
Rheological Property Modification of a Molten-State Polyamide through the Addition of an α-Olefin–Maleic Anhydride Copolymer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
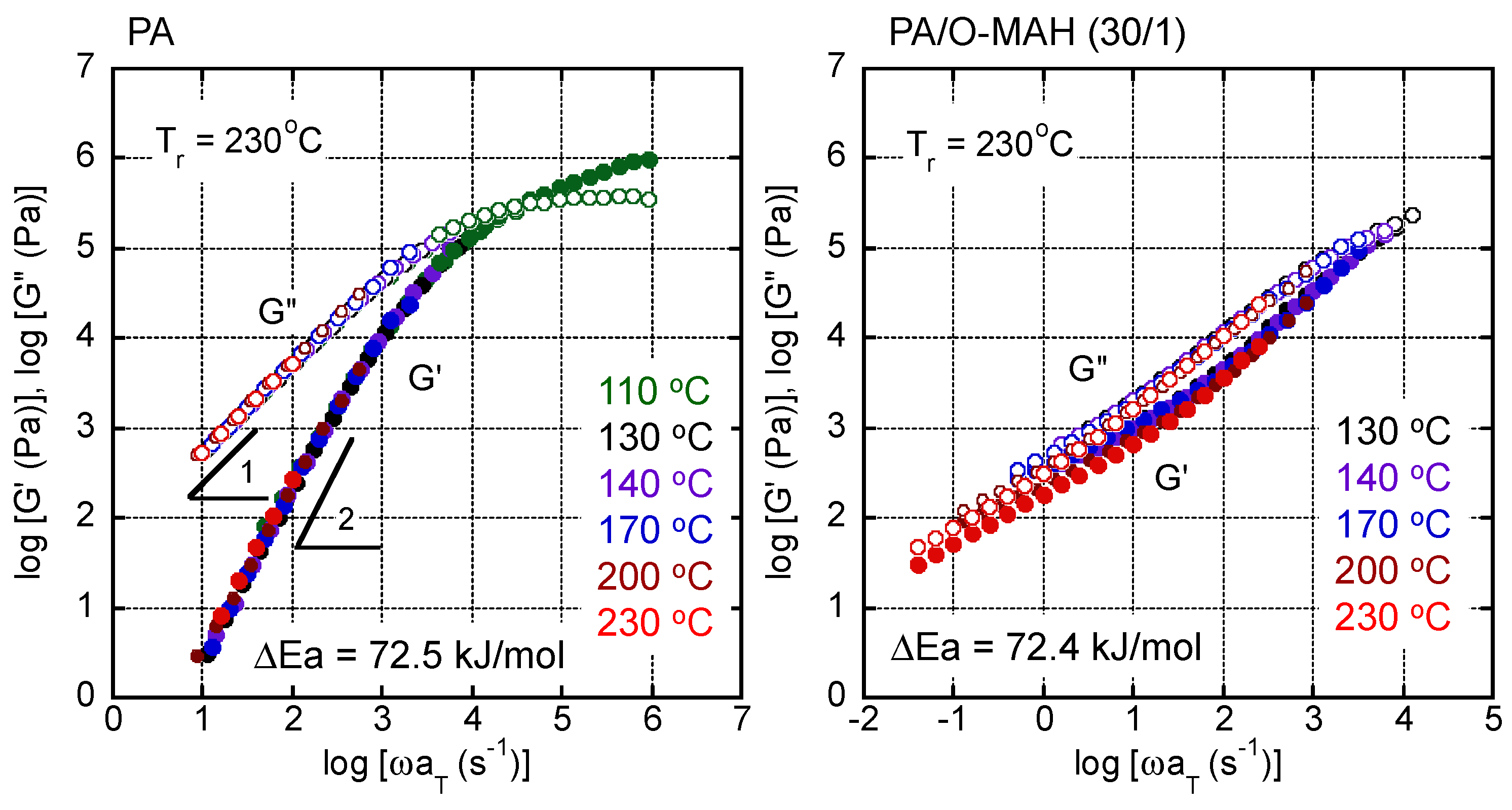
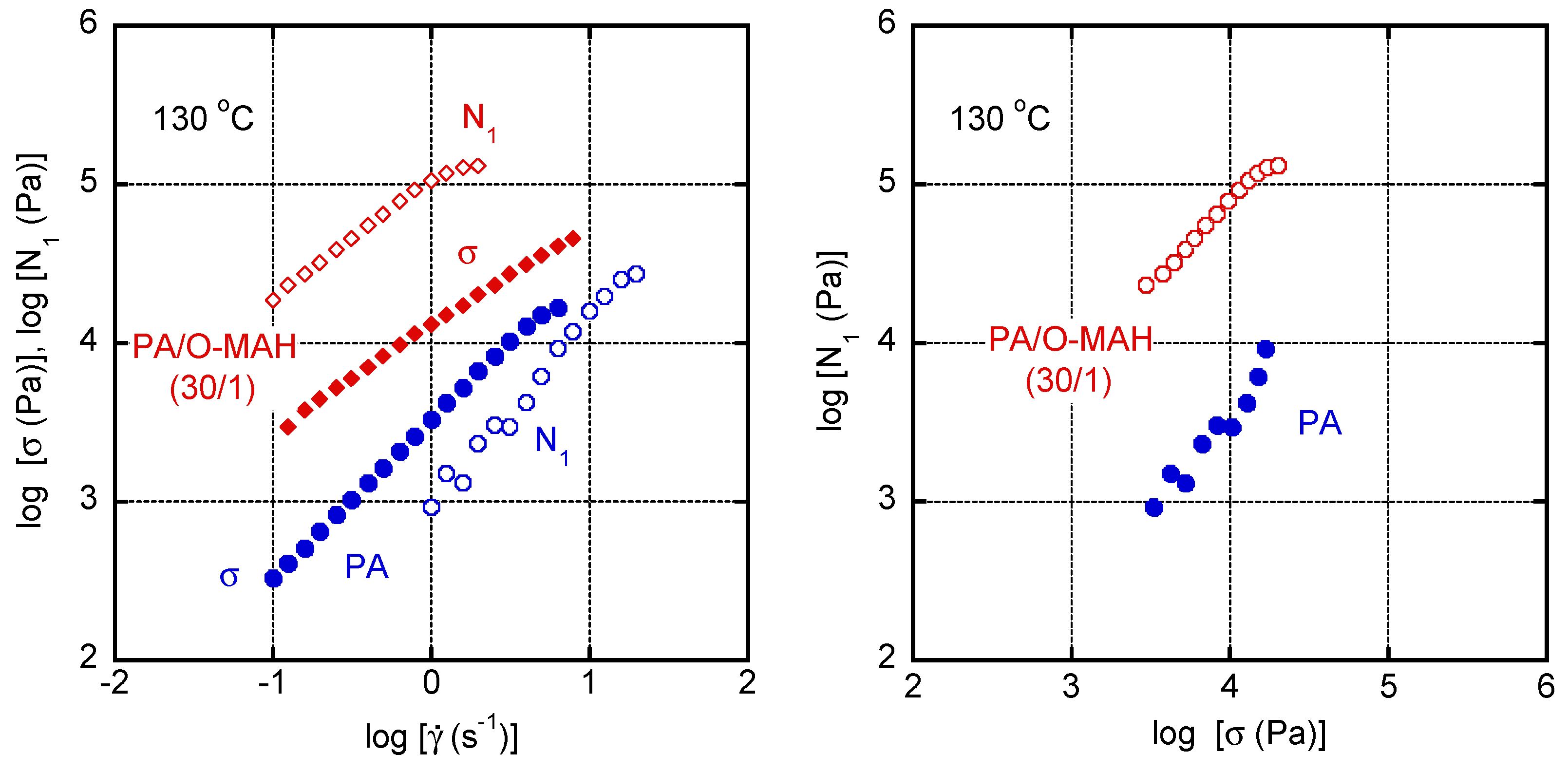
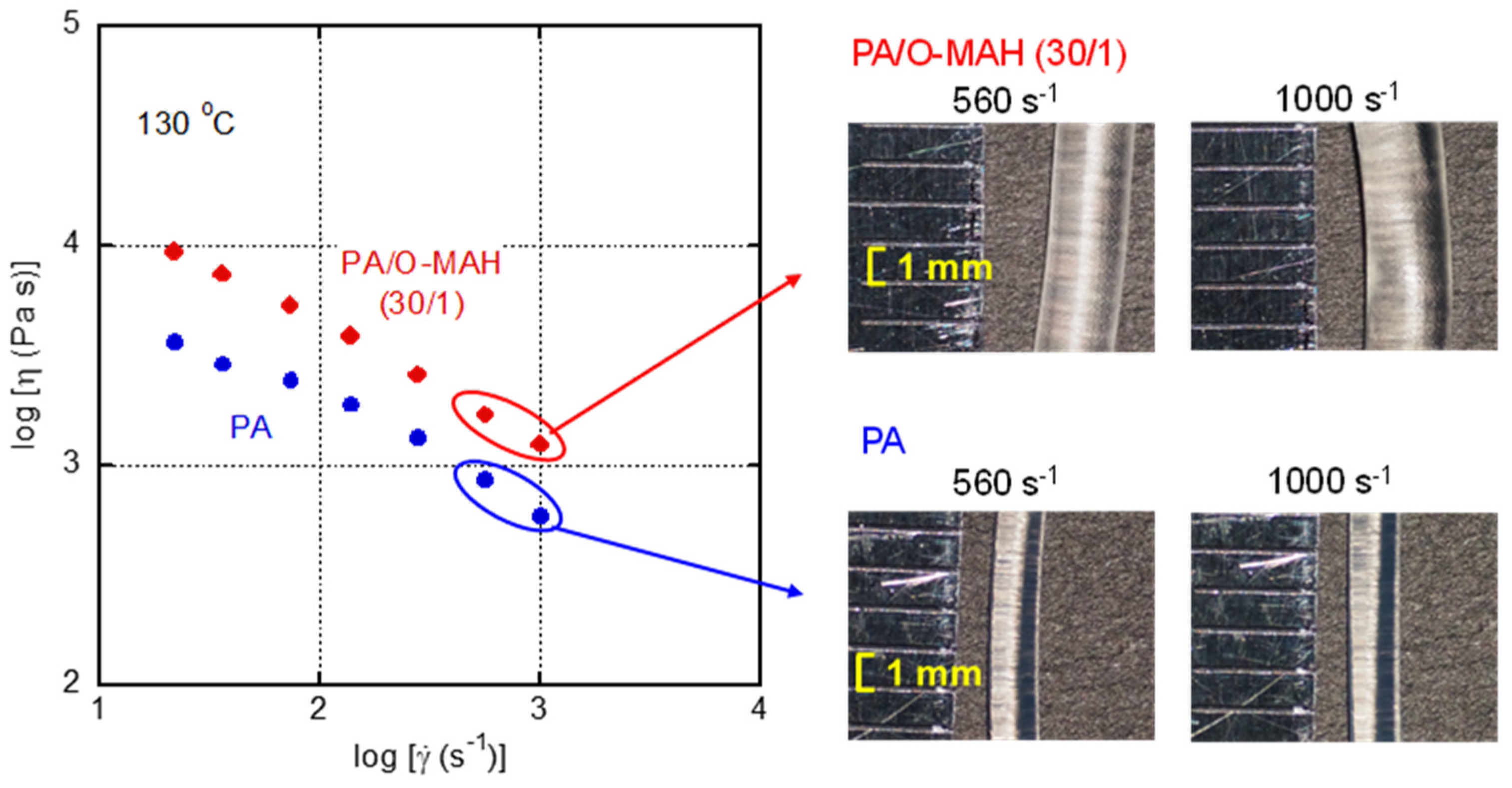
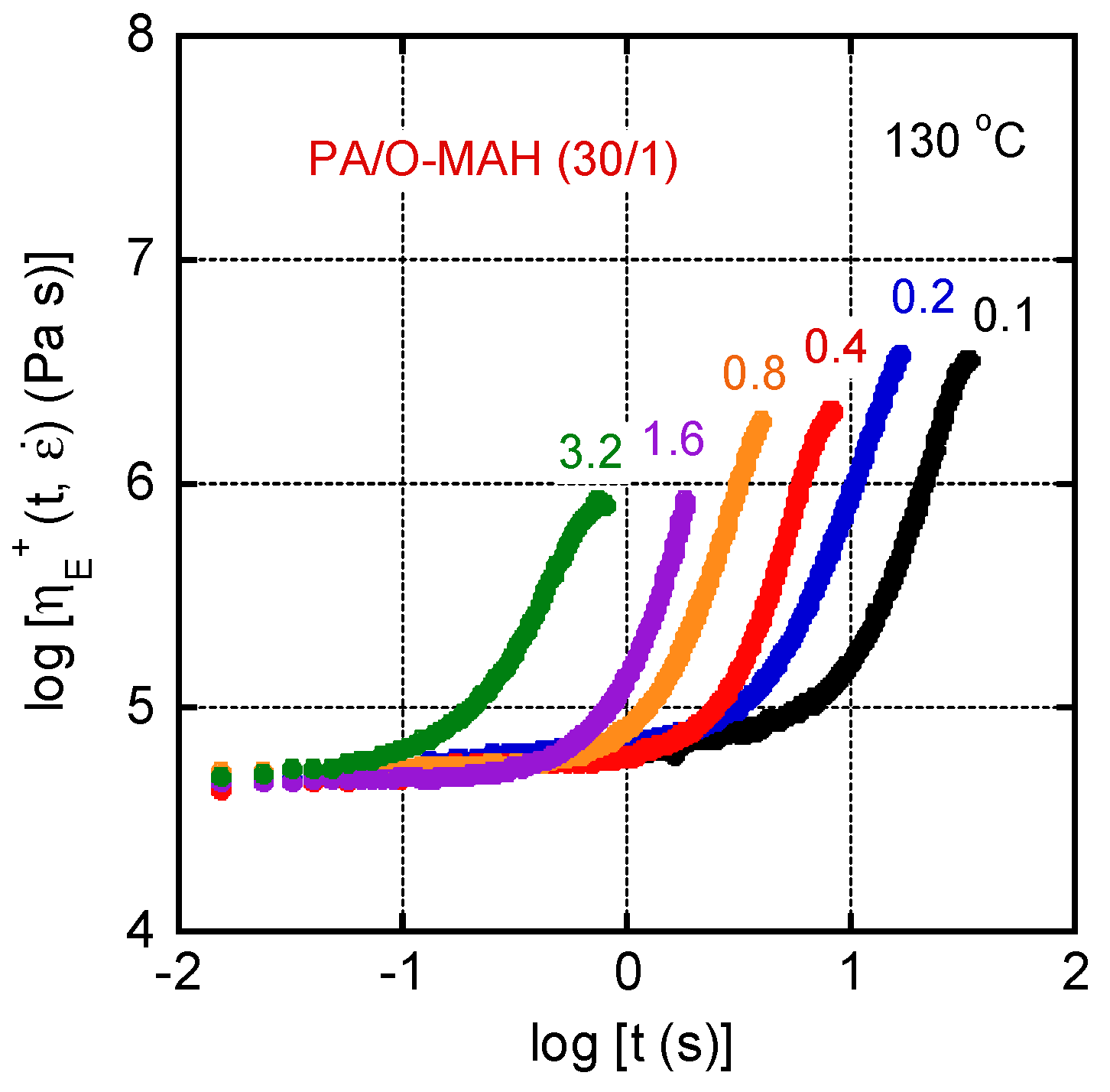
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Münstedt, H. Dependence of the elongational behavior of polystyrene melts on molecular weight and molecular weight distribution. J. Rheol. 1980, 24, 847–867. [Google Scholar] [CrossRef]
- Linster, J.J.; Meissner, J. Melt elongation and structure of linear polyethylene (HDPE). Polym. Bull. 1986, 16, 187–194. [Google Scholar] [CrossRef]
- Sugimoto, M.; Masubuchi, Y.; Takimoto, J.; Koyama, K. Melt rheology of polypropylene containing small amounts of high-molecular-weight chain. 2. Uniaxial and biaxial extensional flow. Macromolecules 2001, 34, 6056–6063. [Google Scholar] [CrossRef]
- Hingmann, R.; Marczinke, B.L. Shear and elongational flow properties of polypropylene melts. J. Rheol. 1994, 38, 573–587. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Todd, D.B.; Gogos, C.G. Rheological properties of LDPE processed by conventional processing machines. Adv. Polym. Technol. 2003, 22, 179–187. [Google Scholar] [CrossRef]
- Wagner, M.H.; Kheirandish, S.; Stange, J.; Münstedt, H. Modeling elongational viscosity of blends of linear and long-chain branched polypropylenes. Rheol. Acta 2006, 46, 211–221. [Google Scholar] [CrossRef]
