Abstract
Iodosilarene derivatives (PhIO, PhI(OAc)2) constitute an important class of oxygen atom transfer reagents in organic synthesis and are often used together with iron-based catalysts. Since the factors controlling the ability of iron centers to catalyze alkane hydroxylation are not yet fully understood, the aim of this report is to develop bioinspired non-heme iron catalysts in combination with PhI(OAc)2, which are suitable for performing C-H activation. Overall, this study provides insight into the iron-based ([FeII(PBI)3(CF3SO3)2] (1), where PBI = 2-(2-pyridyl)benzimidazole) catalytic and stoichiometric hydroxylation of triphenylmethane using PhI(OAc)2, highlighting the importance of reaction conditions including the effect of the co-ligands (para-substituted pyridines) and oxidants (para-substituted iodosylbenzene diacetates) on product yields and reaction kinetics. A number of mechanistic studies have been carried out on the mechanism of triphenylmethane hydroxylation, including C-H activation, supporting the reactive intermediate, and investigating the effects of equatorial co-ligands and coordinated oxidants. Strong evidence for the electrophilic nature of the reaction was observed based on competitive experiments, which included a Hammett correlation between the relative reaction rate (logkrel) and the σp (4R-Py and 4R’-PhI(OAc)2) parameters in both stoichiometric (ρ = +0.87 and +0.92) and catalytic (ρ = +0.97 and +0.77) reactions. The presence of [(PBI)2(4R-Py)FeIIIOIPh-4R’]3+ intermediates, as well as the effect of co-ligands and coordinated oxidants, was supported by their spectral (UV–visible) and redox properties. It has been proven that the electrophilic nature of iron(III)-iodozilarene complexes is crucial in the oxidation reaction of triphenylmethane. The hydroxylation rates showed a linear correlation with the FeIII/FeII redox potentials (in the range of −350 mV and −524 mV), which suggests that the Lewis acidity and redox properties of the metal centers greatly influence the reactivity of the reactive intermediates.
1. Introduction
Catalytic oxidation of alkanes under mild conditions is quite difficult due to their high C-H bond energy. However, these reactions can be catalyzed by various metalloenzymes such as cytochrome P-450, methane monooxygenase, and Rieske dioxygenase, based on which numerous biomimetic oxidation reactions have also been developed [1,2,3,4,5,6,7]. Several efficient biomimetic oxidation reactions have been developed using iron complexes as catalysts with various single oxygen donor oxidants such as PhIO, H2O2, or ROOH [8,9,10,11]. Oxidative functionalization of alkanes into more valuable products (e.g., alcohols, ketones, aldehydes) is of great importance nowadays [12,13,14,15,16,17,18,19]. Complexes of manganese and iron with various ligands such as porphyrins, tetraazaannulenes, phthalocyanines, chlorines, Schiff bases, and triazacyclononanes have been extensively studied as alkane oxidation catalysts [12,20,21,22,23,24,25,26,27,28].
Although great efforts have been made to fine-tune the reactivity of iron–oxo complexes in oxidation reactions by modifying the structural and electronic properties of the metal center, there are few studies in the literature on other reactive intermediates such as FeIII-OIPh, FeIII-OOH, and FeIII-OOR species. In many cases, due to their Janus face, these intermediates not only participate in reactions as precursor complexes of FeIV = O or FeV = O intermediates but can also participate in oxidation processes as active species [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44]. Since the redox properties of catalysts and their intermediates play a special role in redox reactions, such as in hydrogen atom transfer (HAT) and oxygen atom transfer (OAT), it is therefore important to examine what factors influence the reactivity of these complexes. In the case of high valence metal oxo species, it has been shown that the reactivity can be influenced not only by the oxidation and spin state of the metal center(s) but also by the nature of the supporting ligands, including the axial and equatorial co-ligands. [45,46,47,48,49,50,51,52,53,54,55,56,57]. Terminal oxidants, such as the insoluble, polymeric PhIO and the organic solvent-soluble PhI(OAc)2, can be used as mild oxidants in many metal complex-catalyzed oxygenation processes, where their main role is the formation of high-valent metal–oxo intermediates, but in many cases the formation and active role of FeIII-OIPh complexes in the oxidation reactions was also confirmed [29,30,31,32,33,42,43,44].
Previous studies have shown that iron(II) complexes with PBI-type ligands (PBI = 2-(2-pyridyl)benzimidazole) can be used as precursors to produce a number of reactive intermediates believed to be involved in iron-based oxidation processes. With their help, both the [(PBI)4(Solvent)2FeIII2(μ-O2)]4+ [58,59,60,61,62] and the [(PBI)2(Solvent or additives)FeIII(OIPh)]3+ intermediates can be synthesized by the use of H2O2, and PhIO (or PhI(OAc)2), respectively [42,43,44]. The structure of the above intermediates was assumed based on UV–VIS, EPR, and rRaman measurements [43]. Thanks to the ligands, the advantage of these systems is that both the iron(II) precursors and the formed high-valent intermediates have favorable thermal stability, enabling their detailed characterization, the investigation of their spectral and redox properties, and the effect of these characteristics on their reactivity. Considering the results so far, it can be said that the above complexes are suitable as catalysts for the oxygenation of thioanisoles and other compounds containing C-H and C=C bonds with PhIO or its more soluble and mild oxygen source derivative, PhI(OAc)2, or deformylation of various aldehydes in a nucleophilic reaction with H2O2. As a result of previous catalytic studies, it was established that in the case of OAT reactions, such as sulfoxidation and epoxidation, the yield of the reactions can be significantly increased if monodentate pyridines with electron-withdrawing groups are used as equatorial ligands for the catalytically active [(PBI)2(4R-Py)FeIII(OIPh)]3+. Based on the detailed mechanistic studies, the linear free energy (Hammett equations) relationships through the positive ρ-value obtained for the co-ligands indicate that the reactive intermediate of the above reactions is electrophilic [44].
The aim of this report is to study the catalytic and stoichiometric oxidation reactions involving C-H activation using the sample substrate triphenylmethane, namely to study the effect of monodentate 4R-pyridine ligands and 4R’-PhIO (from 4R’-PhI(OAc)2) terminal oxidants with electron-withdrawing and electron donor substituents (Scheme 1). Our further aim is to compare the HAT (this study) and the previously published processes [44] mediated by [(PBI)2(4R-Py)FeIII(OIPh)]3+ intermediates based on the kinetic and catalytic results.
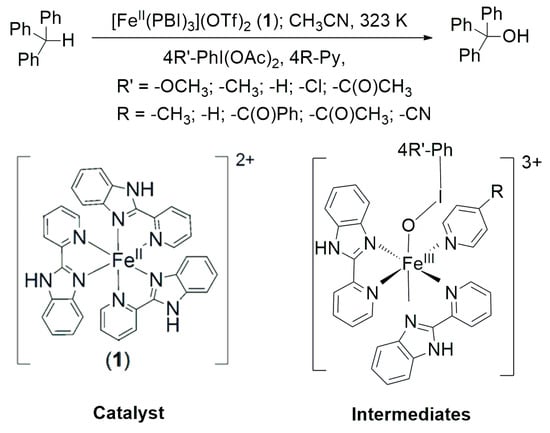
Scheme 1.
[FeII(PBI)3](OTf)2 (1) catalyzed oxidation of triphenylmethane mediated by [(PBI)2(4R-Py)FeIII(4R’-PhIO)]3+ intermediates in the presence of 4R’-PhI(OAc)2 terminal oxidants and 4R-Py co-ligands.
