CrossRef]
- Kaewpanha, M.; Karnjanakom, S.; Guan, G.; Hao, X.; Yang, J.; Abudula, A. Removal of biomass tar by steam reforming over calcined scallop shell supported Cu catalysts. J. Energy Chem. 2017, 26, 660–666. [Google Scholar] [CrossRef]
- Li, D.; Koike, M.; Chen, J.; Nakagawa, Y.; Tomishige, K. Preparation of Ni-Cu/Mg/Al catalysts from hydrotalcite-like compounds for hydrogen production by steam reforming of biomass tar. Int. J. Hydrogen Energy 2014, 39, 10959–10970. [Google Scholar] [CrossRef]
- Zeng, X.; Zhukova, M.; Faniel, S.; Proost, J.; Flandre, D. Structural and Opto-electronic characterization of CuO thin films prepared by DC reactive magnetron sputtering. J. Mater. Sci.-Mater. Electron. 2020, 31, 4563–4573. [Google Scholar] [CrossRef]
- Sun, J.; Wang, H.; Li, Y.; Zhao, M. Porous Co3O4 column as a high-performance lithium anode material. J. Porous Mater. 2021, 28, 889–894. [Google Scholar] [CrossRef]
- Zheng, X.; Zhou, Y.; Liu, X.; Fu, X.; Peng, H.; Lv, S. Enhanced adsorption capacity of MgO/N-doped active carbon derived from sugarcane bagasse. Bioresour. Technol. 2020, 297, 122413. [Google Scholar] [CrossRef] [PubMed]
- Sing, K.S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Chen, K.; Chu, B.; Qin, Q.; Ou, X.; Zhao, R.; Wei, X.; Wu, H.; Li, B.; Dong, L. Effect of reduction on NO+CO reaction performance of CuO-Co3O4 symbiotic oxide porous nanosheet and DFT calculation. Appl. Surf. Sci. 2022, 589, 153052. [Google Scholar] [CrossRef]
- Romar, H.; Lillebø, A.; Tynjälä, P.; Hu, T.; Holmen, A.; Blekkan, E.; Lassi, U. H2-TPR, XPS and TEM study of the reduction of Ru and Re promoted Co/γ-Al2O3, Co/TiO2 and Co/SiC catalysts. J. Mater. Res. 2016, 5, 33–43. [Google Scholar]
- Su, Q.; Gu, L.; Zhong, A.; Yao, Y.; Ji, W.; Ding, W.; Au, C. Layered double hydroxide derived Mg2Al-LDO supported and K-modified Ru catalyst for hydrogen production via ammonia decomposition. Catal. Lett. 2018, 148, 894–903. [Google Scholar] [CrossRef]
- Hu, X.; Wang, W.; Si, R.; Ma, C.; Jia, C. Hydrogen production via catalytic decomposition of NH3 using promoted MgO-supported ruthenium catalysts. Sci. China Chem. 2019, 62, 1625–1633. [Google Scholar] [CrossRef]
- Assaouka, H.T.; Daawe, D.M.; Fomekong, R.L.; Nsangou, I.N.; Kouotou, P.M. Inexpensive and easily replicable precipitation of CuO nanoparticles for low temperature carbon monoxide and toluene catalytic oxidation. Heliyon 2022, 8, e10689. [Google Scholar] [CrossRef]
- Nasir, M.; Patra, N.; Ahmed, M.A.; Shukla, D.; Kumar, S.; Bhattacharya, D.; Prajapat, C.; Phase, D.; Jha, S.; Biring, S. Role of compensating Li/Fe incorporation in Cu0.945Fe0.055-xLixO: Structural, vibrational and magnetic properties. RSC Adv. 2017, 7, 31970–31979. [Google Scholar] [CrossRef]
- Chinnadurai, D.; Rajendiran, R.; Li, O.L.; Prabakar, K. Mn-Co bimetallic phosphate on electrodeposited PANI nanowires with composition modulated structural morphology for efficient electrocatalytic water splitting. Appl. Catal. B 2021, 292, 120202. [Google Scholar] [CrossRef]
