
Location, Location, Location: Establishing Design Principles for New Antibacterials from Ferric Siderophore Transport Systems

Abstract
:
1. Antibiotic Development and Microbial Iron Assimilation
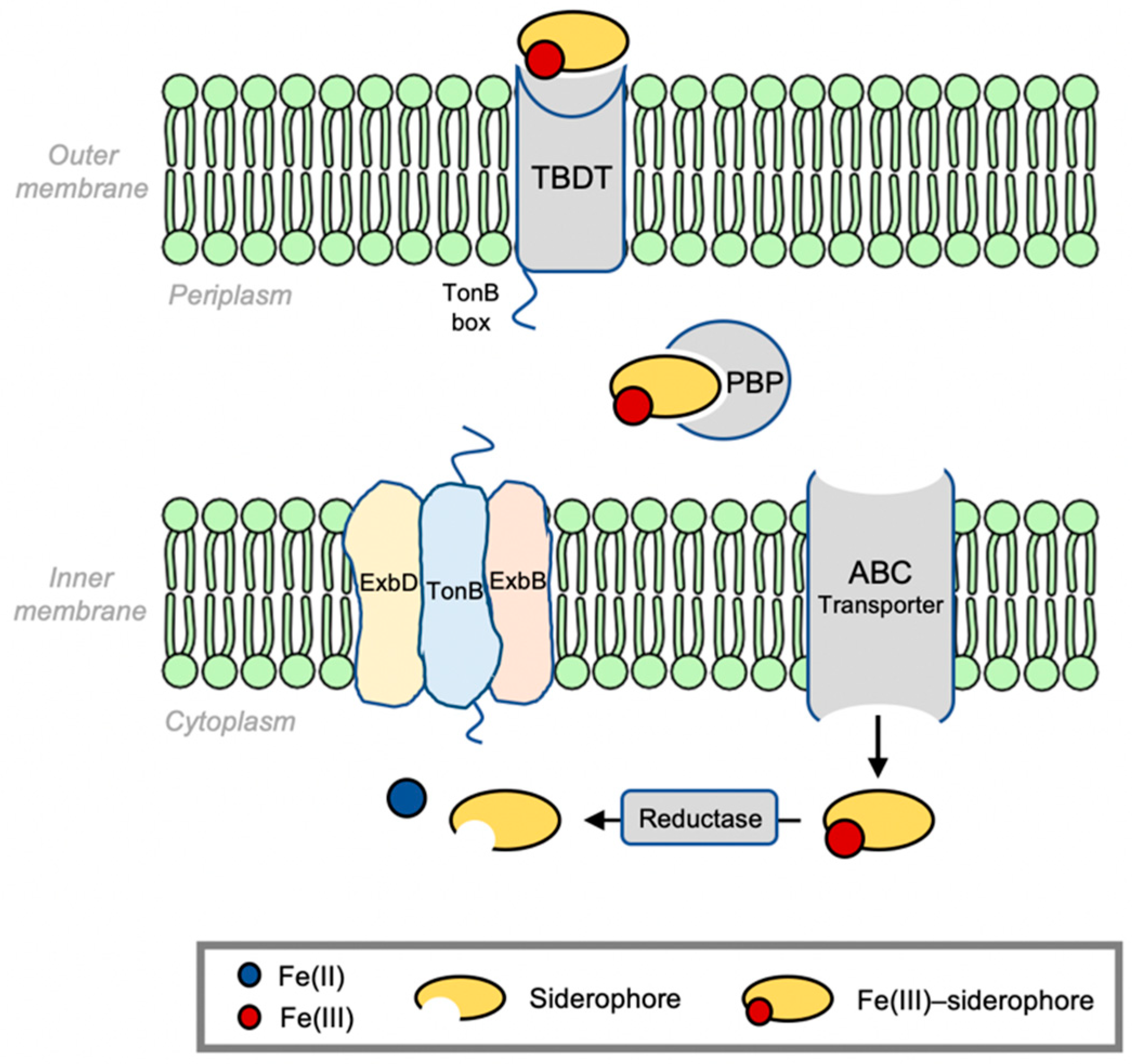
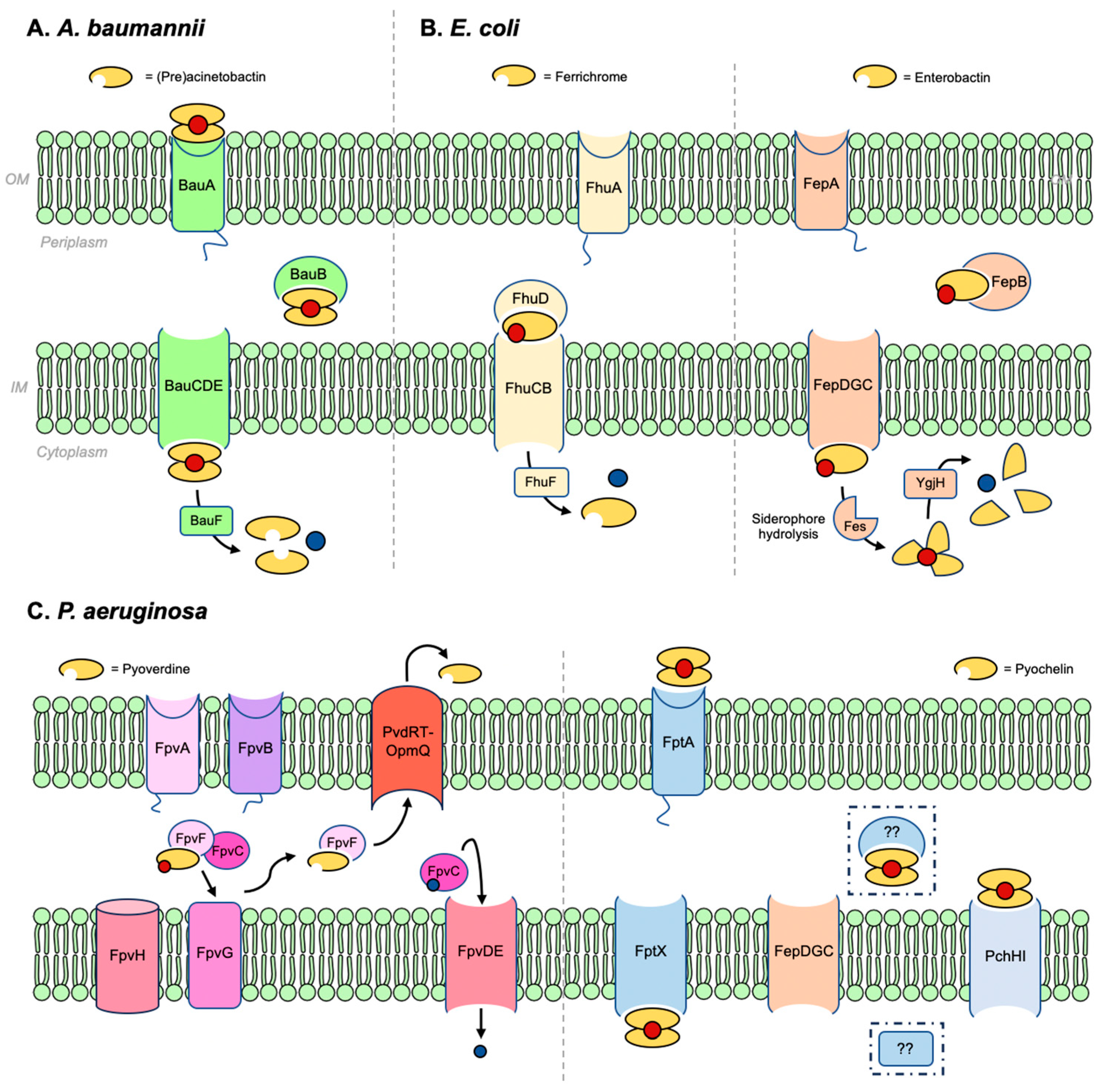
2. Bacterial Infrastructure of Iron Siderophore Transport
2.1. Acinetobacter baumannii
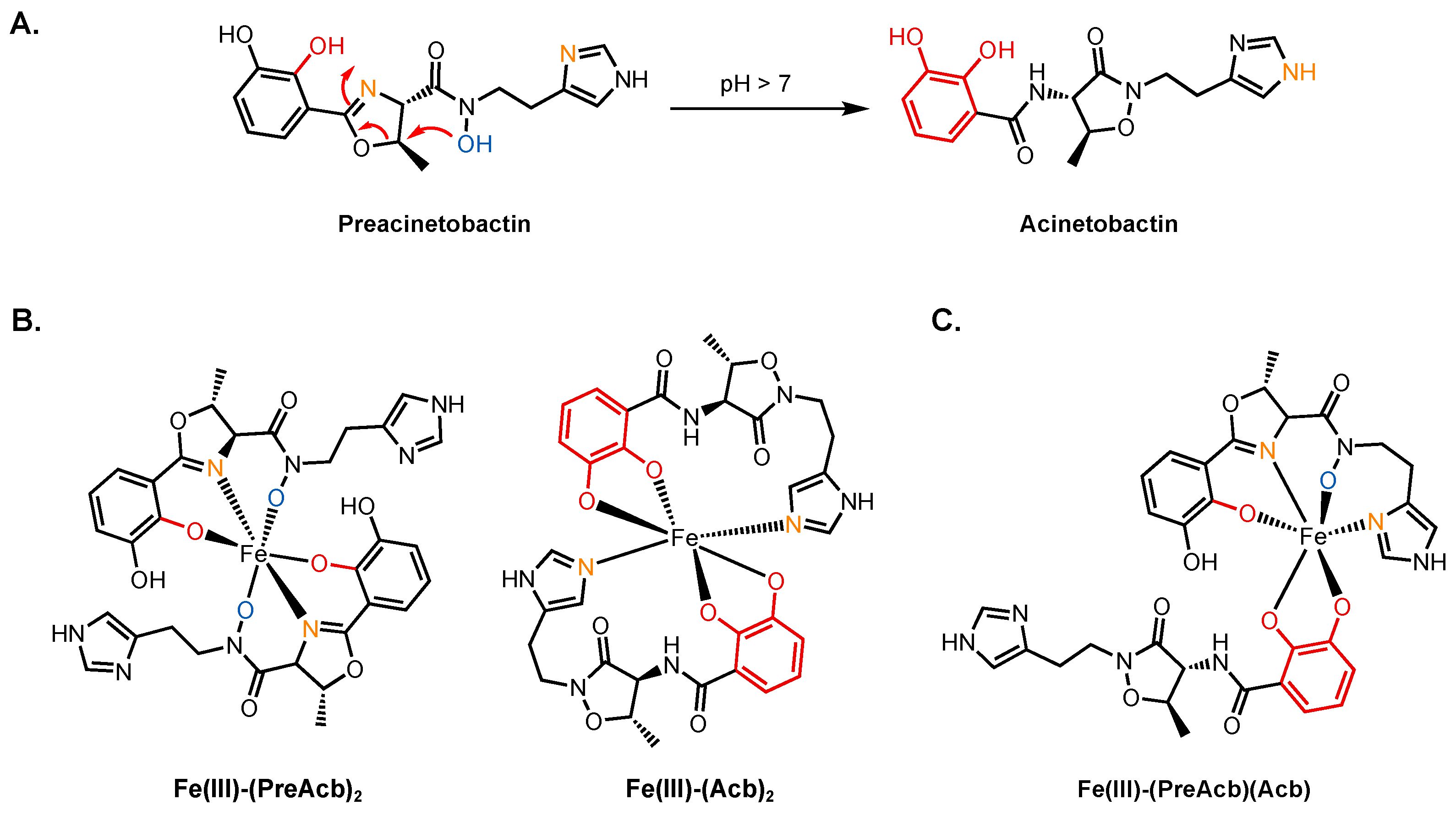
2.1.1. Preacinetobactin and Acinetobactin
2.1.2. Fimsbactins
2.2. Escherichia coli
2.2.1. Ferrichrome and Related Hydroxamates
2.2.2. Enterobactin and Related Catecholates
2.3. Pseudomonas aeruginosa
2.3.1. Pyoverdine and Pyochelin
2.3.2. Enterobactin
3. Siderophore Conjugate Studies
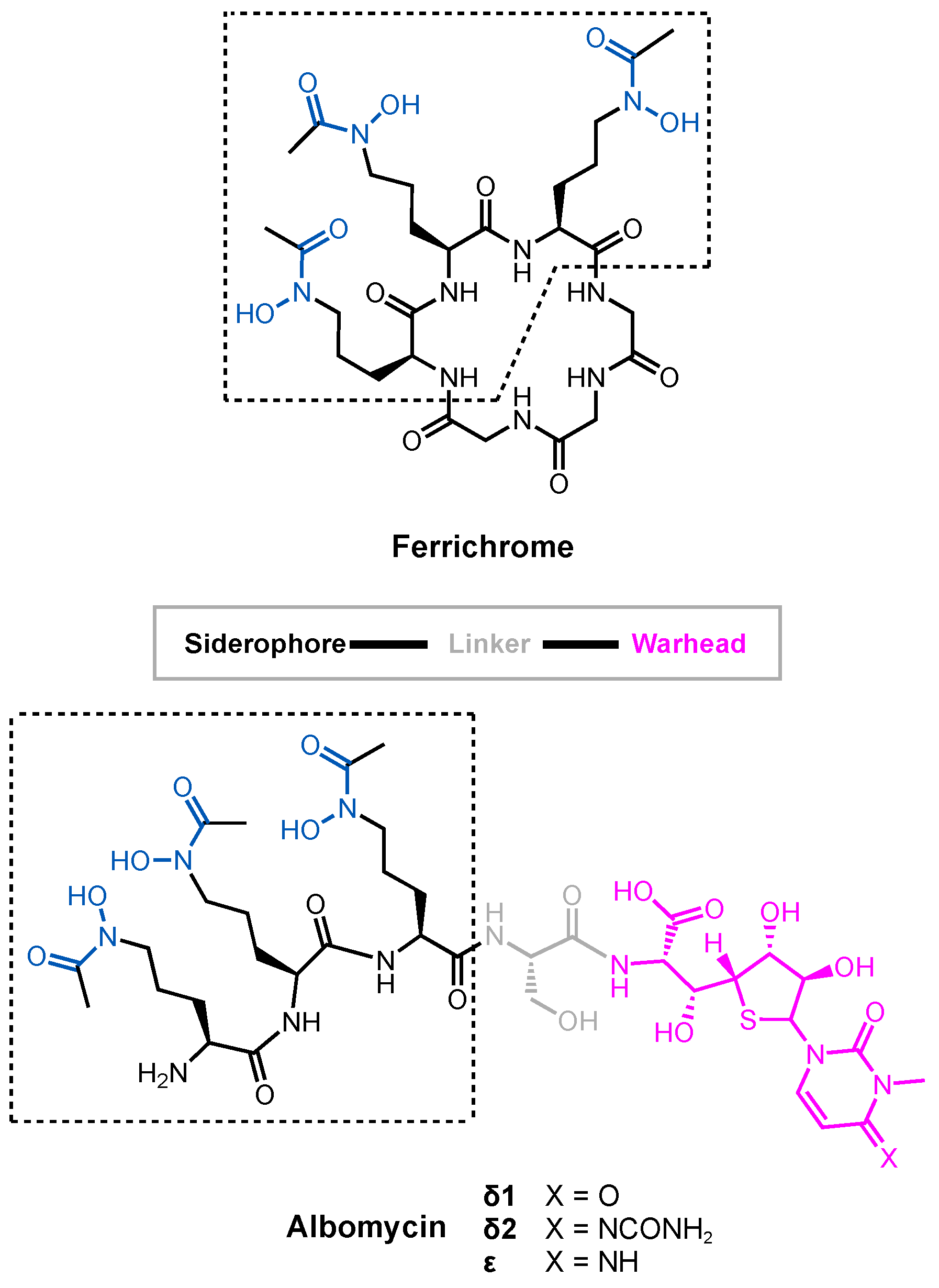
3.1. Albomycin and Hydroxamate Siderophore Conjugates
3.2. Fimsbactin conjugates
3.3. Enterobactin Conjugates and Derivatives
3.4. Catecholate Siderophore Conjugates
3.5. Pyochelin conjugates
4. Cefiderocol
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019. Available online: https://stacks.cdc.gov/view/cdc/82532 (accessed on 17 July 2024).
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An Interna-tional Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Bacterial Priority Pathogens List, 2024: Bacterial Pathogens of Public Health Importance to Guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance. Available online: https://www.who.int/publications/i/item/9789240093461 (accessed on 11 July 2024).
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Hamad, B. The Antibiotics Market. Nat. Rev. Drug Discov. 2010, 9, 675–676. [Google Scholar] [CrossRef]
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Rice, L.B. Progress and Challenges in Implementing the Research on ESKAPE Pathogens. Infect. Control. Hosp. Epidemiol. 2010, 31, S7–S10. [Google Scholar] [CrossRef]
- Giedraitienė, A.; Giedraitienė, A.; Vitkauskienė, A.; Naginienė, R.; Pavilonis, A. Correspondence to Antibiotic Resistance Mechanisms of Clinically Important Bacteria. Medicina 2011, 47, 19. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A.; Silvia Munoz-Price, L. Mechanisms of Disease the New B-Lactamases. N. Engl. J. Med. 2024, 352, 380–391. [Google Scholar] [CrossRef]
- Pucci, M.J.; Dougherty, T.J. Direct Quantitation of the Numbers of Individual Penicillin-Binding Proteins per Cell in Staphylococcus Aureus. J. Bacteriol. 2002, 184, 588–591. [Google Scholar] [CrossRef]
- Tang, S.S.; Apisarnthanarak, A.; Hsu, L.Y. Mechanisms of β-Lactam Antimicrobial Resistance and Epidemiology of Major Community- and Healthcare-Associated Multidrug-Resistant Bacteria. Adv. Drug Deliv. Rev. 2014, 78, 3–13. [Google Scholar] [CrossRef]
- Fukuoka, T.; Ohya, S.; Narita, T.; Katsuta, M.; Iijima, M.; Masuda, N.; Yasuda, H. Activity of the Carbapenem Panipenem and Role of the OprD (D2) Protein in Its Diffusion through the Pseudomonas Aeruginosa Outer Membrane. Antimicrob. Agents Chemother. 1993, 37, 322–327. [Google Scholar] [CrossRef]
- Thomson, J.M.; Bonomo, R.A. The Threat of Antibiotic Resistance in Gram-Negative Pathogenic Bacteria: β-Lactams in Peril! Curr. Opin. Microbiol. 2005, 8, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Deng, Z.; Yan, A. Bacterial Multidrug Efflux Pumps: Mechanisms, Physiology and Pharmacological Exploitations. Biochem. Bioph. Res. Commun. 2014, 453, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Pagès, J.M. Broad-Specificity Efflux Pumps and Their Role in Multidrug Resistance of Gram-Negative Bacteria. FEMS Microbiol. Rev. 2012, 36, 340–363. [Google Scholar] [CrossRef] [PubMed]
- Vaez, H.; Faghri, J.; Isfahani, B.; Moghim, S.; Yadegari, S.; Fazeli, H.; Moghofeei, M.; Safaei, H. Efflux Pump Regulatory Genes Mutations in Multidrug Resistance Pseudomonas Aeruginosa Isolated from Wound Infections in Isfahan Hospitals. Adv. Biomed. Res. 2014, 3, 117. [Google Scholar] [CrossRef]