2. Results and Discussion
Based on previously published results, the complex, [FeII(PBI)3](OTf)2 (1) (OTf = CF3SO3), catalyzes the sulfoxidation of thioanisole and the epoxidation of cis-cyclooctene and cis- and trans-stilbene with PhI(OAc)2 as an oxidant, where the optimal catalytic conditions were as follows: 1:100:300 ratio for catalyst–oxidant–substrate, in acetonitrile at 323 K [62]. Under these conditions, we achieved a conversion value of 35% for thioanisole, 22.46% for cis-cyclooctene, 24.14% for cis-stilbene, and 36.7% for trans-stilbene in the oxygen atom transfer (OAT) processes. Another observation was that the above values could be further increased by adding pyridine ([1]:[Py] = 1:10) as a possible equatorial ligand, namely 81.79%, 36.94%, 36.12%, and 41.15% conversion values were observed for thioanisole, cis-cyclooctene, and cis- and trans-stilbene, respectively, after 4 h. Hammett correlations between relative reaction rates (logkrel) and the σp (4R-Py) parameters, where ρ = +0.16, +0.95, +0.59, and +0.46 for thioanisole, cis-cyclooctene, and cis- and trans-stilbene, respectively, imply the electrophilic character of the OIPh group of the FeIII(OIPh) intermediate and positive charge buildup on the substrates. This proved crucial in controlling the nature of the active oxidizing form, which significantly affected its reactivity in OAT reactions [62]. Based on these results, an electrophilic oxidant, most likely a [(PBI)2(4R-Py)FeIIIOIPh]3+ intermediate, can be assumed as a result of the reaction of 1 with PhIO (PhI(OAc)2) and then with pyridine derivatives. These species show characteristic spectral features (λmax = 700–760 nm, ε = ~1400 M−1 cm−1 with S = ½ low-spin state) [43,45,62].
2.1. Effect of Equatorial Ligands on the FeIIIOIPh-Mediated Oxidation of Triphenylmethane
In this report, we wanted to investigate the effect of equatorial pyridines (4R-py; R = -CH3, -H, -C(O)C6H5, -C(O)CH3, -CN) and the oxidant 4R’-PhI(OAc)2 (R = -OCH3, -CH3, H, -Cl, -C(O)CH3) in catalytic oxidation processes that involve C-H activation, using 1 as catalyst and triphenylmethane as model substrate. In order to compare the systems with OAT and C-H activation, we used the previously observed optimal conditions with a catalyst–additive–oxidant–substrate ratio of 1:10:100:300, in acetonitrile at 323 K for 4 h [62]. The catalyst–co-ligand ratio was chosen to be 1:10 based on the maximum shift of the characteristic λmax of the FeIII(OIPh) species due to pyridine additives [62]. As a mild oxygen source, PhI(OAc)2 has the advantage of not damaging the catalysts and showing no noticeable reactivity with the substrate [63]. According to our previous studies, the formation of the more oxidizing PhIO can be explained by the reaction of PhI(OAc)2 with a trace amount of water [64]. Based on the blank experiments, it can be said that no hydroxylated products were formed in the absence of either the catalyst or PhI(OAc)2. At first glance, it is clear that the catalytic activity of the iron salt [FeII(CH3CN)4(OTf)2] is negligible (conversion = 10.27%). In the case of [FeII(PBI)3](OTf)2 (1), a 2.5-fold increase in activity was observed with a conversion value of 24.71% (TOF = 6.17 h−1) (Figure 1). The results of the comprehensive screening of [Fe(PBI)3](OTf)2 with and without pyridines are shown in Table 1. As expected, triphenylmethanol was the only oxidized product without any other byproducts after 4 h. Based on the obtained conversion values, it can be concluded that the pyridine derivatives have a significant effect on the test reactions. In the case of pyridine (1:py = 1:10), the conversion value increased by 1.5 times (37.12%; TOF = 9.28 h−1). The effect of electron-donating (-CH3) and electron-withdrawing (-CN, -C(O)C6H5, -C(O)CH3) substituents of pyridines on the reactivity was also investigated in competitive experiments, and a significant effect on catalytic oxidation of triphenylmethane was shown (Table 1 and Figure 2a), where the 4-CN-pyridine resulted in the highest (83.00%, TOF = 20.75 h−1), and 4-Me-pyridine gave the lowest conversion with 19.98% (TOF = 4.99 h−1). The effect is remarkable, which can be attributed to the increased electrophilicity of the ([(PBI)2(4R-Py)FeIIIOIPh]3+) species due to the electron-withdrawing pyridine ligand.
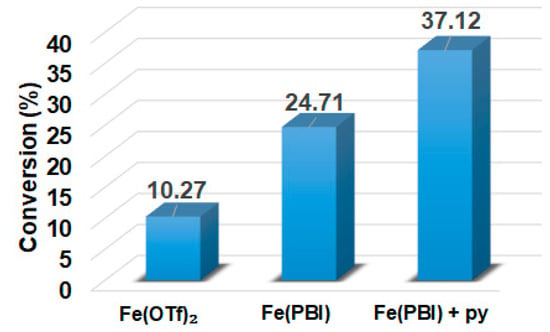
Figure 1.
[FeII(PBI)3](OTf)2-catalyzed hydroxylation of triphenylmethane with PhI(OAc)2 in the absence and presence of para-substituted pyridines in acetonitrile at 323 K: [FeII(OTf)2]0 = 1 × 10−3 M, [1]0 = 1 × 10−3 M, [PhI(OAc)2]0 = 1 × 10−1 M, and [Ph3CH]0 = 3 × 10−1 M, [pyridine]0 = 1 × 10−2 M.

Table 1.
[FeII(PBI)3](OTf)2-catalyzed hydroxylation of triphenylmethane with PhI(OAc)2 in the presence of para-substituted pyridines in acetonitrile at 323 K 1.
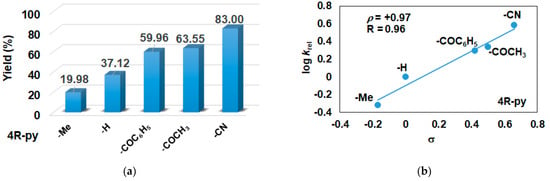
Figure 2.
[FeII(PBI)3](OTf)2-catalyzed hydroxylation of triphenylmethane with PhI(OAc)2 in the presence of para-substituted pyridines in acetonitrile at 323 K: (a) the calculated conversion (=TON) values for para-substituted pyridines. (b) Hammett plot of logkrel against the σp of para-substituted pyridines. [1]0 = 1 × 10−3 M, [PhI(OAc)2]0 = 1 × 10−1 M, [Ph3CH]0 = 3 × 10−1 M, [pyridine]0 = 1 × 10−2 M.
The relative rates (krel) for the [FeII(PBI)3](OTf)2-catalyzed hydroxylation of triphenylmethane in the presence of para-substituted pyridines (4R-Py, R = -CH3, H, -C(O)C6H5, -C(O)CH3 and CN) was determined by measuring the formation of triphenylmethanol by GC. Figure 2b depicts a linear correlation (R = 0.96) of log krel versus Hammett σp constants in the competitive [FeII(PBI)3](OTf)2-catalyzed hydroxylation reaction. The slope (ρ) of the plot is +0.97, clearly indicating that the electron-deficient intermediates are much more reactive than the electron-rich species, which is consistent with the involvement of an electrophilic oxidant, probably a putative [(PBI)2(4R-Py)FeIIIOIPh]3+ intermediate.
Summarizing the results of the competitive experiments and comparing them with the results of the sulfoxidation and epoxidation experiments, it can be said that the value obtained for co-ligands (ρ = +0.97) is much higher than the value obtained for sulfoxidation (ρ = +0.16), which indicates that the oxidation reaction involving C-H activation is much more sensitive to the electrophilic nature of the intermediate as the sulfoxidation reaction described by the direct oxygen transfer (DOT) mechanism [62]. Considering the previous results of the oxidation of cis-cyclooctene (ρ = 0.95) and cis- and trans-stilbene (ρ = 0.59 and +0.46, respectively) under the same conditions, it can be said that both the C-H activation and the epoxidation with nonconcerted ET mechanism are greatly influenced by the electrophilic character of the reactive intermediate (oxidant) [44]. Unfortunately, hydrocarbons with a higher BDE value (such as PhCH3) did not give the reaction under the given conditions.