- Li, Y.; Ma, X.; Lv, W.; Chen, H.; Yu, L.; Ma, Z.; Wang, S.; Li, Y. Efficient Selective Oxidation of 5-Hydroxymethylfurfural with Oxygen over a ZnCrAl Mixed Oxide Catalyst Derived from Hydrotalcite-like Precursor. Ind. Eng. Chem. Res. 2022, 61, 11597–11603. [Google Scholar] [CrossRef]
- Yu, L.; Chen, H.; Wen, Z.; Jin, M.; Ma, Z.; Ma, X.; Sang, Y.; Chen, M.; Li, Y. Highly selective oxidation of 5-hydroxymethylfurfural to 2, 5-diformylfuran over an α-MnO2 catalyst. Catal. Today 2021, 367, 9–15. [Google Scholar] [CrossRef]
- Li, C.; Lyu, Q.; Li, Z.; Yang, S.; Sun, Y.; Tang, X.; Zeng, X.; Lin, L. Hydrogenation of dimethyl 2,5-furandicarboxylate to dimethyl tetrahydrofuran-2,5-dicarboxylate over Ru/HY. Korean J. Chem. Eng. 2023, 40, 2149–2158. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gogoi, P.; Venugopalan, A.T.; Raja, T. A highly efficient and reusable Ru-NaY catalyst for the base free oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic acid. Catal. Today 2021, 375, 145–154. [Google Scholar] [CrossRef]
- Lu, H.; Bai, J.; Fei, Y.; Zhang, X.; Ying, J.; Wang, J.; Ping, C.; Zhou, M. Oxidation of 5-hydroxylmethylfurfural to 2,5-furandicarboxylic acid catalyzed by magnetic MnO2-Fe3O4 composite oxides. J. Fuel Chem. Technol. 2021, 49, 312–321. [Google Scholar] [CrossRef]
- Vandarkuzhali, S.A.A.; Karthikeyan, G.; Pachamuthu, M. Efficient oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over FeNPs@ NH2-SBA-15 catalyst in water. Mol. Catal. 2021, 516, 111951. [Google Scholar] [CrossRef]
- Albonetti, S.; Lolli, A.; Morandi, V.; Migliori, A.; Lucarelli, C.; Cavani, F. Conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Au-based catalysts: Optimization of active phase and metal–support interaction. Appl. Catal. B 2015, 163, 520–530. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Yang, D.; Dong, J.; Cheng, X.; Yu, H. Immobilization of Zn(Ⅱ) and Cu(Ⅱ) in basic magnesium-sulfate-cementitious material system: Properties and mechanism. J. Hazard. 2023, 446, 130720. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.D.D.; de Mello, M.D.; da Silva, M.A.P. Catalytic Oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic Acid over Ru/Al2O3 in a Trickle-bed Reactor. Ind. Eng. Chem. Res. 2018, 58, 128–137. [Google Scholar] [CrossRef]
- Gao, T.; Chen, J.; Fang, W.; Cao, Q.; Su, W.; Dumeignil, F. Ru/MnXCe1OY catalysts with enhanced oxygen mobility and strong metal-support interaction: Exceptional performances in 5-hydroxymethylfurfural base-free aerobic oxidation. J. Catal. 2018, 368, 53–68. [Google Scholar] [CrossRef]
- Wei, Y.; Li, C.; Zhu, C.; Zhang, Y.; Zhu, Z.; Chen, Y.; Li, X.; Yan, Y. Oxygen vacancy and support adsorption synergistic effect in aerobic oxidation of HMF to FDCA: A case study using nitrogen-doped porous carbon supported Bi-CeO2. J. Taiwan Inst. Chem. Eng. 2022, 138, 104439. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Surface Area (m2/g) | Pore Volume (cm3/g) | Average Pore Size (nm) |
