

Do Molecules Tunnel through Nanoporous Graphene?


Abstract
1. Introduction
2. Results and Discussion
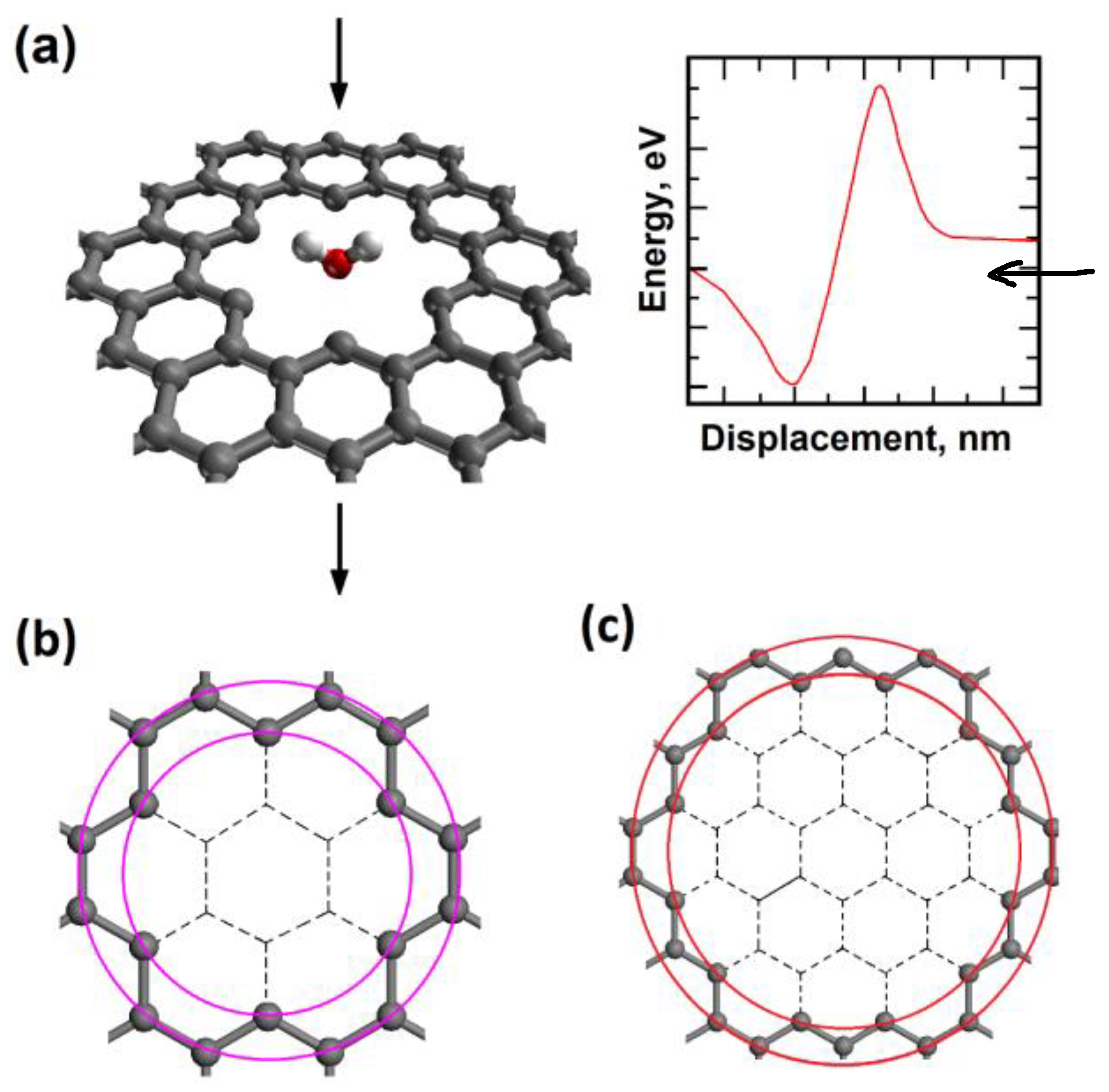
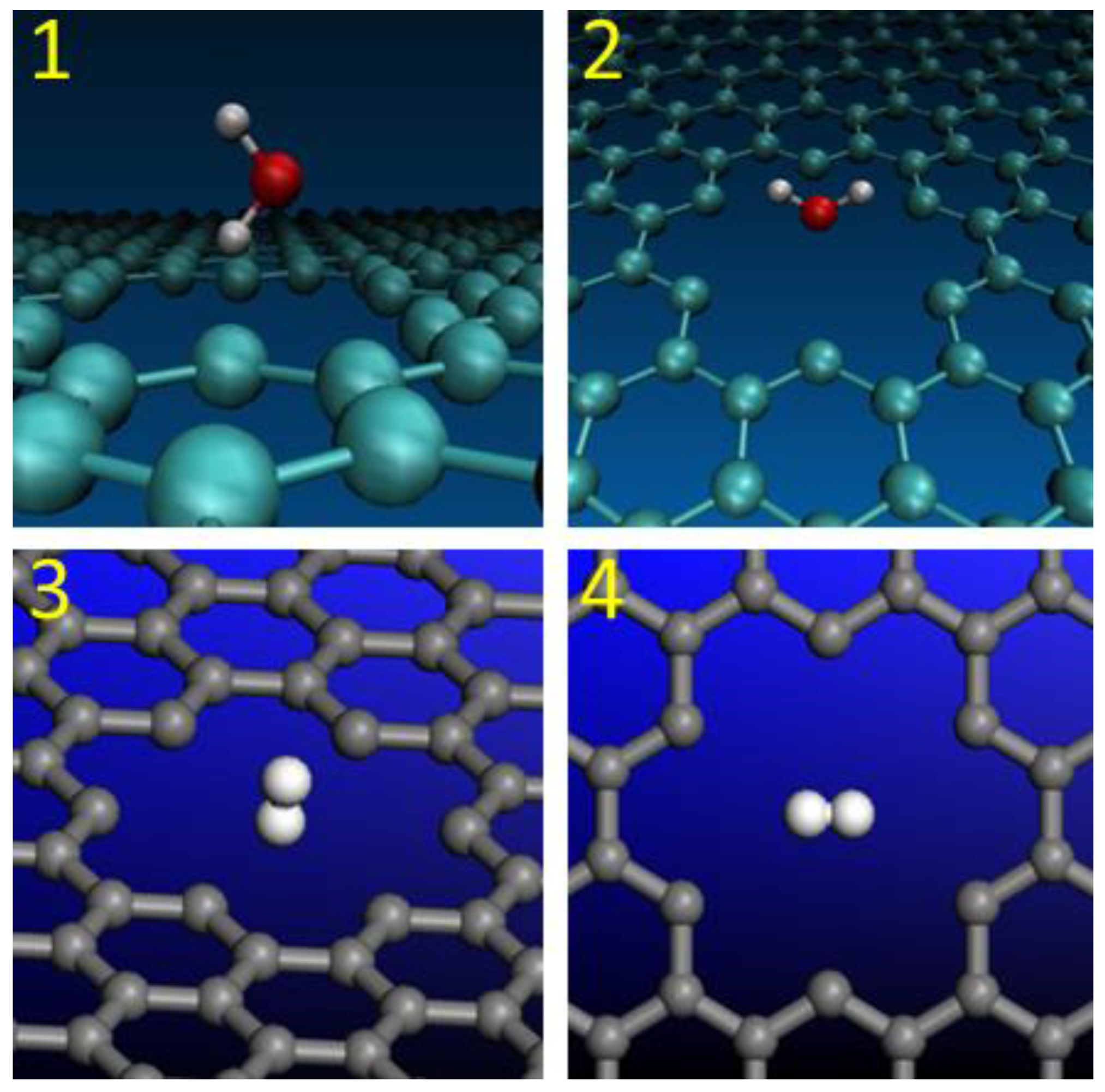
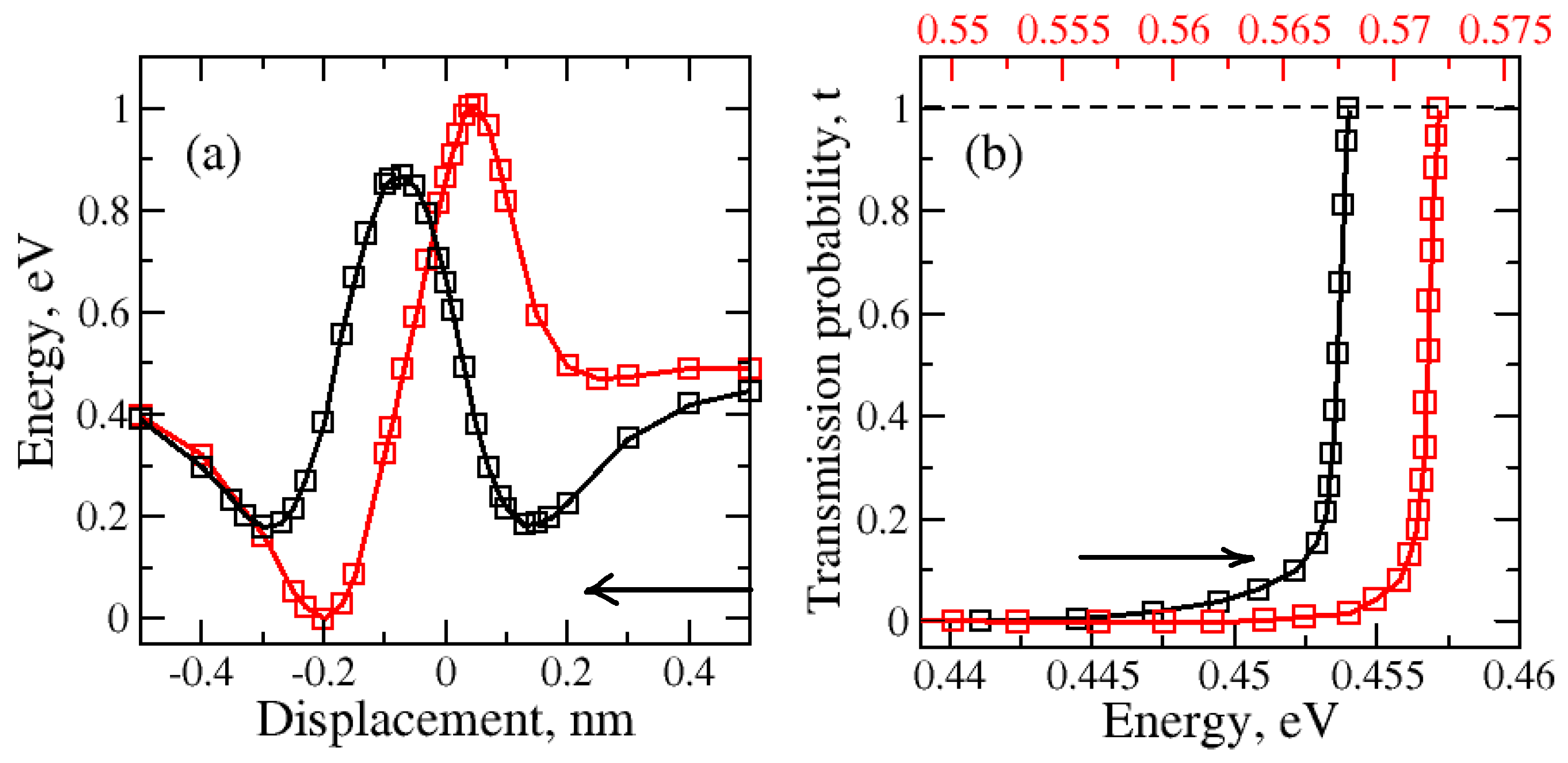
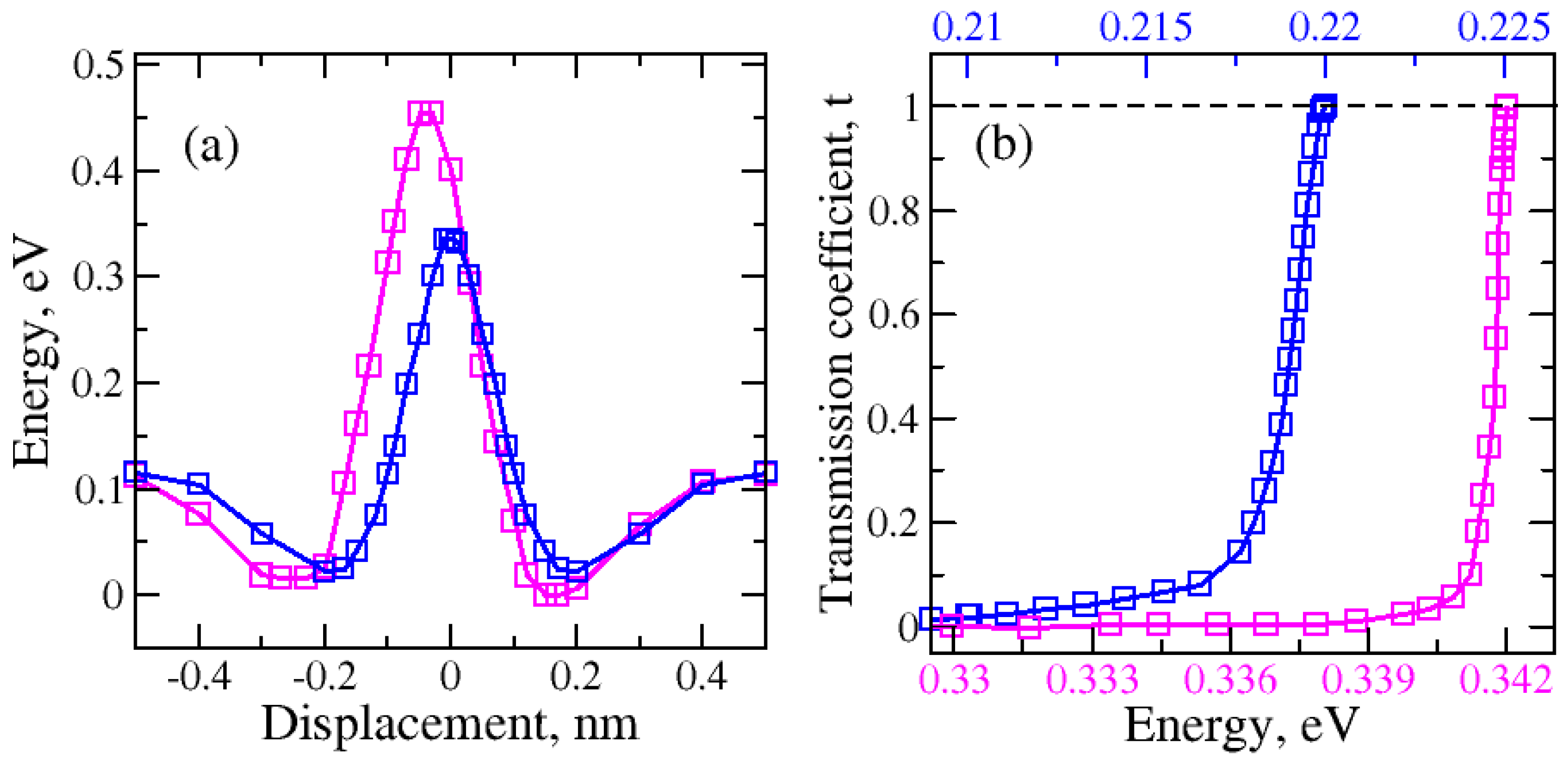
2.1. Nanopore Size and Molecular Orientation Effect on Tunneling Mechanism
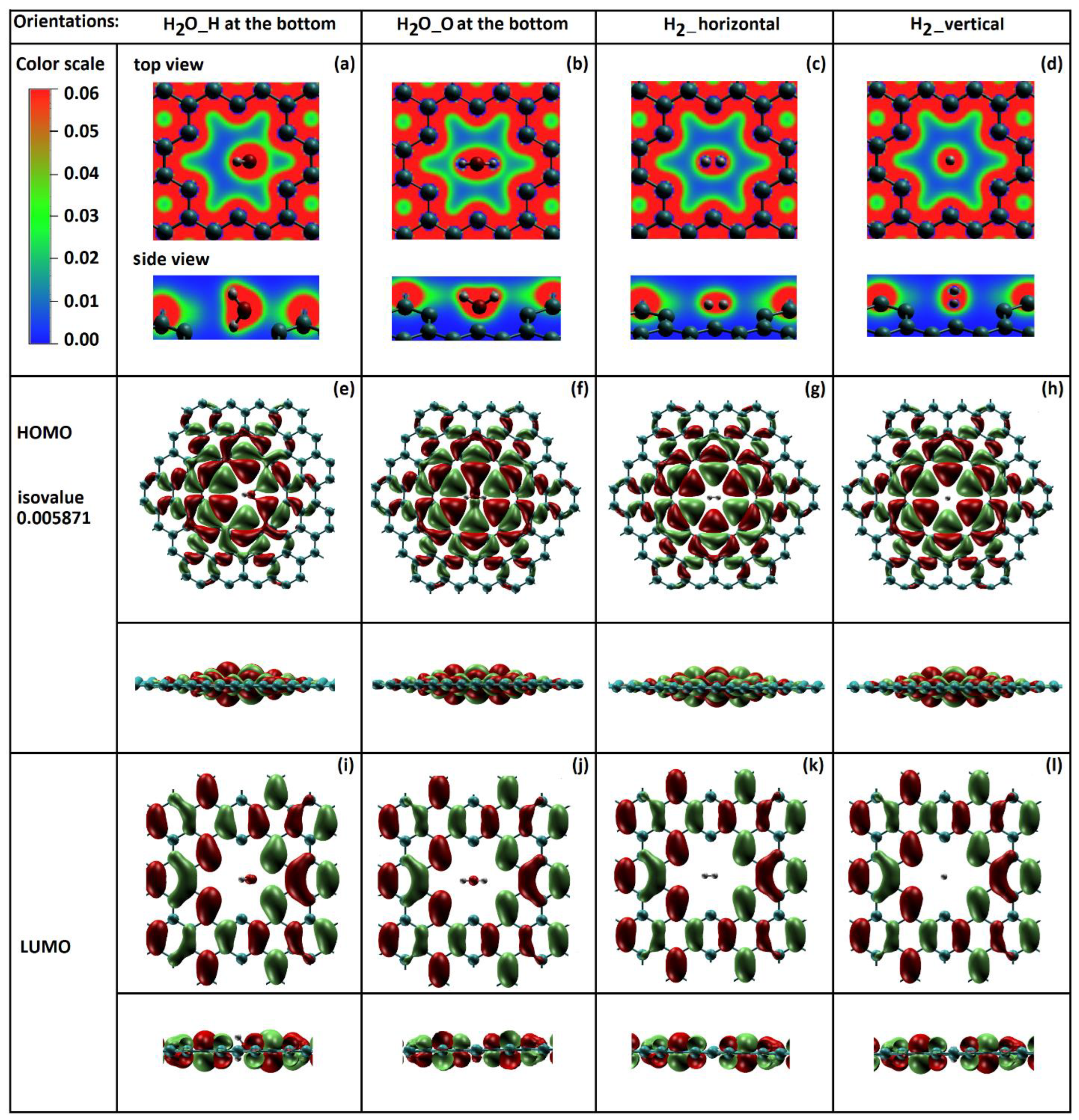
2.2. Total Electron Density of Gas Molecules Inside the Graphene Nanopore
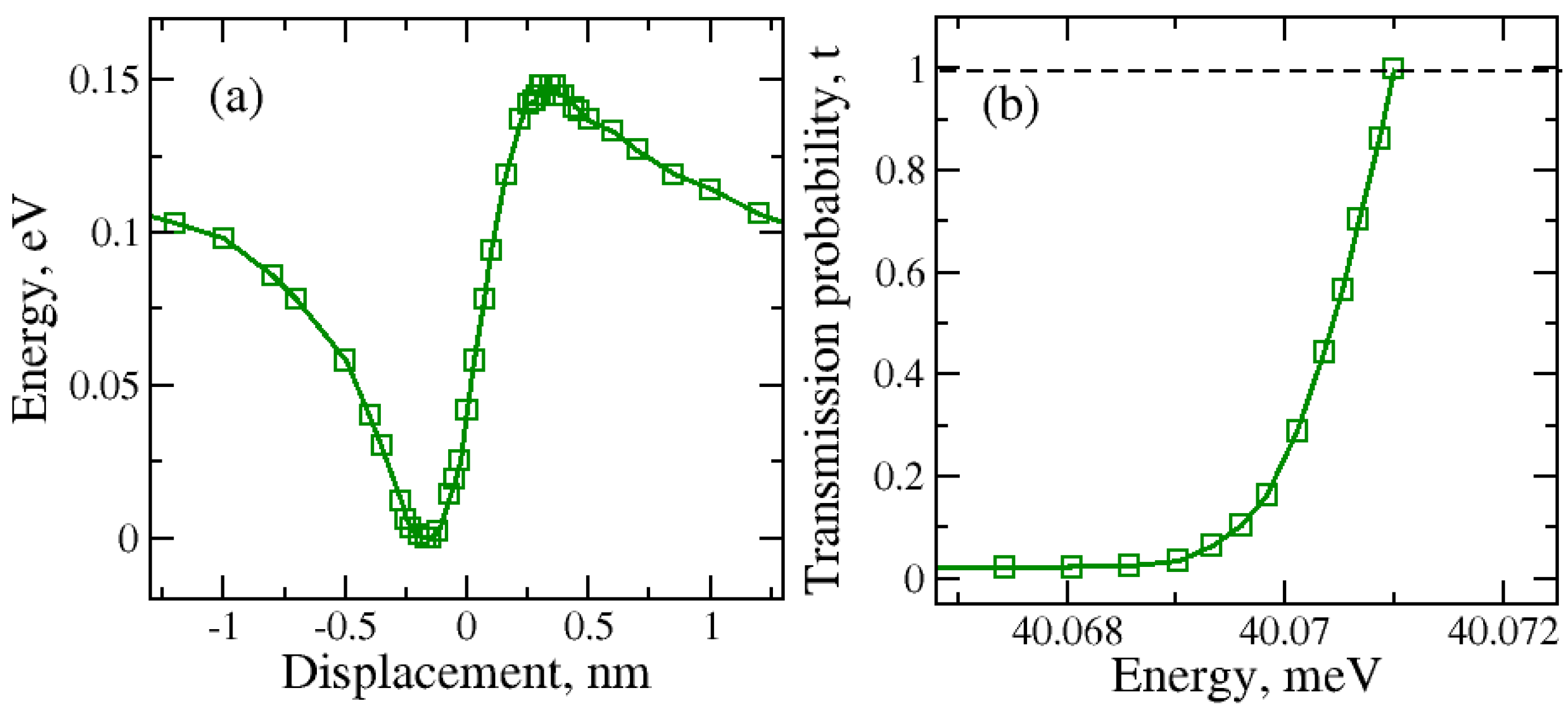
2.3. Molecule Trapping in a Potential Well behind the Graphene Nanopore
3. Models and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shankar, R. Principles of Quantum Mechanics, 2nd ed.; Plenum Press: New York, NY, USA, 1994. [Google Scholar]
- Esaki, L. Long Journey into Quantum Tunneling. Science 1974, 183, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Binnig, G.; Rohrer, H. Scanning tunneling microscopy—from birth to adolescence. Rev. Mod. Phys. 1987, 59, 615–625. [Google Scholar] [CrossRef]