2.2. Effect of Co-Oxidant (4R’-PhI(OAc)2) on the FeIIIOIPh-Mediated Oxidation of Triphenylmethane
As noted earlier, the reaction of 1 with PhIO leads to reactive FeIII(OIPh) formations, suggesting that it may play a key role in the catalytic cycles. This is also supported by the UV–Vis spectrum of the catalytic reaction mixture, where the rise and fall of their characteristic chromophores at λmax = 700–760 nm (depending on the pyridines used as co-ligand) is clearly visible. After that, we wanted to investigate the effect of the electron-donating and electron-withdrawing groups introduced into the co-oxidant (4R’-PhI(OAc)2, where R’ = -OCH3, -CH3, -H, -Cl, and -C(O)CH3) on the catalytic oxidation of triphenylmethane with 1. Before the detailed investigation of the catalytic system, we studied the formation kinetics of FeIIIOIPh-4R’ adducts, with special attention paid to the role of the substituents on the co-oxidant. The rates in the presence of excess of PhI(OAc)2 (10 equivalent) obeyed pseudo-first-order kinetics. The pseudo-first-order rate constants, kobs, in the reaction of 1 with 4R-PhI(OAc)2 (R’ = -OCH3, -CH3, -H, -Cl, and -C(O)CH3) were determined from the absorbance change at 760 nm (Table 2 and Figure 3a). Based on the obtained data, the fastest adduct formation was observed for 4R-PhI(OAc)2 containing an electron-donating substituent (0.637 s−1 for -OCH3), while for the electron-withdrawing group -C(O)Me was the slowest (0.187 s−1). The observed kinetic data were treated based on the Hammett equation, from which the value of ρ was −0.63 (Figure 3b). Since the formation of the adduct in the case of PhIO is orders of magnitude faster than in the case of PhI(OAc)2, based on the negative ρ-value, it can be assumed that the rate-determining step is the formation of PhIO by the proton-assisted hydrolysis of PhI(OAc)2, which means that the higher electron density of the I(OAc)2 group increases the reaction rate.
Table 2.
Formation and self-decay of [(PBI)2(MeCN)FeIII(4R’-PhIO)]3+ intermediates in the in situ reaction of 1 with 4R-PhI(OAc)2 in acetonitrile at 293 K.
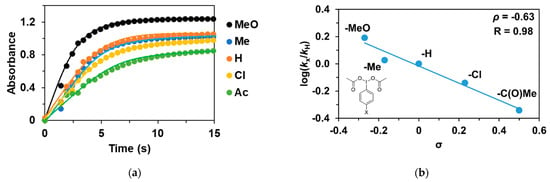
Figure 3.
(a) Formation of [(PBI)2(MeCN)FeIII(4R-PhIO)]3+ intermediates in the in situ reaction of 1 with 4R’-Ph(IOAc)2 in acetonitrile at 293 K monitored at 760 nm. (b) Hammett plot of logkrel against the σp of para-substituted 4R’-Ph(IOAc)2 oxidants. [1]0 = 1 × 10−3 M, [4R’-PhI(OAc)2]0 = 1 × 10−2 M.
To investigate the effect of the oxidant, the catalytic oxidation of triphenylmethane was carried out with 1 and 4R’-PhI(OAc)2 (R’ = -OCH3, -CH3, -H, -Cl, and -C(O)CH3) derivatives under the conditions used for the study of the effect of co-ligands earlier (catalyst–oxidant–substrate ratio is 1:100:300 in acetonitrile at 323 K).
As evident in Table 3, the catalytic oxidation of triphenylmethane was affected by the substituents on the oxidant, 4R’-PhI(OAc)2 used. For example, the 4-C(O)CH3-PhI(OAc)2 resulted in the highest (45.75%), and the 4- CH3O-PhI(OAc)2 gave the lowest conversion value with 13.87% for the hydroxylation of triphenylmethane (Figure 4a). The effect is also remarkable and can be traced back to the increased electrophilicity of the [(PBI)2(CH3CN)FeIIIOIPh-4R’]3+ species, due to the electron-withdrawing substituents of the PhIO oxidant, which is consistent with the results obtained for pyridine co-ligands.
Table 3.
[FeII(PBI)3](OTf)2-catalyzed hydroxylation of triphenylmethane with 4R’-PhI(OAc)2 in acetonitrile at 323 K 1.
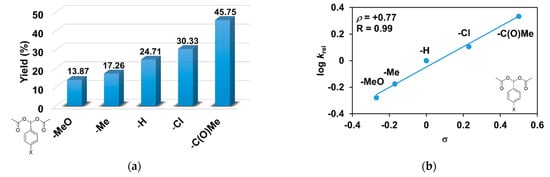
Figure 4.
[FeII(PBI)3](OTf)2-catalyzed hydroxylation of triphenylmethane with 4R’-PhI(OAc)2 in acetonitrile at 323 K: (a) the calculated conversion (=TON) values for 4R’-PhI(OAc)2. (b) Hammett plot of logkrel against the σp of 4R-PhI(OAc)2. [1]0 = 1 × 10−3 M, [4R’-PhI(OAc)2]0 = 1 × 10−1 M, and [Ph3CH]0 = 3 × 10−1 M.
The relative rates (krel) for the [FeII(PBI)3](OTf)2-catalyzed hydroxylation of triphenylmethane with substituted iodosobenzene diacetates (4R-PhI(OAc)2, R = -OCH3, -CH3, -H, -Cl, and -C(O)CH3) was determined by measuring the formation of triphenylmethanol by GC. Figure 4b shows a linear correlation (R = 0.99) of log krel versus Hammett σp constants in the competitive reactions (Figure 4b). The slope (ρ) of the plot is +0.77, which means that the reaction of electron-deficient intermediates is much more favored. This value is similar to the value obtained for substituted pyridines (ρ = +0.97), which confirms the importance of the electrophilic feature of the [(PBI)2(CH3CN)FeIIIOIPh-4R’]3+ intermediate.
2.3. Effect of Equatorial Ligands on the FeIIIOIPh-Mediated Stoichiometric Oxidation of Triphenylmethane
As a possible elementary step of the catalytic reaction, we wanted to investigate the stoichiometric oxidation of the in situ formed iron(III)-iodosylbenzene adduct with triphenylmethane, with particular attention paid to the effect of pyridine co-ligands. Based on our previous results [62], for the full formation of [(PBI)2(4R-Py)FeIlIOIPh]3+ (R = -CH3, -H, C(O)CH3, -C(O)Ph, and -CN) in the reaction of 1 with PhI(OAc)2, the optimal amount of co-ligands (10 equivalents) was determined by titration. The λmax values of the [(PBI)2(4R-Py)FeIlIOIPh]3+ complexes are shown in Table 4 [62]. The observed hypochromic shift in the near IR region clearly indicated the coordination of the pyridine ligands, as well as their electronic effect on the electrophilicity of the active species, based on the observed trend of the Hammett correlation between the λmax−1 (at 700–760 nm) and the Hammett constants (σp) [62]. These results imply that the effect of the ligands appears in the redox properties of the intermediates, as well as in their redox potential values, which may be consistent with our previous spectroscopic results.
Table 4.
Summary of kinetic data for the stoichiometric oxidation of triphenylmethane with [(PBI)2FeIII(OIPh)(4R-Py)] intermediates generated in situ by the reaction of 1 with Ph(IOAc)2 and 4R-Py derivatives in acetonitrile at 293 K 1.