|---|---|---|---|
| Cu1-Co2-O | 1.3 | 0.0034 | 10.87 |
| Cu1-Co1-O | 4.0 | 0.0190 | 18.84 |
| Cu2-Co1-O | 6.6 | 0.0268 | 15.76 |
| MgO | 38.8 | 0.1544 | 16.72 |
| Cu1-Co2-O·MgO | 39.3 | 0.1264 | 2.73 |
| Cu1-Co1-O·MgO | 35.9 | 0.0677 | 7.49 |
| Cu2-Co1-O·MgO | 29.9 | 0.1070 | 3.83 |
| Ru4/Cu1-Co2-O·MgO | 44.4 | 0.2041 | 1.16 |
| Ru4/Cu1-Co1-O·MgO | 55.9 | 0.2193 | 15.68 |
| Ru4/Cu2-Co1-O·MgO | 63.4 | 0.3358 | 2.45 |
| Sample | Basicity (mmol/g) | |||
|---|---|---|---|---|
| Low Basicity (<250 °C) | Moderate Basicity (250–400 °C) | High Basicity (>400 °C) | Total | |
| Cu1-Co1-O | 0.001 | 0.001. | 0 | 0.002 |
| Cu1-Co1-O·MgO | 0.002 | 0.055 | 0.058 | 0.115 |
| Ru4/Cu1-Co1-O | 0.005 | 0.043 | 0.040 | 0.088 |
| Ru4/Cu1-Co1-O·MgO | 0.009 | 0.132 | 0.152 | 0.293 |
| Entry | Sample | Conv. (%) | YFDCA (%) | YHFCA (%) | YFFCA (%) |
|---|---|---|---|---|---|
| 1 | Ru4/MgO | 100 | 59.3 | 1.2 | 1.4 |
| 2 | Ru4/CuxOy·MgO | 100 | 43.1 | 0.2 | 0.6 |
| 3 | Ru4/CoxOy·MgO | 100 | 18.6 | 1.1 | 1.5 |
| 4 | Ru4/Cu1-Co2-O·MgO | 100 | 22.4 | 1.2 | 1.8 |
| 5 | Ru4/Cu2-Co1-O·MgO | 100 | 33.7 | 0.5 | 1.3 |
| 6 | Ru4/Cu1-Co1-O·MgO | 100 | 78.6 | 1.3 | 1.4 |
| 7 | Ru4/Cu1-Co1-O·MgO (2 mmol) | 100 | 17.9 | 0.6 | 0.5 |
| 8 | Ru4/Cu1-Co1-O·MgO (4 mmol) | 100 | 42.0 | 0.3 | 0.6 |
| 9 | Ru4/Cu1-Co1-O·MgO (8 mmol) | 100 | 38.9 | 1.2 | 1.4 |
| 10 | Ru4/Cu1-Co1-O | 100 | 10.6 | 0.8 | 0.3 |
| 11 | Cu1-Co1-O·MgO | 72.7 | 3.5 | 2.2 | 25.7 |
| 12 | Ru1/Cu1-Co1-O·MgO | 84.8 | 30.2 | 15.3 | 22.6 |
| 13 | Ru2/Cu1-Co1-O·MgO | 100 | 35.4 | 1.7 | 2.5 |
| 14 | Ru3/Cu1-Co1-O·MgO | 100 | 71.0 | 4.3 | 2.7 |
| 15 | Ru5/Cu1-Co1-O·MgO | 100 | 63.7 | 9.1 | 2.0 |
| Temperature (°C) | Conv. (%) | FDCA Yield (%) | TON 1 | TOF 2 (h−1) |
|---|---|---|---|---|
| 90 | 98.4 | 27.3 | 1.71 | 0.14 |
| 100 | 100 | 49.8 | 3.11 | 0.26 |
| 110 | 100 | 78.6 | 4.91 | 0.41 |
| 120 | 100 | 86.1 | 5.38 | 0.45 |
| 130 | 100 | 70.4 | 4.40 | 0.37 |
| Sample | [Mg2+] (mg/L) | [Run+] (mg/L) |
|---|---|---|
| Ru4/MgO | 216.373 | 0.083 |
| Ru4/Cu1-Co1-O·MgO | 113.106 | 0.015 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Chu, G.; Wang, S.; Ma, J.; Wang, C. Base-Free Oxidation of HMF to FDCA over Ru/Cu-Co-O·MgO under Aqueous Conditions. Molecules 2024, 29, 3213. https://doi.org/10.3390/molecules29133213
Zhang S, Chu G, Wang S, Ma J, Wang C. Base-Free Oxidation of HMF to FDCA over Ru/Cu-Co-O·MgO under Aqueous Conditions. Molecules. 2024; 29(13):3213. https://doi.org/10.3390/molecules29133213
Chicago/Turabian StyleZhang, Shuang, Guoning Chu, Sai Wang, Ji Ma, and Chengqian Wang. 2024. "Base-Free Oxidation of HMF to FDCA over Ru/Cu-Co-O·MgO under Aqueous Conditions" Molecules 29, no. 13: 3213. https://doi.org/10.3390/molecules29133213