- Schweizer, H.P.; Schweizer, H.P. Efflux as a Mechanism of Resistance to Antimicrobials in Pseudomonas Aeruginosa and Related Bacteria: Unanswered Questions. Genet. Mol. Res. 2003, 2, 48–62. [Google Scholar] [PubMed]
- Laverty, G.; Gorman, S.P.; Gilmore, B.F. Biomolecular Mechanisms of Pseudomonas aeruginosa and Escherichia coli Biofilm Formation. Pathogens 2014, 3, 596–632. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo, J.L.; Patel, R. The Challenge of Treating Biofilm-Associated Bacterial Infections. Clin. Pharmacol. Ther. 2007, 82, 204–209. [Google Scholar] [CrossRef]
- Sun, D. Pull in and Push out: Mechanisms of Horizontal Gene Transfer in Bacteria. Front. Microbiol. 2018, 9, 2154. [Google Scholar] [CrossRef]
- Venkateswaran, P.; Vasudevan, S.; David, H.; Shaktivel, A.; Shanmugam, K.; Neelakantan, P.; Solomon, A.P. Revisiting ESKAPE Pathogens: Virulence, Resistance, and Combating Strategies Focusing on Quorum Sensing. Front. Cell Infect. Microbiol. 2023, 13, 1159798. [Google Scholar] [CrossRef]
- Evstatiev, R.; Gasche, C. Iron Sensing and Signalling. Gut 2012, 61, 933–952. [Google Scholar] [CrossRef]
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial Iron Homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Raymond, K.N.; Dertz, E.A. Biochemical and Physical Properties of Siderophores. In Iron Transport in Bacteria; Crosa, J.H., Mey, A.R., Payne, S.M., Eds.; Wiley: Hoboken, NJ, USA, 2004; ISBN 9781683672050. [Google Scholar]
- Krewulak, K.D.; Vogel, H.J. Structural Biology of Bacterial Iron Uptake. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1781–1804. [Google Scholar] [CrossRef]
- Noinaj, N.; Guillier, M.; Barnard, T.J.; Buchanan, S.K. TonB-Dependent Transporters: Regulation, Structure, and Function. Annu. Rev. Microbiol. 2010, 64, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Fanucci, G.E.; Lee, J.Y.; Cafiso, D.S. Spectroscopic Evidence That Osmolytes Used in Crystallization Buffers Inhibit a Conformation Change in a Membrane Protein. Biochemistry 2003, 42, 13106–13112. [Google Scholar] [CrossRef]
- Fanucci, G.E.; Cadieux, N.; Kadner, R.J.; Cafiso, D.S. Competing Ligands Stabilize Alternate conformations of the Energy Coupling Motif of a TonB-Dependent Membrane Transporter. Proc. Natl. Acad. Sci. USA 2003, 100, 11382–11387. [Google Scholar] [CrossRef] [PubMed]
- Guerinot, M.L. Microbial Iron Transport. Annu. Rev. Microbiol. 1994, 48, 743–772. [Google Scholar] [CrossRef]
- Berntsson, R.P.A.; Smits, S.H.J.; Schmitt, L.; Slotboom, D.J.; Poolman, B. A Structural Classification of Substrate-Binding Proteins. FEBS Lett. 2010, 584, 2606–2617. [Google Scholar] [CrossRef]
- Thomas, C.; Tampé, R. Multifaceted Structures and Mechanisms of ABC Transport Systems in Health and Disease. Curr. Opin. Struct. Biol. 2018, 51, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Seyffer, F.; Tampé, R. ABC Transporters in Adaptive Immunity. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 449–460. [Google Scholar] [CrossRef]
- Locher, K.P. Mechanistic Diversity in ATP-Binding Cassette (ABC) Transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef]
- Wilkins, R.G. Kinetics and Mechanisms of Reactions of Transition Metal Complexes, 2nd ed.; VCH: Weinheim, Germany, 1991; ISBN 9783527600823. [Google Scholar]
- Raines, D.J.; Sanderson, T.J.; Wilde, E.J.; Duhme-Klair, A.-K. Siderophores. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Waltham, MA, USA, 2015. [Google Scholar]
- Valentino, H.; Korasick, D.A.; Bohac, T.J.; Shapiro, J.A.; Wencewicz, T.A.; Tanner, J.J.; Sobrado, P. Structural and Biochemical Characterization of the Flavin-Dependent Siderophore-Interacting Protein from Acinetobacter Baumannii. ACS Omega 2021, 6, 18537–18547. [Google Scholar] [CrossRef] [PubMed]
- Trindade, I.B.; Rollo, F.; Todorovic, S.; Catarino, T.; Moe, E.; Matias, P.M.; Piccioli, M.; Louro, R.O. The Structure of a Novel Ferredoxin—FhuF, a Ferric-Siderophore Reductase from E. coli K-12 with a Novel 2Fe-2S Cluster Coordination. bioRxiv 2023. [Google Scholar] [CrossRef]