To determine the half-wave potentials (E1/2) of the in situ-generated [(PBI)2(4R-Py)FeIlIOIPh]3+ (R = -CH3, -H, C(O)CH3, -C(O)Ph, and -CN) complexes, cyclic voltammetric measurements (CV) were also performed in acetonitrile. Cyclic voltammograms of [(PBI)2(4R-Py)FeIlIOIPh]3+ show a single reversible one-electron oxidation wave with peak separations similar to ferrocene in the same solution (Table 5, Figure 5a). The peaks belong to the FeIII/FeII redox couple. By applying the Randles Sevcik equation, their reaction is found reversible and occurred under diffusion control system (Figure S1–S19) [65,66,67]. The half-wave potentials (E1/2) of the generated [(PBI)2(4R-Py)FeIlIOIPh]3+ (R = -CH3, -H, C(O)CH3, -C(O)Ph, and -CN) complexes become more negative in the following sequence: R is 4-Me-Py (−350 mV) > Py (−440.4 mV) > 4-PhC(O)-Py (−500.6 mV) > 4-MeC(O)-Py (−523.2 mV) > 4-CN-Py (−524 mV). The E1/2 spans a 174 mV range from −524 mV (R = 4-CN-Py) to −350 mV (X = 4-Me-Py) vs. Fc/Fc+), indicating that replacement of the CH3CN with electron-rich pyridines results in a remarkable increase in the redox potential value. Furthermore, the E1/2 potentials correlate well with the Hammett σp constants of the 4R-Py (Figure 5b) as well as the λmax values.
Table 5.
Redox properties (CV data) of the [(PBI)2FeIII(OIPh)(4R-Py)] intermediates generated in situ by the reaction of 1 with Ph(IOAc)2 in acetonitrile at 293 K 1.
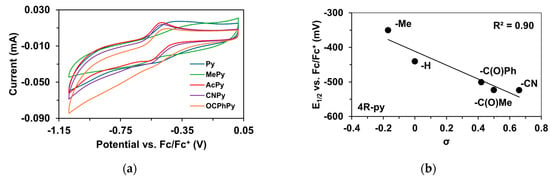
Figure 5.
Redox properties of the [(PBI)2FeIII(OIPh)(4R-Py)] intermediates generated in situ by the reaction of 1 with Ph(IOAc)2 in acetonitrile at 293 K. (a) Cyclic voltammograms of [(PBI)2FeIII(OIPh)(4R-Py)] intermediates. (b) E1/2 vs. σ values for [(PBI)2[(4R-Py)FeIII(OIPh)]3+ intermediates. Conditions: [[FeII(PBI)3](OTf)2]0 = 1 × 10−3 M, [PhI(OAc)2]0= 2 × 10−3 M, [4R-Py]0 = 1 × 10−2 M, in (0.1 M TBAClO4) CH3CN (10 cm3), scan rate: 1500 mV/s.
The reactivity of the in situ-formed [(PBI)2(4R-Py)FeIlIOIPh]3+ (R = -CH3, -H, C(O)CH3, -C(O)Ph, -CN) species was investigated in stoichiometric hydroxylation reactions with triphenylmethane. The reaction was carried out in CH3CN at 293 K with an excess of Ph3CH substrate, and the time courses for the decay of the in situ-generated [(PBI)2(4R-Py)FeIlIOIPh]3+ intermediates were monitored at their characteristic near-IR wavelengths (λmax) in the range of 700–760 nm (Figure 6). The decay process resulted in the formation of triphenylmethanol as main products (Yield ~80%/[1] based on GC) without any side-products based on GC-Ms analysis. All reactions follow general second-order kinetics, in first-order Ph3CH and [(PBI)2(4R-Py)FeIlIOIPh]3+ complexes (Figure 7a). Electron-releasing substituents in the pyridine co-ligand reduce the rate of oxidation. Correlations with σ (ρ = +0.87, Figure 7b) and with E1/2 (slope −3.94 V−1, Figure 8a and Figure S20–S28) and ν (Figure 8b) are evidence for an electrophilic mechanism. In the presence of PhIO and Py, the formation of pyridin-N-oxide (PyO) and its participation as a co-ligand in the reaction cannot be ruled out. When the stoichiometric reaction was performed in the presence of 10 equivalents of PyO, a much smaller hypsochromic shift (10 nm) and a nearly three-fold rate (kobs = 3.14 × 10−3 s−1; (kobs = kselfdecay + k2[PH3CH] and kobs >>> kselfdecay) compared to pyridine (kobs = 1.33 × 10−3 s−1) were observed (Figure 9), consistent with a ν vs logkrel relationship (Table 4, Figure 8b).
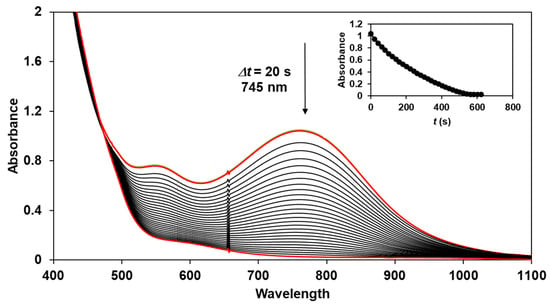
Figure 6.
Decrease in absorbance of [(PBI)2[(4Ac-Py)FeIII(OIPh)]3+ intermediate in the stoichiometric oxidation of triphenylmethane at 293 K in acetonitrile. Inset: time course of the decay monitored at 745 nm. [1]0 = 1 × 10−3 M, [PhI(OAc)2]0 = 1.2 × 10−3 M, [triphenylmethane]0 = 1 × 10−1 M, [4Ac-Py]0 = 1 × 10−2 M.
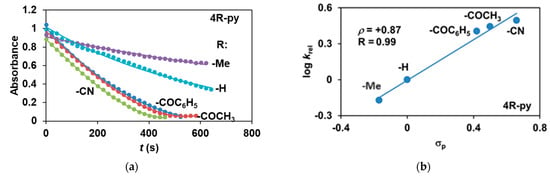
Figure 7.
Stoichiometric hydroxylation of triphenylmethane with [(PBI)2(4R-Py)FeIII(OIPh)]3+ intermediates generated in situ by the reaction of 1 with Ph(IOAc)2 and 4R-Py derivatives in acetonitrile at 293 K: (a) monitoring the decrease in absorbance of [(PBI)2(4R-Py)FeIII(OIPh)]3+ intermediates by UV–Vis spectroscopy at 723–760 nm over time at 293 K. (b) Hammett plot of logkrel against the σp of 4R-Py. [1]0 = 1 × 10−3 M, [PhI(OAc)2]0 = 1.2 × 10−3 M, [Ph3CH]0 = 1 × 10−1 M, [4R-Py]0 = 1 × 10−2 M.
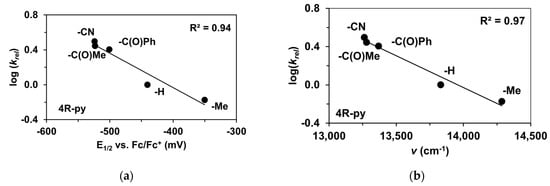
Figure 8.
Stoichiometric hydroxylation of triphenylmethane with [(PBI)2(4R-Py)FeIII(OIPh)]3+ intermediates generated in situ by the reaction of [FeII(PBI)3](OTf)2 with Ph(IOAc)2 and 4R-Py derivatives in acetonitrile at 293 K: (a) log(krel) against E1/2 for [(PBI)2(4R-Py)FeIII(OIPh)]3+ intermediates. (b) log(krel) against ν for [(PBI)2(4R-Py)FeIII(OIPh)]3+ intermediates. [1]0 = 1 × 10−3 M, [PhI(OAc)2]0 = 1.2 × 10−3 M, [Ph3CH]0 = 1 × 10−1 M, [4R-Py]0 = 1 × 10−2 M.