- Yokohara, T.; Nobukawa, S.; Yamaguchi, M. Rheological properties of polymer composites with flexible fine fibers. J. Rheol. 2011, 55, 1205–1218. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Fukuda, K.; Yokohara, T.; Ali, M.A.B.M.; Nobukawa, S. Modification of rheological properties under elongational flow by addition of polymeric fine fibers. Macromol. Mater. Eng. 2012, 297, 654–658. [Google Scholar] [CrossRef]
- Rizvi, A.; Andalib, Z.K.M.; Park, C.B. Fiber-spun polypropylene/polyethylene terephthalate microfibrillar composites with enhanced tensile and rheological properties and foaming ability. Polymer 2017, 110, 139–148. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Miyata, H. Strain hardening behavior in elongational viscosity for binary blends of linear polymer and crosslinked polymer. Polym. J. 2000, 32, 164–170. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Suzuki, K. Rheological properties and foam processability for blends of linear and crosslinked polyethylenes. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 2159–2167. [Google Scholar] [CrossRef]
- Tsou, A.H.; Lopez-Barron, C.R.; Jiang, P.; Crowther, D.J.; Zeng, Y. Bimodal poly(ethylene-cb-propylene) comb block copolymers from serial reactors: Synthesis and applications as processability additives and blend compatibilizers. Polymer 2016, 104, 72–82. [Google Scholar] [CrossRef]
- Lopez-Barron, C.R.; Tsou, A.H. Strain hardening of polyethylene/polypropylene blends via interfacial reinforcement with poly(ethylene-cb-propylene) comb block copolymer. Macromolecules 2017, 50, 2986–2995. [Google Scholar] [CrossRef]
- Fujii, Y.; Nishikawa, R.; Phulkerd, P.; Yamaguchi, M. Modifying the rheological properties of polypropylene under elongational flow by adding polyethylene. J. Rheol. 2019, 63, 11–18. [Google Scholar] [CrossRef]