- Bunch, J.S.; Verbridge, S.S.; Alden, J.S.; van der Zande, A.M.; Parpia, J.M.; Craighead, H.G.; McEuen, P.L. Impermeable Atomic Membranes from Graphene Sheets. Nano Lett. 2008, 8, 2458–2462. [Google Scholar] [CrossRef] [PubMed]
- O’Hern, S.C.; Boutilier, M.S.H.; Idrobo, J.C.; Song, Y.; Kong, J.; Laoui, T.; Atieh, M.; Karnik, R. Selective Ionic Transport through Tunable Subnanometer Pores in Single-Layer Graphene Membranes. Nano Lett. 2014, 14, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Kausar, A.; Ahmad, I.; Aldaghri, O.; Ibnaouf, K.H.; Eisa, M.H.; Lam, T.D. Potential of nanoporous graphene and functionalized nanoporous graphene derived nanocomposites for environmental membranes—A review. Nanocomposites 2024, 10, 152–172. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Cao, S.; Wang, Z.; Yu, C.; Wu, C.; Li, G. Defect torsion angle of bilayer porous graphene membrane regulates the gas separation performance. Int. J. Hydrog. Energy 2024, 70, 341–346. [Google Scholar] [CrossRef]
- Villalobos, L.F.; Babu, D.J.; Hsu, K.J.; Van Goethem, C.; Agrawal, K.V. Gas Separation Membranes with Atom-Thick Nanopores: The Potential of Nanoporous Single-Layer Graphene. Acc Mater Res. 2022, 3, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Tanugi, D.; Grossman, J.C. Water Desalination across Nanoporous Graphene. Nano Lett. 2012, 12, 3602–3608. [Google Scholar] [CrossRef] [PubMed]
- Sumedh, P.S.; Smirnov, S.N.; Vlassiouk, I.V.; Raymond, R.U.; Gabriel, M.V.; Sheng, D.; Shannon, M.M. Water desalination using nanoporous single-layer graphene. Nat. Nanotechnol. 2015, 10, 459–464. [Google Scholar]
- Ali, I.; Hasan, S.Z.; Garcia, H.; Danquah, M.K.; Imanova, G. Recent advances in graphene-based nano-membranes for desalination. Chem. Eng. J. 2024, 483, 149108. [Google Scholar] [CrossRef]
- Boutilier, M.S.H.; Sun, C.; Hern, C.O.; Hadjiconsttantinou, N.G.; Karnik, R. Implication of Permeation through Intrinsic Defects in Graphene on the Design of Defect-Tolerant Membranes for Gas Separation. ACS Nano 2014, 8, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Boutilier, M.S.H.; Au, H.; Poesio, P.; Bai, B.; Karnik, R.; Hadjiconsttantinou, N.G. Mechanism of