- Miethke, M.; Hou, J.; Marahiel, M.A. The Siderophore-Interacting Protein YqjH Acts as a Ferric Reductase in Different Iron Assimilation Pathways of Escherichia coli. Biochemistry 2011, 50, 10951–10964. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.; Zeder-Lutz, G.; Hagege, A.; Celia, H.; Pattus, F. The Metal Dependence of Pyoverdine Interactions with Its Outer Membrane Receptor FpvA. J. Bacteriol. 2008, 190, 6548–6558. [Google Scholar] [CrossRef] [PubMed]
- Ganne, G.; Brillet, K.; Basta, B.; Roche, B.; Hoegy, F.; Gasser, V.; Schalk, I.J. Iron Release from the Siderophore Pyoverdine in Pseudomonas Aeruginosa Involves Three New Actors: FpvC, FpvG, and FpvH. ACS Chem. Biol. 2017, 12, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Chakraborty, R.; Smith, B.S.; Esser, L.; van der Helm, D.; Deisenhofer, J. Structural Basis of Gating by the Outer Membrane Transporter FecA. Science 2002, 295, 1715–1719. [Google Scholar] [CrossRef]
- Reynolds, D.M.; Schatz, A.; Waksman, S.A. Grisein, a New Antibiotic Produced by a Strain of Streptomyces Griseus. Proc. Soc. Exptl. Biol. Med. 1947, 64, 50–54. [Google Scholar] [CrossRef]
- Zhu, W.; Winter, M.G.; Spiga, L.; Hughes, E.R.; Chanin, R.; Mulgaonkar, A.; Pennington, J.; Maas, M.; Behrendt, C.L.; Kim, J.; et al. Xenosiderophore Utilization Promotes Bacteroides Thetaiotaomicron Resilience during Colitis. Cell Host Microbe 2020, 27, 376–388.e8. [Google Scholar] [CrossRef] [PubMed]
- Benz, G.; Schröder, T.; Kurz, J.; Wünsche, C.; Karl, W.; Steffens, G.; Pfitzner, J.; Schmidt, D. Constitution of the Deferriform of the Albomycins Δ1, Δ2 and ε. Angew. Chem. Int. Ed. Engl. 1982, 21, 527–528. [Google Scholar] [CrossRef]
- Braun, V.; Pramanik, A.; Gwinner, T.; Köberle, M.; Bohn, E. Sideromycins: Tools and Antibiotics. BioMetals 2009, 22, 3–13. [Google Scholar] [CrossRef]
- Gause, G.F. Recent Studies on Albomycin, a New Antibiotic. Br. Med. J. 1955, 2, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chakravorty, S.; Yang, T.; Russo, T.A.; Newton, S.M.; Klebba, P.E. Siderophore-Mediated Iron Acquisition by Klebsiella Pneumoniae. J. Bacteriol. 2024, 206, e0002424. [Google Scholar] [CrossRef]
- Sebeny, P.J.; Riddle, M.S.; Petersen, K. Acinetobacter Baumannii Skin and Soft-Tissue Infection Associated with War Trauma. Clin. Infect. Dis. 2008, 47, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.C.; Wareham, D.W. Multidrug-Resistant Acinetobacter Baumannii: Mechanisms of Virulence and Resistance. Int. J. Antimicrob. Agents 2010, 35, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Liou, M.L.; Chen, K.H.; Yeh, H.L.; Lai, C.Y.; Chen, C.H. Persistent Nasal Carriers of Acinetobacter Baumannii in Long-Term-Care Facilities. Am. J. Infect. Control. 2017, 45, 723–727. [Google Scholar] [CrossRef]
- De Vos, D.; Pirnay, J.P.; Bilocq, F.; Jennes, S.; Verbeken, G.; Rose, T.; Keersebilck, E.; Bosmans, P.; Pieters, T.; Hing, M.; et al. Molecular Epidemiology and Clinical Impact of Acinetobacter Calcoaceticus-Baumannii Complex in a Belgian Burn Wound Center. PLoS ONE 2016, 11, e0156237. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, G.M.; Peleg, A.Y. Insights into Acinetobacter Baumannii Pathogenicity. IUBMB Life 2011, 63, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Garnacho-Montero, J.; Ortiz-Leyba, C.; Fernández-Hinojosa, E.; Aldabó-Pallás, T.; Cayuela, A.; Marquez-Vácaro, J.A.; Garcia-Curiel, A.; Jiménez-Jiménez, F.J. Acinetobacter Baumannii Ventilator-Associated Pneumonia: Epidemiological and Clinical Findings. Intensive Care Med. 2005, 31, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Anstey, N.M.; Currie, B.J.; Hassell, M.; Palmer, D.; Dwyer, B.; Seifert, H. Community-Acquired Bacteremic Acinetobacter Pneumonia in Tropical Australia Is Caused by Diverse Strains of Acinetobacter Baumannii, with Carriage in the Throat in at-Risk Groups. J. Clin. Microbiol. 2002, 40, 685–686. [Google Scholar] [CrossRef]