- Kugimoto, D.; Kouda, S.; Yamaguchi, M. Modification of poly(lactic acid) rheological properties using ethylene–vinyl acetate copolymer. J. Polym. Environ. 2021, 29, 121–129. [Google Scholar] [CrossRef]
- Jordan, A.M.; Lee, B.; Kim, K.; Ludtke, E.; Lhost, O.; Jaffer, S.A.; Bates, F.S.; Macosko, C.W. Rheology of polymer multilayers: Slip in shear, hardening in extension. J. Rheol. 2019, 63, 751–761. [Google Scholar] [CrossRef]
- Aharoni, S.M. n-Nylons: Their Synthesis, Structure, and Properties; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Deopura, B.L.; Alagirusamy, R.; Joshi, M.; Gupta, B. Polyesters and Polyamides; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- García, J.M.; García, F.C.; Serna, F.; de la Peña, J.L. High-performance aromatic polyamides. Prog. Polym. Sci. 2010, 35, 623–686. [Google Scholar] [CrossRef]
- Xu, M.; Lu, J.; Zhao, j.; Wei, L.; Liu, T.; Zhao, L.; Park, C.B. Rheological and foaming behaviors of long-chain branched polyamide 6 with controlled branch length. Polymer 2021, 224, 123730. [Google Scholar] [CrossRef]
- Xanthos, M. Reactive Extrusion: Principles and Practice; Hanser Publishers: Munich, Germany, 1992. [Google Scholar]
- Lu, C.; Chen, T.; Zhao, X.; Ren, X.; Cai, X. Chemical modification of polyamide-6 by chain extension with 2,2′-bis(2-oxazoline). J. Polym. Sci. Part B Polym. Phys. 2007, 45, 1976–1982. [Google Scholar] [CrossRef]
- Van Ruymbeke, E.; Slot, J.J.M.; Kapnistos, K.; Steeman, P.A.M. Structure and rheology of branched polyamide 6 polymers from their reaction recipe. Soft Matter 2013, 9, 6921–6935. [Google Scholar] [CrossRef]