Molecular Permeation through Nanoporous Graphene Membranes. Langmuir 2013, 30, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Koenig, S.P.; Wang, L.; Pellegrino, J.; Bunch, J.S. Selective molecular sieving through porous graphene. Nat. Nanotechnol. 2012, 7, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Li, J.; Zhang, J.; Su, G.; Li, X.; Zhao, Y. Separation of Hydrogen and Nitrogen Gases with Porous Graphene Membrane. J. Phys. Chem. 2011, 115, 23261–23266. [Google Scholar] [CrossRef]
- Jiang, D.; Cooper, V.R.; Dai, S. Porous Graphene as the Ultimate Membrane for Gas Separation. Nano Lett. 2009, 9, 4019–4024. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Benck, J.D.; Eatmon, Y.; Blankschtein, D.; Strano, M.S. Stable, Temperature-Dependent Gas Mixture Permeation and Separation through Suspended Nanoporous Single-Layer Graphene Membranes. Nano Lett. 2018, 18, 5057–5069. [Google Scholar] [CrossRef]
- Viana, D.; Walston, S.T.; Masvidal-Codina, E.; Illa, X.; Rodríguez-Meana, B.; Del Valle, J.; Hayward, A.; Dodd, A.; Loret, T.; Prats-Alfonso, E.; et al. Nanoporous graphene-based thin-film microelectrodes for in vivo high-resolution neural recording and stimulation. Nat. Nanotechnol. 2024, 19, 514–523. [Google Scholar] [CrossRef]
- Yamamoto, M.; Goto, S.; Tang, R.; Yamazaki, K. Toward three-dimensionally ordered nanoporous graphene materials: Template synthesis, structure, and applications. Chem. Sci. 2024, 15, 1953–1965. [Google Scholar] [CrossRef]
- Lv, H.; Yao, Y.; Yuan, M.; Chen, G.; Wang, Y.; Rao, L.; Li, S.; Kara, U.I.; Dupont, R.L.; Zhang, C.; et al. Functional nanoporous graphene superlattice. Nat. Commun. 2024, 15, 1295. [Google Scholar] [CrossRef]
- Karla, A.; Gorde, S.; Hummer, G. Osmotic water transport through carbon nanotube membranes. Proc. Natl. Acad. Sci. USA 2003, 100, 10175–10180. [Google Scholar]
- Majumder, M.; Chopra, N.; Andrews, R.; Hinds, B.J. Enhanced flow in carbon nanotubes. Nature 2005, 438, 44. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.K.; Park, H.G.; Wang, Y.; Stadermann, M.; Artyukhin, A.B.; Grigoropoulos, C.P.; Noy, A.; Bakajin, O. FastMassTransport