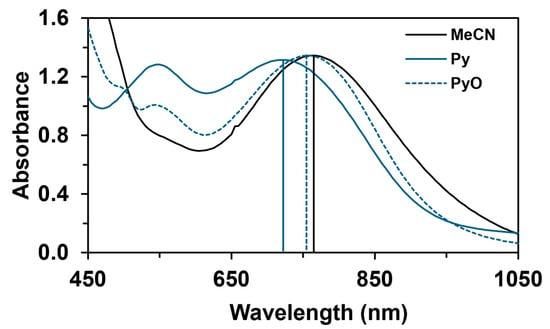
Figure 9.
UV–Vis spectral properties of [(PBI)2[(X)FeIII(OIPh)]3+ (X = CH3CN, Py, PyO) intermediates at 293 K in acetonitrile. [1]0 = 1 × 10−3 M, [PhI(OAc)2]0 = 1.2 × 10−3 M, [triphenylmethane]0 = 1 × 10−1 M, [X]0 = 1 × 10−2 M.
Finally, the tests were also extended to the effect of the oxidizing agent (PhI(OAc)2). Para-substituted (4R’-PhI(OAc)2, R’ = -CH3, -OCH3, -H, -Cl, -C(O)CH3) oxidants were used for the competition (Hammett correlation) stoichiometric reactions. The effect of the substituents of the 4R’-PhI(OAc)2 oxidants on the relative reactivity was studied for triphenylmethane and a remarkable effect on the stoichiometric hydroxylation reactions was observed (Table 6 and Figure 10a). Electron-withdrawing groups such as -C(O)CH3 gave significantly higher rate (kobs = 9.22 × 10−3 s−1) than electron-donating groups such as -OCH3 (kobs = 1.78 × 10−3 s−1). The Hammett treatments of relative reactivities (log krel) versus Hammett σp constants in the hydroxylation reactions gave ρ-values of +0.92 (Figure 10b), which is further evidence of the electrophilic behavior of the (PBI)2(4R-Py)FeIIIOIPh) intermediates. This value is almost identical to the results observed for the pyridine-containing stoichiometric (ρ = +0.87) and catalytic reactions (ρ = +0.97), and is comparable for the 4R-PhI(OAc)2-containing catalytic processes (ρ = +0.77).
Table 6.
Stoichiometric hydroxylation of triphenylmethane with [(PBI)2(MeCN)FeIII(4R-PhIO)]3+ intermediates generated in situ by the reaction of 1 with 4R-Ph(IOAc)2 oxidants in acetonitrile at 293 K 1.
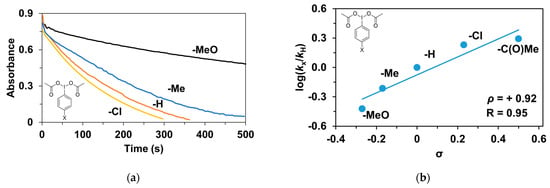
Figure 10.
Stoichiometric hydroxylation of triphenylmethane with [(PBI)2(MeCN)FeIII(OIPh-4R)]3+ (R = -Cl, -H, -Me, -OMe) intermediates generated in situ by the reaction of 1 with 4R-Ph(IOAc)2 (R = -Cl, -H, -Me, -OMe) oxidants in acetonitrile at 293 K: (a) Monitoring the decrease in absorbance of [(PBI)2(CH3CN)FeIII(OIPh-4R)]3+ intermediates by UV–Vis spectroscopy at 760 nm over time at 293 K. (b) Hammett plot of logkrel against the σp of [(PBI)2(CH3CN)FeIII(OIPh-4R)]3+intermediates. [[FeII(PBI)3](OTf)2]0 = 1 × 10−3 M, [4R-PhI(OAc)2]0 = 1.2 × 10−3 M, [Ph3CH]0 = 1 × 10−1 M.
3. Materials and Methods
All materials including 4-substituted pyridines (4-R-Py; R = 4-CH3, 4-H, 4-C(O)Ph, 4-C(O)CH3, 4-CN), PBI ligand, PhI(OAc)2, and substrates (Ph3CH) were obtained from Aldrich Chemical Co. and used without further purification unless otherwise noted. Solvents (CH3CN, pyridines) were purified according to published procedures, distilled, and stored under argon. [Fe(PBI)3](OTf)2 was synthesized according to methods in the literature [54]. The UV–Visible spectra were obtained with an Agilent 8453 (Budapest, Hungary) diode-array spectrophotometer using quartz cells. GC and GC-MS analyses were performed on an Agilent 7820A (Budapest, Hungary) equipped with a flame ionization detector and a 30 m Equity-1 column, and a Shimadzu QP2010SE (Budapest, Hungary) equipped with a secondary electron multiplier detector with a conversion dynode and a 30 m HP5MS column, respectively. Cyclic voltammetric experiments were carried out using an SP-150 potentiostat, using EC-Lab V11.41 software. During the measurements we used a three-electrode setup: we used a 3.0 mm diameter glassy-carbon electrode as a working electrode, a Pt wire as counter electrode, and an Ag/AgNO3 reference-electrode (https://www.biologic.net/accessory/small-reference-electrodes/; https://www.biologic.net/documents/re-7n-re-7sn-internal-solution-preparation-2/; 12 January 2024) with an acetonitrile solution containing 0.01 mol/dm3 silver(I)nitrate (AgNO3) and 0.1 mol/dm3 tetrabutylammonium perchlorate. The potentials were referred to the Fc/Fc+ couple.
Ligand exchange and stoichiometric oxidation reactions by UV-Vis spectroscopy:
All reactions were performed in a 1.0 cm UV cuvette and followed by monitoring UV–Vis spectral changes (increase for the formation of [(PBI)2(CH3CN)FeIlIOIPh-4R]3+ at 760 nm, and decrease in [(PBI)2(CH3CN)FeIlIOIPh-4R]3+ or [(PBI)2(4R-Py)FeIlIOIPh]3+ in the stoichiometric hydroxylation of triphenylmethane at 700–760 nm) in the reaction solutions. Rate constants (kobs) were determined under pseudo-first-order conditions (e.g., [Ph3CH]/[1] > 100 or [4R-PhI(Oc)2]/[1] > 10) by fitting the absorbance changes at 700–760 nm in CH3CN at 293 K). Reactions were run at least in triplicate, and the data reported are the average of the reactions.
Stoichiometric reaction: In a typical reaction, [FeII(PBI)3](OTf)2 (1 ×10−3 M) was dissolved in acetonitrile (2 mL), and/or (diacetoxyiodo)benzene (1.2 × 10−3 M) derivatives in acetonitrile was added to generate the reactive [(PBI)2(CH3CN)FeIlIOIPh-4R]3+ species. To generate the [(PBI)2(4R-Py)FeIlIOIPh]3+ species, 10 equivalents of 4R-Py (1 × 10−2 M) was added to the [(PBI)2(CH3CN)FeIlIOIPh]3+ intermediate. After the full formation of [(PBI)2(CH3CN)FeIlIOIPh-4R]3+ or [(PBI)2(4R-Py)FeIlIOIPh]3+, triphenylmethane (1 × 10−1 M) was added to the solution. The reactions were monitored with a UV–Vis spectrophotometer at 700–760 nm at 293 K. Reaction rates (kobs) were calculated using Biochemical Analysis Software (version B.05.01) for Agilent ChemStation.