- Bouchékif, H.; Tunc, D.; Le Coz, C.; Deffieux, A.; Desbois, P.; Carlotti, S. Controlled synthesis of crosslinked polyamide 6 using a bis-monomer derived from cyclized lysine. Polymer 2014, 55, 5991–5997. [Google Scholar] [CrossRef]
- Seo, Y.P.; Seo, Y. Effect of molecular structure change on the melt rheological properties of a polyamide (Nylon 6). ACS Omega 2018, 3, 16549–16555. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Lin, X.; Luo, C.; Xiao, W. The application of a tri-functional epoxy resin as a crosslinking agent in extruded polyamide-6 foam. Polym. Sci. Ser. B 2019, 61, 574–581. [Google Scholar]
- Ozmen, S.C.; Ozkoc, G.; Serhatli, E. Thermal, mechanical and physical properties of chain extended recycled polyamide 6 via reactive extrusion: Effect of chain extender types. Polym. Degrad. Stab. 2019, 162, 76–84. [Google Scholar]
- Cai, J.; Liu, Z.; Cao, B.; Guan, X.; Liu, S.; Zhao, J. Simultaneous improvement of the processability and mechanical properties of polyamide-6 by chain extension in extrusion. Ind. Eng. Chem. Res. 2020, 59, 14334–14343. [Google Scholar] [CrossRef]
- Li, S.; Jiang, T.; Zeng, X.; Zhu, N.; Shen, C.; Gong, W.; Zhang, C.; He, L. The effect of α-olefin–maleic anhydride copolymer on the rheological and crystalline properties and microcellular foaming behavior of polyamide 6. Polymers 2013, 15, 2056. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, B.K.; Jeong, H.M. Morphological, thermal and rheological properties of the blends polypropylene/nylon-6, polypropylene/nylon-6/(maleic anhydride-g-polypropylene) and (maleic anhydride-g-polypropylene)/nylon-6. Eur. Polym. J. 1990, 26, 131–136. [Google Scholar] [CrossRef]