- Wisplinghoff, H.; Bischoff, T.; Tallent, S.M.; Seifert, H.; Wenzel, R.P.; Edmond, M.B. Nosocomial Bloodstream Infections in US Hospitals: Analysis of 24,179 Cases from a Prospective Nationwide Surveillance Study. Clin. Infect. Dis. 2004, 309, 309–317. [Google Scholar] [CrossRef]
- Briggs, S.; Ellis-Pegler, R.; Raymond, N.; Thomas, M.; Wilkinson, L. Gram-Negative Bacillary Meningitis after Cranial Surgery of Trauma in Adults. Scand. J. Infect. Dis. 2004, 36, 165–173. [Google Scholar] [CrossRef]
- Penwell, W.F.; Arivett, B.A.; Actis, L.A. The Acinetobacter Baumannii EntA Gene Located Outside the Acinetobactin Cluster Is Critical for Siderophore Production, Iron Acquisition and Virulence. PLoS ONE 2012, 7, e36493. [Google Scholar] [CrossRef]
- Gaddy, J.A.; Actis, L.A.; Arivett, B.A.; Mcconnell, M.J.; Rafael, L.R.; Pachón, J. Role of Acinetobactin-Mediated Iron Acquisition Functions in the Interaction of Acinetobacter Baumannii Strain ATCC 19606T with Human Lung Epithelial Cells, Galleria Mellonella Caterpillars, and Mice. Infect. Immun. 2012, 80, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.A.; Wencewicz, T.A. Acinetobactin Isomerization Enables Adaptive Iron Acquisition in Acinetobacter Baumannii through PH-Triggered Siderophore Swapping. ACS Infect. Dis. 2016, 2, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter Baumannii: Emergence of a Successful Pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Stubbings, W.; Wisplinghoff, H.; Seifert, H. Activity of the Investigational Fluoroquinolone Finafloxacin against Ciprofloxacin-Sensitive and -Resistant Acinetobacter Baumannii Isolates. Antimicrob. Agents Chemother. 2010, 54, 1613–1615. [Google Scholar] [CrossRef]
- Song, W.Y.; Jeong, D.; Kim, J.; Lee, M.W.; Oh, M.H.; Kim, H.J. Key Structural Elements for Cellular Uptake of Acinetobactin, a Major Siderophore of Acinetobacter Baumannii. Org. Lett. 2017, 19, 500–503. [Google Scholar] [CrossRef]
- Bohac, T.J.; Fang, L.; Giblin, D.E.; Wencewicz, T.A. Fimsbactin and Acinetobactin Compete for the Periplasmic Siderophore Binding Protein BauB in Pathogenic Acinetobacter Baumannii. ACS Chem. Biol. 2019, 14, 674–687. [Google Scholar] [CrossRef]
- Mihara, K.; Tanabe, T.; Yamakawa, Y.; Funahashi, T.; Nakao, H.; Narimatsu, S.; Yamamoto, S. Identification and Transcriptional Organization of a Gene Cluster Involved in Biosynthesis and Transport of Acinetobactin, a Siderophore Produced by Acinetobacter Baumannii ATCC 19606T. Microbiology 2004, 150, 2587–2597. [Google Scholar] [CrossRef]
- Moynié, L.; Serra, I.; Scorciapino, M.A.; Oueis, E.; Page, M.G.; Ceccarelli, M.; Naismith, J.H. Preacinetobactin Not Acinetobactin Is Essential for Iron Uptake by the BauA Transporter of the Pathogen Acinetobacter Baumannii. eLife 2018, 7, e42270. [Google Scholar] [CrossRef]
- Song, W.Y.; Kim, H.J. Current Biochemical Understanding Regarding the Metabolism of Acinetobactin, the Major Siderophore of the Human Pathogen: Acinetobacter Baumannii, and Outlook for Discovery of Novel Anti-Infectious Agents Based Thereon. Nat. Prod. Rep. 2020, 37, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.C.; Bohac, T.J.; Shapiro, J.A.; Giblin, D.E.; Wencewicz, T.A.; Gulick, A.M. Crystal Structure of the Siderophore Binding Protein BauB Bound to an Unusual 2:1 Complex between Acinetobactin and Ferric Iron. Biochemistry 2018, 57, 6653–6661. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.A.; Wencewicz, T.A. Structure-Function Studies of Acinetobactin Analogs. Metallomics 2017, 9, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.M.; Pierce, S.; Angeles-Boza, A.M.; Peczuh, M.W. Synthesis and Characterization of Preacinetobactin and 5-Phenyl Preacinetobactin. Molecules 2022, 27, 3688. [Google Scholar] [CrossRef]
- Proschak, A.; Lubuta, P.; Grün, P.; Löhr, F.; Wilharm, G.; De Berardinis, V.; Bode, H.B. Structure and Biosynthesis of Fimsbactins A-F, Siderophores from Acinetobacter Baumannii and Acinetobacter Baylyi. ChemBioChem 2013, 14, 633–638. [Google Scholar] [CrossRef]