Catalytic oxidations: [FeII(PBI)3](OTf)2 (1 × 10−3 M) was dissolved in acetonitrile (3 mL), and (diacetoxyiodo)benzene derivatives (1 × 10−1 M), substrate (3 × 10−1 M) (triphenylmethane), and/or different para-substituted pyridines (1 × 10−2 M) were added to the solution. Bromobenzene (1 × 10−1 M) was used as an internal standard. The mixture was stirred for 4 h at 323 K, the solution was freed from the metal catalyst using an Al2O3-filled column, the products from the diluted solutions were identified by GC-MS, and the yields were determined by GC.
Product of triphenylmethane oxidation: triphenylmethanol’s GC-MS: (m/z) relative intensities were 260.10 (M+, 14.07); 184.05 (9.15); 183.05 (63.15); 182.10 (15.27); 165.10 (10.26); 155.10 (17.23); 154.10 (28.54); 106.10 (10.08); 105.05 (100); 77.05 (69.30); and 51.05 (16.62).
4. Conclusions
The aim of this work was to generate an iodosylarene–iron(III) ([(PBI)2(CH3CN)FeIlIOIPh]3+) intermediate with a labile site, thus the effect of nitrogen-containing co-ligands (4R-Py, R = -CH3, -H, C(O)CH3, -C(O)Ph, -CN) for their spectral properties and reactivity in CH-activation reactions can be tested by ligand exchange reactions. In a similar way, we wanted to investigate the effect of substituted 4R’-PhI(OAc)2 (R’ = -OMe, -Me, -H, -Cl, -C(O)Me) derivatives as oxidizing agents in both stoichiometric and catalytic oxidation reactions towards triphenylmethane. It was found that both pyridine and iodosylbenzene derivatives as ligands (oxidants) can induce a hypsochromic shift in the complexes [(PBI)2(4R-Py)FeIlIOIPh]3+ and [(PBI)2(CH3CN)FeIlIOIPh-4R’]3+, which is consistent and proportional to the electron density of the metal center. The linear correlation between λmax−1 (at 700–760 nm) and the Hammett constants (σp) of [(PBI)2(4R-Py)FeIlIOIPh]3+ indicates that the observed hypsochromic shift (blue shift) of the absorption bands can be attributed to the electronic effect of the co-ligands (4R-Py) and the electrophilicity of the active intermediate. Based on Hammett correlations between the logkrel and the σp (4R-Py and 4R’-PhI(OAc)2) parameters, strong evidence for the electrophilic nature of the reactive species was observed for both stoichiometric (ρ = +0.87 and +0.92, respectively) and catalytic (ρ = +0.97 and +0.77, respectively) reactions. Furthermore, the redox potential values of [(PBI)2(4R-Py)FeIlIOIPh]3+ intermediates correlate very well with the hydroxylation rate of the triphenylmethane. The highest reactivity increase was achieved by the intermediates [(PBI)2(4R-Py)FeIlIOIPh]3+ and [(PBI)2(CH3CN)FeIlIOIPh-4R’]3+ containing an electron-withdrawing group. Based on the competitive experiments, similar results were observed in both the catalytic and stoichiometric reactions, which suggests that the electrophilic [(PBI)2(4R-Py)FeIIIOIPh-4R’]3+ intermediate is formed as the reactive species responsible for the oxidation reactions. These results clearly indicate that the reactive form of the hydroxylation reactions is the iron(III)–iodosylarene adduct (Scheme 2, Pathway a), and not the iron(V)oxo complex (Scheme 2 Pathway b).
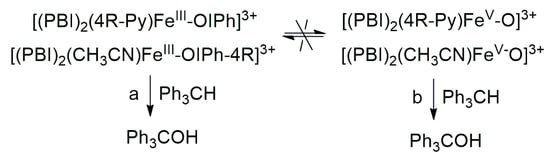
Scheme 2.
Proposed mechanistic pathways for the iron(III)–iodosylarene ([(PBI)2[(4R-Py)FeIII(OIPh-4R)]3+) mediated oxidation of triphenylmethane.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29163842/s1.
Author Contributions
Resources, P.T.; writing—original draft preparation and supervision, J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or Supplementary Materials.
Acknowledgments
Financial support of the Hungarian National Research, Development and Innovation Fund, OTKA K142212 (J.K.), TKP2021-NKTA-21 (J.K.), and ÚNKP-23-3 (P.T) New National Excellence Program of the Ministry for Culture and Innovation are gratefully acknowledged. Special thanks to Dóra Lakk-Bogáth for the GC-MS measurements.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Barry, S.M.; Challis, G.L. Mechanism and Catalytic Diversity of Rieske Non-Heme Iron-Dependent Oxygenases. ACS Catal. 2013, 3, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Tinberg, C.E.; Lippard, S.J. Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Res. 2011, 44, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Costas, M. Selective C–H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. [Google Scholar] [CrossRef]
- Liu, W.; Groves, J.T. Manganese Catalyzed C–H Halogenation. Acc. Chem. Res. 2015, 48, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Tucci, F.J. Rosenzweig, Direct Methane Oxidation by Copper- and Iron-Dependent Methane Monooxygenases. Chem. Rev. 2024, 124, 1288–1320. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Xu, K.; Gao, Z.-H.; Zhu, Z.-H.; Ye, C.; Zhao, B.; Luo, S.; Ye, S.; Zhou, Y.-G.; Xu, S.; et al. Biomimetic asymmetric catalysis. Sci. China. Chem 2023, 66, 1553–1633. [Google Scholar]
- Masoudian, S.; Yahyaei, H. Oxidation of alcohols with hydrogen peroxide catalyzed by supported Fe(III) porphyrins. Indian J. Chem. 2011, 50A, 1002–1005. [Google Scholar]
- Nam, W.; Park, S.-E.; Lim, I.K.; Lim, M.H.; Hong, J.; Kim, J. First direct evidence for stereospecific olefin epoxidation and alkane hydroxylation by an oxoiron (IV) porphyrin complex. J. Am. Chem. Soc. 2003, 125, 14674–14675. [Google Scholar] [CrossRef]
- Ozaki, S.-I.; Roach, M.P.; Matsui, T.; Watanabe, Y. Investigations of the roles of the distal heme environment and the proximal heme iron ligand in peroxide activation by heme enzymes via molecular engineering of myoglobin. Acc. Chem. Res. 2001, 34, 818–825. [Google Scholar] [CrossRef]
- Pariyar, A.; Bose, S.; Biswas, A.N.; Barman, S.; Bandyopadhyay, B. A non-heme cationic Fe(iii)-complex intercalated in montmorillonite K-10: Synthesis, characterization and catalytic alkane hydroxylation with H2O2 at room temperature. Cat. Sci. Tech. 2014, 4, 3180–3185. [Google Scholar] [CrossRef]
- Shilov, A.E.; Shul’pin, G.B. Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes; Kluwer: Boston, MA, USA, 2000. [Google Scholar]
- Shul’pin, G.B. Hydrocarbon Oxygenations with Peroxides Catalyzed by Metal Compounds. Mini-Rev. Org. Chem. 2009, 6, 95–104. [Google Scholar] [CrossRef]