Through Sub-2-Nanometer Carbon Nanotubes. Science 2006, 312, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Chatzichristos, A.; Hassan, J. Current Understanding of water properties inside carbon nanotubes. Nanomaterials 2022, 12, 174. [Google Scholar] [CrossRef] [PubMed]
- Thiemann, F.L.; Schran, C.; Rowe, P.; Muller, E.A.; Michaelides, A. Water flow in single-walled nanotubes: Oxygen makes it slip, hydrogen makes it stick. ACS Nano 2022, 16, 10775–10782. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, D. From endohedral complexes to endohedral fullerene covalent derivatives: A density functional theory prognosis of chemical transformation of water endofullerene H2O@C60 upon its compression. J. Phys. Chem. C 2013, 117, 1178–1182. [Google Scholar] [CrossRef]
- Pizzagalli, L. First principles molecular dynamics calculations of the mechanical properties of endofullerenes containing noble gas atoms or small molecules. Phys. Chem. Chem. Phys. 2022, 24, 9449–9458. [Google Scholar] [CrossRef] [PubMed]
- Chiricotto, M.; Martelli, F.; Guinta, G.; Carbone, P. Role of long-range electrostatic interactions and local topology of the hydrogen bond network in the wettability of fully and partially wetted single and multilayer graphene. J. Phys. Chem. C 2021, 125, 6367–6377. [Google Scholar] [CrossRef]
- Xu, Y.; Tian, B.; Fang, S.; Guo, W.; Zhang, Z. Probing the interaction of water molecules with oxidized graphene by first principles. J. Phys. Chem. C 2021, 125, 4580–4587. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Comm. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Wang, C.L.; Zhang, W.B.; Sun, H.J.; Van Horn, R.M.; Kulkarni, R.R.; Tsai, C.C.; Hsu, C.S.; Lotz, B.; Gong, X.; Cheng, S.Z. A Supramolecular “Double-Cable” Structure with a 12944 Helix in a Columnar Porphyrin-C60 Dyad and its Application in Polymer Solar Cells. Adv. Energy Mater. 2012, 2, 1375–1382. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.C.; Nocedal, J. On the Limited Memory BFGS Method for Large Scale Optimization. Math. Prog. 1989, 45, 503–528. [Google Scholar] [CrossRef]