- Gordon, D.M.; Cowling, A. The Distribution and Genetic Structure of Escherichia coli in Australian Vertebrates: Host and Geographic Effects. Microbiology 2003, 149, 3575–3586. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.D. The Indigenous Gastrointestinal Microflora. Trends Microbiol. 1994, 4, 430–435. [Google Scholar] [CrossRef]
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The Population Genetics of Commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef]
- Terlizzi, M.E.; Gribaudo, G.; Maffei, M.E. UroPathogenic Escherichia coli (UPEC) Infections: Virulence Factors, Bladder Responses, Antibiotic, and Non-Antibiotic Antimicrobial Strategies. Front. Microbiol. 2017, 8, 1566. [Google Scholar] [CrossRef]
- Totsika, M.; Gomes Moriel, D.; Idris, A.; Rogers, B.A.; Wurpel, D.J.; Phan, M.-D.; Paterson, D.L.; Schembri, M.A. Uropathogenic Escherichia coli Mediated Urinary Tract Infection. Curr. Drug Targets 2012, 13, 1386–1399. [Google Scholar] [CrossRef]
- Ratledge, C.; Dover, L.G. Iron Metabolism in Pathogenic Bacteria. Annu. Rev. Microbiol. 2000, 54, 881–941. [Google Scholar] [CrossRef] [PubMed]
- Hantke, K.; Nicholson, G.; Rabsch, W.; Winkelmann, G. Salmochelins, Siderophores of Salmonella Enterica and Uropathogenic Escherichia coli Strains, Are Recognized by the Outer Membrane Receptor IroN. Proc. Natl. Acad. Sci. USA 2003, 100, 3677–3682. [Google Scholar] [CrossRef] [PubMed]
- Carniel, E. The Yersinia High-Pathogenicity Island: An Iron-Uptake Island. Microbes Infect. 2001, 3, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R. Virulence Factors in Escherichia coli Urinary Tract Infection. Clin. Microbiol. Rev. 1991, 4, 80–128. [Google Scholar] [CrossRef] [PubMed]
- Fecker, L.; Braun, V. Cloning and Expression of the Fhu Genes Involved in Iron(III)-Hydroxamate Uptake by Escherichia coli. J. Bacteriol. 1983, 156, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Hantke, K. Identification of an Iron Uptake System Specific for Coprogen and Rhodotorulic Acid in Escherichia coli K12. Mol. Gen. Genet. 1983, 191, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Neilands, J.B. A Crystalline Organo-Iron Pigment from a Rust Fungus (Ustilago Sphaerogena). J. Am. Chem. Soc. 1952, 74, 4846–4847. [Google Scholar] [CrossRef]
- Hesseltine, C.W.; Pidacks, C.; Whitehill, A.R.; Bohonos, N.; Hutchings, B.L.; Williams, J.H. Coprogen, a new growth factor for coprophilic fungi. J. Am. Chem. Soc. 1952, 74, 1362. [Google Scholar] [CrossRef]
- Bickel, H.; Hall, G.E.; Keller-Schierlein, W.; Prelog, V.; Vischer, E.; Wettstein, A. Stoffwechselprodukte von Actinomyceten. 27. Mitteilung. Über Die Konstitution von Ferrioxamin B. Helv. Chim. Acta 1960, 43, 2129–2138. [Google Scholar] [CrossRef]
- Anderegg, G.; L’Eplattenier, F.; Schwarzenbach, G. Hydroxamatkomplexe III. Eisen(III)-Austausch Zwischen Sideraminen Und Komplexonen. Diskussion Der Bildungskonstanten Der Hydroxamatkomplexe. Helv. Chim. Acta 1963, 46, 1409–1422. [Google Scholar] [CrossRef]
- van der Helm, D.; Baker, J.R.; Eng-Wilmot, D.L.; Hossain, B.; Loghry, R.A. Crystal Structure of Ferrichrome and a Comparison with the Structure of Ferrichrome A. J. Am. Chem. Soc. 1980, 102, 4224–4231. [Google Scholar] [CrossRef]
- Carrano, C.J.; Raymond, K.N. Coordination Chemistry of Microbial Iron Transport Compounds. 10. Characterization of the Complexes of Rhodotorulic Acid, a Dihydroxamate Siderophore. J. Am. Chem. Soc. 1978, 100, 5371–5374. [Google Scholar] [CrossRef]
- Stefanska, A.L.; Cassels, R.; Ready, S.J.; Warr, S.R. SB-203207 and SB-203208, Two Novel Isoleucyl TRNA Synthetase Inhibitors from a Streptomyces Sp. I. Fermentation, Isolation and Properties. J. Antibiot. 2000, 53, 357–363. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Hofmann, E.; Coulton, J.W.; Diederichs, K.; Welte, W. Siderophore-Mediated Iron Transport: Crystal Structure of FhuA with Bound Lipopolysaccharide. Science 1998, 282, 2215–2220. [Google Scholar] [CrossRef]