- Shilov, A.E.; Shul’pin, G.B. Activation of C–H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C-H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Fokin, A.A.; Schreiner, P.R. Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev. 2002, 102, 1551–1593. [Google Scholar] [CrossRef] [PubMed]
- Sen, A. Catalytic Functionalization of Carbon−Hydrogen and Carbon−Carbon Bonds in Protic Media. Acc. Chem. Res. 1998, 31, 550–557. [Google Scholar] [CrossRef]
- Crabtree, R.H. Organometallic alkane CH activation. J. Organomet. Chem. 2004, 689, 4083–4091. [Google Scholar] [CrossRef]
- Crabtree, R.H. Alkane C–H activation and functionalization with homogeneous transition metal catalysts: A century of progress—A new millennium in prospect. J. Chem. Soc. Dalton Trans. 2001, 2437–2450. [Google Scholar] [CrossRef]
- Dolphin, D.; Traylor, T.G.; Xie, L.Y. Polyhaloporphyrins: Unusual Ligands for Metals and Metal-Catalyzed Oxidations. Acc. Chem. Res. 1997, 30, 251–259. [Google Scholar] [CrossRef]
- Montanari, F. Biomimetic oxygenations catalyzed by metalloporphyrins and metalloporphinoids bearing co-catalytic functions. Pure Appl. Chem. 1994, 66, 1519–1526. [Google Scholar] [CrossRef][Green Version]
- Friedermann, G.R.; Halma, M.; Castro, K.A.D.F.; Benedito, F.L.; Doro, F.G.; Drechsel, S.M.; Mangrich, A.S.; Assis, M.D.; Nakagaki, S. Intermediate species generatedfrom halogenated manganese porphyrins electr ochemically and in homogeneous catalysis ofalkane oxidation. Appl. Catal. A Gen. 2006, 308, 172–181. [Google Scholar] [CrossRef]
- De Vos, D.E.; Jacobs, P.A. Heterogenization of Mn an Fe complex oxidation catalysts. Catal. Today 2000, 57, 105–114. [Google Scholar] [CrossRef]
- Lindsay Smith, J.R.; Shul’pin, G.B. Efficient stereoselective oxygenation of alkanes by peroxyacetic acid or hydrogen peroxide and acetic acid catalysed by a manganese(IV) 1,4,7-trimethyl-1,4,7-triazacyclononane complex. Tetrahedron Lett. 1998, 39, 4909–4912. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K.; Lin, C.-C. Polymer-supported Schiff base complexes in oxidation reactions. Coord. Chem. Rev. 2009, 253, 1926–1946. [Google Scholar] [CrossRef]
- Correa, R.J.; Salomao, G.C.; Olsen, M.H.N.; Filho, L.C.; Drago, V.; Fernandes, C.; Antunes, O.A.C. Catalytic activity of Mn-III(Salen) complexes encapsulated in and FeIII(Salen) zeolite Y. Appl. Catal. A Gen. 2008, 336, 35–39. [Google Scholar] [CrossRef]
- Mardani, H.R.; Golchoubian, H. Selective and efficient C-H oxidation of alkanes with hydrogen peroxide catalyzed by a manganese(III) Schiff base complex. J. Mol. Catal. A Chem. 2006, 259, 197–200. [Google Scholar] [CrossRef]
- Vinhado, F.S.; Gandini, M.E.F.; Iamamoto, Y.; Silva, A.M.G.; Simoes, M.M.Q.; Neves, M.G.P.M.S.; Tomé, A.C.; Rebelo, S.L.H.; Pereira, A.M.V.M.; Cavaleiro, J.A.S. Novel Mn (III) chlorins as versatile catalysts for oxyfunctionalisation of hydrocarbons under homogeneous conditions. J. Mol. Catal. A Chem. 2005, 239, 138–143. [Google Scholar] [CrossRef]
- Lennartson, A.; McKenzie, C.J. An Iron(III) Iodosylbenzene Complex: A Masked Non-Heme FeVO. Angew. Chem. Int. Ed. 2012, 51, 6767–6770. [Google Scholar] [CrossRef]
- Wang, C.; Kurahashi, T.; Fujii, H. Highly Reactive Nonheme Iron(III) Iodosylarene Complexes in Alkane Hydroxylation and Sulfoxidation Reactions. Angew. Chem. Int. Ed. 2014, 53, 6388–6392. [Google Scholar]
- Wang, B.; Lee, Y.M.; Seo, M.S.; Nam, W. Mononuclear Nonheme Iron(III)-Iodosylarene and High-Valent Iron-Oxo Complexes in Olefin Epoxidation Reactions. Angew. Chem. Int. Ed. 2015, 54, 11740–11744. [Google Scholar] [CrossRef]
- Kang, Y.; Li, X.-X.; Cho, K.-B.; Sun, W.; Xia, C.; Nam, W.; Wang, Y. Mutable Properties of Nonheme Iron(III)–Iodosylarene Complexes Result in the Elusive Multiple-Oxidant Mechanism. J. Am. Chem. Soc. 2017, 139, 7444–7447. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Pandey, B.; Rajaraman, G. Comparative oxidative ability of iron(III)-iodosylarene vs. high-valent iron(IV/V)-oxo species: Is lower oxidation state a key to enhance selectivity in organic transformations? J. Indian Chem. Soc. 2019, 96, 825–836. [Google Scholar]
- Wang, C.; Kurahashi, T.; Inomata, K.; Hada, M.; Fujii, H. Oxygen-Atom Transfer from Iodosylarene Adducts of a Manganese(IV) Salen Complex: Effect of Arenes and Anions on I(III) of the Coordinated Iodosylarene. Inorg. Chem. 2013, 52, 9557–9566. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kurahashi, T.; Fujii, H. Structure and Reactivity of an Iodosylarene Adduct of a Manganese(IV)-Salen Complex. Angew. Chem. 2012, 124, 7929–7931. [Google Scholar] [CrossRef]
- Guo, M.; Dong, H.; Li, J.; Cheng, B.; Huang, Y.-Q.; Feng, Y.-Q.; Lei, A. Spectroscopic Observation of Iodosylarene Metalloporphyrin Adducts and Manganese(V)-Oxo Porphyrin Species in a Cytochrome P450 Analogue. Nat. Commun. 2012, 3, 1190. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Lee, Y.-M.; Fukuzumi, S.; Nam, W. Biomimetic metal-oxidant adducts as active oxidants in oxidation reactions. Coord. Chem. Rev. 2021, 435, 213807. [Google Scholar] [CrossRef]
- Guo, M.; Lee, Y.-M.; Seo, M.S.; Kwon, Y.-J.; Li, X.-X.; Ohta, T.; Kim, W.-S.; Sarangi, R.; Fukuzumi, S.; Nam, W. Mn(III)-Iodosylarene Porphyrins as an Active Oxidant in Oxidation Reactions: Synthesis, Characterization, and Reactivity Studies. Inorg. Chem. 2018, 57, 10232–10240. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Choi, S.K.; Lim, M.H.; Rohde, J.-U.; Kim, I.; Kim, J.; Kim, C.; Que, L., Jr. Reversible formation of iodosylbenzene-iron porphyrin intermediates in the reaction of oxoiron(IV) porphyrin pi-cation radicals and iodobenzene. Angew. Chem. Int. Ed. 2003, 42, 109–111. [Google Scholar] [CrossRef]
- Hill, E.A.; Kelty, M.L.; Filatov, A.S.; Anderson, J.S. Isolable iodosylarene and iodoxyarene adducts of Co and their O-atom transfer and C–H activation reactivity. Chem. Sci. 2018, 9, 4493–4499. [Google Scholar] [CrossRef]
- Jeong, D.; Ohta, T.; Cho, J.J. Structure and Reactivity of a Mononuclear Nonheme Manganese(III)–Iodosylarene Complex. Am. Chem. Soc. 2018, 140, 16037–16041. [Google Scholar] [CrossRef]
- Török, P.; Lakk-Bogáth, D.; Kaizer, J. Stoichiometric Alkane and Aldehyde Hydrtoxylation Reactions Mediated by In Situ Generated Iron(III)-Iodosylbenzene Adduct. Molecules 2023, 28, 1855. [Google Scholar] [CrossRef] [PubMed]