- Nishio, T.; Suzuki, Y.; Kojima, K.; Kakugo, M. Morphology of maleic anhydride grafted polypropylene and polyamide alloy produced by reactive processing. J. Polym. Eng. 1991, 10, 123–150. [Google Scholar] [CrossRef]
- Grossman, R.F.; Lutz, J.T., Jr. Polymer Modifiers and Additives (Plastics Engineering); CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Sundararaj, U.; Macosko, C.W. Drop breakup and coalescence in polymer blends: The effects of concentration and compatibilization. Macromolecules 1995, 28, 2647–2657. [Google Scholar]
- Hua, G.H.; Hoppe, S.; Feng, L.F.; Fonteix, C. Reactive Compounding. In Mixing and Compounding of Polymers, 2nd ed.; Manas-Zloczower, I., Ed.; Hanser: Munich, Germany, 2009; Chapter 27. [Google Scholar]
- Achhammer, B.G.; Reinhart, F.W.; Kline, G.M. Mechanism of the degradation of polyamides. J. Appl. Polym. Sci. 1951, 1, 301–332. [Google Scholar]
- Esmaeili, N.; Kandola, B.K.; Ebdon, J.R.; Horrocks, A.R. Comparison of the effects of antimony trioxide and zinc, calcium and copper stannates on the thermal degradation of polyamide-6,6. Polym. Degrad. Stab. 2023, 214, 110402. [Google Scholar] [CrossRef]
- Hirschberg, V.; Rodrigue, D. Recycling of polyamides: Processes and conditions. J. Polym. Sci. 2023, 61, 1937–1958. [Google Scholar] [CrossRef]
- Kurima, A.; Nguyen, T.A.; Kinashi, K.; Sakai, W.; Tsutsumi, N. Direct observation of the thermo-oxidative degradation of PA66 by spin-trapping ESR analysis. Polym. Degrad. Stab. 2023, 215, 110429. [Google Scholar] [CrossRef]
- Laun, H.M.; Schmidt, G. Rheotens tests and viscoelastic simulations related to high-speed spinning of Polyamide 6. J. Non-Newton. Fluid Mech. 2015, 222, 45–55. [Google Scholar] [CrossRef]