- Haas, H. Fungal Siderophore Metabolism with a Focus on Aspergillus Fumigatus. Nat. Prod. Rep. 2014, 31, 1266–1276. [Google Scholar] [CrossRef]
- Killmann, H.; Benz, R.; Braun, V. Conversion of the FhuA Transport Protein into a Diffusion Channel through the Outer Membrane of Escherichia coli. EMBO J. 1993, 12, 3007–3016. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, A.D.; Coulton, J.W.; Diederichs, K.; Welte, W.; Braun, V.; Fiedler, H. Crystal Structure of the Antibiotic Albomycin in Complex with the Outer Membrane Transporter FhuA. Protein Sci. 2000, 9, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Killmann, H.; Herrmann, C.; Wolff, H.; Braun, V. Identification of a New Site for Ferrichrome Transport by Comparison of the FhuA Proteins of Escherichia coli, Salmonella Paratyphi B, Salmonella Typhimurium, and Pantoea agglomerans. J. Bacteriol. 1998, 180, 3845–3852. [Google Scholar] [CrossRef]
- Renshaw, J.C.; Robson, G.D.; Trinci, A.P.J.; Wiebe, M.G.; Livens, F.R.; Collison, D.; Taylor, R.J. Fungal Siderophores: Structures, Functions and Applications. Mycol. Res. 2002, 106, 1123–1142. [Google Scholar] [CrossRef]
- Grinter, R.; Lithgow, T. Determination of the Molecular Basis for Coprogen Import by Gram-Negative Bacteria. IUCrJ 2019, 6, 401–411. [Google Scholar] [CrossRef]
- Matzanke, B.F.; Miller, G.I.; Raymond, K.N. Hydroxamate siderophore mediated iron uptake in E. coli: Stereospecific recognition of ferric rhodotorulic acid. Biochem. Biophys. Res. Commun. 1984, 121, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.E.; Ku, S.-Y.; Dougan, D.R.; Vogel, H.J.; Tari, L.W. The Structure of the Ferric Siderophore Binding Protein FhuD Complexed with Gallichrome. Nat. Struct. Biol. 2000, 7, 287–291. [Google Scholar]
- Clarke, T.E.; Braun, V.; Winkelmann, G.; Tari, L.W.; Vogel, H.J. X-ray Crystallographic Structures of the Escherichia coli Periplasmic Protein FhuD Bound to Hydroxamate-Type Siderophores and the Antibiotic Albomycin. J. Biol. Chem. 2002, 277, 13966–13972. [Google Scholar] [CrossRef]
- Hu, W.; Zheng, H. Cryo-EM Reveals Unique Structural Features of the FhuCDB Escherichia coli Ferrichrome Importer. Commun. Biol. 2021, 4, 1383. [Google Scholar] [CrossRef]
- Pravda, L.; Sehnal, D.; Toušek, D.; Navrátilová, V.; Bazgier, V.; Berka, K.; Vařeková, R.S.; Koča, J.; Otyepka, M. MOLEonline: A Web-Based Tool for Analyzing Channels, Tunnels and Pores (2018 Update). Nucleic Acids Res. 2018, 46, W368–W373. [Google Scholar] [CrossRef]
- Matzanke, B.F.; Berner, I.; Bill, E.; Trautwein, A.X.; Winkelmann, G. Transport and Utilization of Ferrioxamine-E-Bound Iron in Erwinia Herbicola (Pantoea agglomerans). Biol. Metals 1991, 4, 181–185. [Google Scholar] [CrossRef]
- Matzanke, B.F.; Bill, E.; Trautwein, A.X.; Winkelmann, G. Siderophores as iron storage compounds in the yeasts rhodotorula minuta and ustilago sphaerogena detected by In Vivo mossbauer spectroscopy. Hyperfine Interact 1990, 58, 2359–2364. [Google Scholar] [CrossRef]
- Rauscher, L.; Expert, D.; Matzanke, B.F.; Trautwein, A.X. Chrysobactin-Dependent Iron Acquisition in Erwinia Chrysanthemi. Functional Study of a Homolog of the Escherichia coli Ferric Enterobactin Esterase. J. Biol. Chem. 2002, 277, 2385–2395. [Google Scholar] [CrossRef]
- Pollack, J.R.; Neilands, J.B. Enterobactin, an iron transport comkxind from salmonella typhimurium. Biochem. Biophys. Res. Commun. 1970, 38, 989–992. [Google Scholar] [CrossRef]
- O’Brien, I.G.; Gibson, F. The Structure of Enterochelin and Related 2,3-Dihydroxy-N-Benzoylserine Conjugates from Escherichia coli. Biochim. Biophys. Acta 1970, 215, 393–402. [Google Scholar] [CrossRef]
- Poole, K.; Young, L.; Neshat, S. Enterobactin-Mediated Iron Transport in Pseudomonas aeruginosa. J. Bacteriol. 1990, 172, 6991–6996. [Google Scholar] [CrossRef]
- Loomis, L.D.; Raymond, K.N. Solution Equilibria of Enterobactin and Metal-Enterobactin Complexes. Inorg. Chem. 1991, 30, 906–911. [Google Scholar] [CrossRef]