- Morse, P.M.; Feshbach, H. Methods of Theoretical Physics, 1st ed.; McGraw Hill: New York, NY, USA, 1953. [Google Scholar]
- Buldum, A.; Ciraci, S. Controlled lateral and perpendicular motion of atoms on surfaces. Phys. Rev. B 1996, 54, 2175. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | De, (eV) | n |
|---|---|---|
| 1 | 0.2650 | 50 |
| 2 | 0.3978 | 70 |
| 3 | 0.1154 | 12 |
| 4 | 0.0962 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barabanova, L.; Buldum, A. Do Molecules Tunnel through Nanoporous Graphene? Molecules 2024, 29, 3306. https://doi.org/10.3390/molecules29143306
Barabanova L, Buldum A. Do Molecules Tunnel through Nanoporous Graphene? Molecules. 2024; 29(14):3306. https://doi.org/10.3390/molecules29143306
Chicago/Turabian StyleBarabanova, Liudmyla, and Alper Buldum. 2024. "Do Molecules Tunnel through Nanoporous Graphene?" Molecules 29, no. 14: 3306. https://doi.org/10.3390/molecules29143306
APA StyleBarabanova, L., & Buldum, A. (2024). Do Molecules Tunnel through Nanoporous Graphene? Molecules, 29(14), 3306. https://doi.org/10.3390/molecules29143306