- Luo, M.; Liu, B.; Li, T.; Xu, J.; Zhu, N.; Zeng, X.; Jiang, T.; He, L. Effect of viscosity on foaming behavior and surface quality of foamed polyamide 6. J. Appl. Polym. Sci. 2023, 140, e54588. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Wagner, M.H. Impact of processing history on rheological properties for branched polypropylene. Polymer 2006, 47, 3629–3635. [Google Scholar] [CrossRef]
- Ganesan, M.; Nagaraaj, P. Recent developments in dehydration of primary amides to nitriles. Org. Chem. Front. 2020, 7, 3792–3814. [Google Scholar] [CrossRef]
- Born, M.; Wolf, E. Principles of Optics, 7th ed.; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Takahashi, S.; Okada, H.; Yamaguchi, M. Optical properties of polymer blends composed of poly(methyl methacrylate) and ethylene-vinyl acetate copolymer. Eur. Polym. J. 2012, 48, 974–980. [Google Scholar] [CrossRef]
- Keane, J.; Stein, R.S. The scattering of light from thin polymer films. II. Scattering from polyethylene. J. Polym. Sci. 1956, 20, 327–350. [Google Scholar] [CrossRef]
- Norris, F.H.; Stein, R.S. The scattering of light from thin polymer films. IV. Scattering from oriented polymers. J. Polym. Sci. 1958, 22, 87–114. [Google Scholar] [CrossRef]
- Tenma, M.; Yamaguchi, M. Structure and properties of injection-molded polypropylene with sorbitol-based clarifier. Polym. Eng. Sci. 2007, 47, 1441–1446. [Google Scholar] [CrossRef]
- Menczel, J.D.; Prime, R.B. Thermal Analysis of Polymers; Wiley: New York, NY, USA, 2009. [Google Scholar]
- Greenberg, S.A.; Alfey, T. Side chain crystallization of n-alkyl polymethacrylates and polyacrylates. J. Am. Chem. Sci. 1954, 76, 6280–6285. [Google Scholar] [CrossRef]