- Usher, K.C.; Özkan, E.; Gardner, K.H.; Deisenhofer, J. The Plug Domain of FepA, a TonB-Dependent Transport Protein from Escherichia coli, Binds Its Siderophore in the Absence of the Transmembrane Barrel Domain. Proc. Natl. Acad. Sci. USA 2001, 98, 10676–10681. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, S.K.; Smith, B.S.; Venkatramani, L.; Xia, D.; Esser, L.; Palnitkar, M.; Chakraborty, R.; van der Helm, D.; Deisenhofer, J. Crystal Structure of the Outer Membrane Active Transporter FepA from Escherichia coli. Nat. Struct. Biol. 1999, 6, 56–63. [Google Scholar] [CrossRef]
- Cao, Z.; Qi, Z.; Sprencel, C.; Newton, S.M.C.; Klebba, P.E. Aromatic Components of Two Ferric Enterobactin Binding Sites in Escherichia coli FepA. Mol. Microbiol. 2000, 37, 1306–1317. [Google Scholar] [CrossRef]
- Rabsch, W.; Voigt, W.; Reissbrodt, R.; Tsolis, R.M.; Bäumler, A.J. Salmonella typhimurium IroN and FepA Proteins Mediate Uptake of Enterobactin but Differ in Their Specificity for Other Siderophores. J. Bacteriol. 1999, 181, 3610–3612. [Google Scholar] [CrossRef]
- Nikaido, H.; Rosenberg, E.Y. Cir and Fiu Proteins in the Outer Membrane of Escherichia coli Catalyze Transport of Monomeric Catechols: Study with 1-Lactam Antibiotics Containing Catechol and Analogous Groups. J. Bacteriol. 1990, 172, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Agarwal, V.; Tikhonov, A.; Metlitskaya, A.; Severinov, K.; Nair, S.K. Structure and Function of a Serine Carboxypeptidase Adapted for Degradation of the Protein Synthesis Antibiotic Microcin C7. Proc. Natl. Acad. Sci. USA 2012, 109, 4425–4430. [Google Scholar] [CrossRef]
- Critchley, L.A.; Basker, M.J.; Edmoodson, R.A.; Knott, S.J. Uptake of a Catecholic Cephalosporin by the Iron Transport System of Escherichia coli. J. Antimicrob. Chemother. 1991, 28, 377–388. [Google Scholar] [CrossRef]
- Braun, V.; Braun, M. Active Transport of Iron and Siderophore Antibiotics. Curr. Opin. Microbiol. 2002, 5, 194–201. [Google Scholar] [CrossRef]
- Destoumieux-Garzón, D.; Peduzzi, J.; Thomas, X.; Djediat, C.; Rebuffat, S. Parasitism of Iron-Siderophore Receptors of Escherichia coli by the Siderophore-Peptide Microcin E492m and Its Unmodified Counterpart. BioMetals 2006, 19, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.K.; Francis, C.L.; McIntosh, M.A. Molecular Analysis of the Escherichia coli Ferric Enterobactin Receptor FepA. J. Biol. Chem. 1990, 265, 14536–14543. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.R.; Pickett, C.L.; Earhart, C.F. Two Fep Genes Are Required for Ferrienterochelin Uptake in Escherichia coli K-12. J. Bacteriol. 1983, 155, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Grinter, R.; Lithgow, T. The Structure of the Bacterial Iron-Catecholate Transporter Fiu Suggests That It Imports Substrates via a Two-Step Mechanism. J. Biol. Chem. 2019, 294, 19523–19534. [Google Scholar] [CrossRef]
- Konisky, J.; Cowell, B.S. Interaction of Colicin Ia with Bacterial Cells. J. Biol. Chem. 1972, 24, 6524–6529. [Google Scholar] [CrossRef]
- Konisky, J. Characterization of Colicin Ia and Colicin Ib. J. Biol. Chem. 1972, 247, 3750–3755. [Google Scholar] [CrossRef] [PubMed]
- Wiener, M.; Freymann, D.; Ghosh, P.; Stroud, R.M. Crystal Structure of Colicin Ia. Nature 1997, 385, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Stroud, R.M.; Reiling, K.; Wiener, M. Douglas Freymann Ion-Channel-Forming Colicins. Curr. Opin. Struct. Biol. 1998, 8, 525–533. [Google Scholar] [CrossRef]
- Jakes, K.S.; Kienker, P.K.; Finkelstein, A. Channel-Forming Colicins: Translocation (and Other Deviant Behaviour) Associated with Colicin Ia Channel Gating. Q. Rev. Biophys. 1999, 32, 189–205. [Google Scholar] [CrossRef]
- Buchanan, S.K.; Lukacik, P.; Grizot, S.; Ghirlando, R.; Ali, M.M.U.; Barnard, T.J.; Jakes, K.S.; Kienker, P.K.; Esser, L. Structure of Colicin I Receptor Bound to the R-Domain of Colicin Ia: Implications for Protein Import. EMBO J. 2007, 26, 2594–2604. [Google Scholar] [CrossRef]