- Lakk-Bogáth, D.; Szávuly, M.; Török, P.; Kaizer, J. Catalytic and Stoichiometric Baeyer-Villiger Oxidation Mediated by Nonheme Peroxi-Diiron(III), Acylperoxo, and Iodosylbenzene Iron(III) Intermediates. Molecules 2022, 27, 2814. [Google Scholar] [CrossRef]
- Török, P.; Lakk-Bogáth, D.; Kaizer, J. Mechanisms of Sulfoxidation and Epoxidation Mediated by Iron(III)-Iodosylbenzene Adduct: Electron-Transfer vs Oxygen-Transfer Mechanism. Molecules 2023, 28, 4745. [Google Scholar] [CrossRef]
- Kumar, R.; Pandey, B.; Sen, A.; Ansari, M.; Sharma, S.; Rajaraman, G. Role of oxidation state, ferryl-oxygen, and ligand architecture on the reactivity of popular high-valent FeIV=O species: A theoretical perspective. Coord. Chem. Rev. 2020, 419, 213397. [Google Scholar] [CrossRef]
- Guo, M.; Corona, T.; Ray, K.; Nam, W. Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent Sci. 2018, 5, 13–28. [Google Scholar] [CrossRef]
- Roy, L. Theoretical Identification of the Factors Governing the Reactivity of C−H Bond Activation by Non-Heme Iron(IV)-Oxo Complexes. ChemPlusChem 2019, 84, 893–906. [Google Scholar] [CrossRef]
- Lee, J.L.; Ross, D.L.; Barman, S.K.; Ziller, J.W.; Borovik, A.S. C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes. Inorg. Chem. 2021, 60, 13759–13783. [Google Scholar] [CrossRef] [PubMed]
- Warm, K.; Paskin, A.; Kuhlmann, U.; Bill, E.; Swart, M.; Haumann, M.; Dau, H.; Hildebrandt, P.; Ray, K. A Pseudotetrahedral Terminal Oxoiron(IV) Complex: Mechanistic Promiscuity in C-H Bond Oxidation Reactions. Angew. Chem. Int. Ed. 2021, 60, 6752–6756. [Google Scholar] [CrossRef]
- Kupper, C.; Mondal, B.; Serrano-Plana, J.; Klawitter, I.; Neese, F.; Costas, M.; Ye, S.; Meyer, F. Nonclassical Single-State Reactivity of an Oxo-Iron(IV) Complex Confined to Triplet Pathways. J. Am. Chem. Soc. 2017, 139, 8939–8949. [Google Scholar] [CrossRef]
- Deutscher, J.; Gerschel, P.; Warm, K.; Kuhlmann, U.; Mebs, S.; Haumann, M.; Dau, H.; Hildebrandt, P.; Apfel, U.P.; Ray, K. A Bioinspired Oxoiron(IV) Motif Supported on a N2S2 macrocyclic Ligand. Chem. Commun. 2021, 57, 2947–2950. [Google Scholar] [CrossRef]
- Mandal, D.; Mallick, D.; Shaik, S. Kinetic Isotope Effect Determination Probes the Spin of the Transition State, Its Stereochemistry, and Its Ligand Sphere in Hydrogen Abstraction Reactions of Oxoiron(IV) Complexes. Acc. Chem. Res. 2018, 51, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Eguía, B.N.; Serrano-Plana, J.; Company, A.; Costas, M. Catalytic O2 activation with Synthetic Models of α-Ketoglutarate Dependent Oxygenases. Chem. Commun. 2020, 56, 14369–14372. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, G.; Alili, A.; Barman, P.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Interplay Between Steric and Electronic Effects: A Joint Spectroscopy and Computational Study of Nonheme Iron(IV)-Oxo Complexes. Chem. Eur. J. 2019, 25, 5086–5098. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, W.; Draksharapu, A.; Banerjee, S.; Young, V.G., Jr.; Fan, R.; Guo, Y.; Ozerov, M.; Nehrkorn, J.; Krzystek, J.; Telser, J.; et al. Crystallographic Evidence for a Sterically Induced Ferryl Tilt in a Non-Heme Oxoiron(IV) Complex that Makes it a Better Oxidant. Angew. Chem. Int. Ed. 2018, 57, 9387–9391. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, G.; Lee, C.W.Z.; Nag, S.S.; Alili, A.; Cantu Reinhard, F.G.; Kumar, D.; Sastri, C.V.; de Visser, S.P. Dramatic rate-Enhancement of oxygen atom transfer by an Iron(IV)-oxo species by equatorial ligand field perturbations. Dalton Trans. 2018, 47, 14945–14957. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Ganguly, G.; Malinkin, S.O.; Demeshko, S.; Meyer, F.; Nordlander, E.; Paine, T.K. A Mononuclear Nonheme Iron(IV)- Oxo Complex of a Substituted N4Py Ligand: Effect of Ligand Field on Oxygen Atom Transfer and C-H Bond Cleavage Reactivity. Inorg. Chem. 2019, 58, 1862–1876. [Google Scholar] [CrossRef] [PubMed]
- Pap, J.S.; Draksharapu, A.; Giorgi, M.; Browne, W.R.; Kaizer, J.; Speier, G. Stabilisation of μ-peroxido-bridged Fe(III) intermediates with non-symmetric bidentate N-donor ligands. Chem. Commun. 2014, 50, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Török, P.; Lakk-Bogáth, D.; Unjaroen, D.; Browne, W.R.; Kaizer, J. Effect of monodentate heterocycle co-ligands on the m-1,2-peroxo-diiron(III) mediated aldehyde deformylation reactions. J. Inorg. Biochem. 2024, 258, 112620. [Google Scholar] [CrossRef] [PubMed]
- Török, P.; Unjaroen, D.; Csendes, F.V.; Giorgi, M.; Browne, W.R.; Kaizer, J. A nonheme peroxo-diiron(III) complex exhibiting both nucleophilic and electrophilic oxidation of organic substrares. Dalton Trans. 2021, 50, 7181–7185. [Google Scholar] [CrossRef]
- Kripli, B.; Csendes, V.F.; Török, P.; Speier, G.; Kaizer, J. Stoichiometric Aldehyde Mediated by Nucleophilic Peroxo-diiron(III) Complex as a Functional Model of Aldehyde Deformylating Oxygenase. Chem. Eur. J. 2019, 25, 14290–14294. [Google Scholar] [CrossRef]
- Lakk-Bogáth, D.; Pintarics, D.; Török, P.; Kaizer, J. Influence of Equatorial Co-Ligands on the Reactivity of LFeIIIOIPh. Molecules 2024, 29, 58. [Google Scholar] [CrossRef] [PubMed]
- Adam, W.; Hajra, S.; Herderich, M.; Saha-Moller, C.R. A highly chemoselective oxidation of alcohols to carbonyl products with iodosobenzene diacetate mediated by chromium(III)(salen) complexes: Synthetic and mechanistic aspects. Org. Lett. 2000, 2, 2773–2776. [Google Scholar] [CrossRef] [PubMed]
- In, J.H.; Park, S.E.; Song, R.; Nam, W. Iodobenzene diacetate as an efficient terminal oxidant in iron(III) porphyrin complex-catalyzed oxygenation reactions. Inorg. Chim. Acta 2003, 343, 373–376. [Google Scholar] [CrossRef]
- Khurshid, S.; Mohiuddin, S.; Maqsood, Z. Investigations of Ferrous Thiamine Complex by Cyclic Voltammetry. Int. Res. J. Pure Appl. Chem. 2014, 4, 718–726. [Google Scholar] [CrossRef]
- Gaillard, M.; Kanso, H.; Denat, F.; Calas-Blanchard, C.; Inguimbert, N.; Noguer, T. Fe(III)-DOTA/Fe(III)-NOTA Complexes: Attractive Alternative Markers for Future Electrochemical Biosensors. J. Electrochem. Soc. 2020, 167, 117502. [Google Scholar] [CrossRef]
- Santos, M.A.; Simoes Goncalves, M.L. Electrochemistry of iron(III) tris-hydroxamate complexes: Kinetic and thermodynamic studies. Electrochim. Acta 1992, 37, 205–209. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).