- Russell, K.E.; McFaddin, D.C.; Hunter, B.K.; Heyding, R.D. Crystallization of side chains in copolymers of ethylene and 1-alkenes. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 2447–2458. [Google Scholar] [CrossRef]
- Janicek, M.; Cermak, R.; Obadai, M.; Piel, C.; Ponizil, P. Ethylene copolymers with crystallizable side chains. Macromolecules 2011, 44, 6759–6766. [Google Scholar] [CrossRef]
- Kitphaitun, S.; Takeshita, H.; Nomura, H. Analysis of ethylene copolymers with long-chain α-olefins (1-dodecene, 1-tetradecene, 1-hexadecene): A transition between mein chain crystallization and side chain crystallization. ACS Omega 2022, 7, 6900–6910. [Google Scholar] [CrossRef]
- Hayakawa, R.; Wada, Y. Theory of the local mode relaxation in the glassy state of polymers. J. Polym. Sci. Part B Polym. Phys. 1974, 12, 2119–2134. [Google Scholar] [CrossRef]
- Ishisaki, A.; Kawagoe, M. Examination of the time-water content superposition on the dynamic viscoelasticity of moistened polyamide 6 and epoxy. J. Appl. Polym. Sci. 2004, 93, 560–567. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; Wiley: Hoboken, NJ, USA, 1980. [Google Scholar]
- Wu, S. Chain structure and entanglement. J. Polym. Sci. Part B Polym. Phys. 1989, 27, 723–741. [Google Scholar] [CrossRef]
- Raju, V.R.; Rachapudy, H.; Graessley, W.W. Properties of amorphous and crystallizable hydrocarbon polymers. IV. Melt rheology of linear and star-branched hydrogenated polybutadiene. J. Polym. Sci. Part B Polym. Phys. 1979, 17, 1223–1235. [Google Scholar] [CrossRef]
- Carella, J.M.; Gotro, J.T.; Graessley, W.W. Thermorheological effects of long-chain branching in entangled polymer melts. Macromolecules 1986, 19, 659–667. [Google Scholar] [CrossRef]
- Meller, M.; Luciani, A.; Sarioglu, A.; Manson, J.A.E. Flow through a convergence. Part 1: Critical conditions for unstable flow. Polym. Eng. Sci. 2002, 42, 611–633. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Miyata, H.; Tan, V.; Gogos, C.G. Relation between molecular structure and flow instability for ethylene/α-olefin copolymers. Polymer 2002, 43, 5249–5255. [Google Scholar] [CrossRef]
- Allal, A.; Vergnes, B. Molecular design to eliminate sharkskin defect for linear polymers. J. Non-Newton. Fluid Mech. 2007, 146, 45–50. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, X.; Do, Q.-V.; Narita, T.; Yamaguchi, M.; Yamaguchi, M. Rheological Property Modification of a Molten-State Polyamide through the Addition of an α-Olefin–Maleic Anhydride Copolymer. Molecules 2024, 29, 3730. https://doi.org/10.3390/molecules29163730
Mei X, Do Q-V, Narita T, Yamaguchi M, Yamaguchi M. Rheological Property Modification of a Molten-State Polyamide through the Addition of an α-Olefin–Maleic Anhydride Copolymer. Molecules. 2024; 29(16):3730. https://doi.org/10.3390/molecules29163730
Chicago/Turabian StyleMei, Xianzhu, Quoc-Viet Do, Takaaki Narita, Misaki Yamaguchi, and Masayuki Yamaguchi. 2024. "Rheological Property Modification of a Molten-State Polyamide through the Addition of an α-Olefin–Maleic Anhydride Copolymer" Molecules 29, no. 16: 3730. https://doi.org/10.3390/molecules29163730
APA StyleMei, X., Do, Q.-V., Narita, T., Yamaguchi, M., & Yamaguchi, M. (2024). Rheological Property Modification of a Molten-State Polyamide through the Addition of an α-Olefin–Maleic Anhydride Copolymer. Molecules, 29(16), 3730. https://doi.org/10.3390/molecules29163730