- Stephens, D.L.; Choe, M.D.; Earhart, C.F. Escherichia coli Periplasmic Protein FepB Binds Ferrienterobactin. Microbiology 1995, 141, 1647–1654. [Google Scholar] [CrossRef]
- Chenault, S.S.; Earhart, C.F. Organization of Genes Encoding Membrane Proteins of the Escherichia coli Ferrienterobactin Permease. Mol. Microbiol. 1991, 5, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Chenault, S.S.; Earhart, C.F. Identification of Hydrophobic Proteins FepD and FepG of the Escherichia coli Ferrienterobactin Permease. J. Gen. Microbiol. 1992, 138, 21–67. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, N.; Yue, Y.; Liu, X.; Huang, Y.; Gu, L.; Xu, S. An Unusual Crystal Structure of Ferric-Enterobactin Bound FepB Suggests Novel Functions of FepB in Microbial Iron Uptake. Biochem. Biophys. Res. Commun. 2016, 478, 1049–1053. [Google Scholar] [CrossRef]
- Wyckoff, E.E.; Valle, A.-M.; Smith, S.L.; Payne, S.M. A Multifunctional ATP-Binding Cassette Transporter System from Vibrio Cholerae Transports Vibriobactin and Enterobactin. J. Bacteriol. 1999, 181, 7588–7596. [Google Scholar] [CrossRef]
- Sprencel, C.; Cao, Z.; Qi, Z.; Scott, D.C.; Montague, M.A.; Ivanoff, N.; Xu, J.; Raymond, K.M.; Newton, S.M.C.; Klebba, P.E. Binding of Ferric Enterobactin by the Escherichia coli Periplasmic Protein FepB. J. Bacteriol. 2000, 182, 5359–5364. [Google Scholar] [CrossRef]
- Brickman, T.J.; Mcintosh, M.A. Overexpression and Purification of Ferric Enterobactin Esterase from Escherichia coli. Demonstration of Enzymatic Hydrolysis of Enterobactin and Its Iron Complex. J. Biol. Chem. 1992, 267, 12350–12355. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Fischbach, M.A.; Liu, D.R.; Walsh, C.T. In Vitro Characterization of Salmochelin and Enterobactin Trilactone Hydrolases IroD, IroE, and Fes. J. Am. Chem. Soc. 2005, 127, 11075–11084. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; La Rosa, R.; Bartell, J.A.; Marvig, R.L.; Haagensen, J.A.J.; Sommer, L.M.; Molin, S.; Johansen, H.K. Pseudomonas Aeruginosa Adaptation and Evolution in Patients with Cystic Fibrosis. Nat. Rev. Microbiol. 2021, 19, 331–342. [Google Scholar] [CrossRef]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas Aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef]
- del Cendra, M.M.; Torrents, E. Pseudomonas Aeruginosa Biofilms and Their Partners in Crime. Biotechnol. Adv. 2021, 49, 107734. [Google Scholar] [CrossRef]
- Botelho, J.; Grosso, F.; Peixe, L. Antibiotic Resistance in Pseudomonas Aeruginosa—Mechanisms, Epidemiology and Evolution. Drug Resist. Updat. 2019, 44, 100640. [Google Scholar] [CrossRef]
- Haque, M.; Sartelli, M.; McKimm, J.; Bakar, M.A. Health Care-Associated Infections—An Overview. Infect. Drug Resist. 2018, 11, 2321–2333. [Google Scholar] [CrossRef]
- Feng, W.; Sun, F.; Wang, Q.; Xiong, W.; Qiu, X.; Dai, X.; Xia, P. Epidemiology and Resistance Characteristics of Pseudomonas Aeruginosa Isolates from the Respiratory Department of a Hospital in China. J. Glob. Antimicrob. Resist. 2017, 8, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.F.; Kvitko, B.; He, S.Y. Pseudomonas Syringae: What It Takes to Be a Pathogen. Nat. Rev. Microbiol. 2018, 16, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H.; Nikaido, K.; Harayama, S. Identification and Characterization of Porins in Pseudomonas Aeruginosa. J. Biol. Chem. 1991, 266, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, W. Penetration of Beta-Lactam Antibiotics into Their Target Enzymes in Pseudomonas Aeruginosa: Comparison of a Highly Sensitive Mutant with Its Parent Strain. Antimicrob. Agents Chemother. 1980, 18, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Blomquist, K.C.; Nix, D.E. A Critical Evaluation of Newer β-Lactam Antibiotics for Treatment of Pseudomonas Aeruginosa Infections. Ann. Pharmacother. 2021, 55, 1010–1024. [Google Scholar] [CrossRef]
- Cox, C.D.; Rinehart, K.L.; Mooret, M.L.; Cook, J.C. Pyochelin: Novel Structure of an Iron-Chelating Growth Promoter for Pseudomonas Aeruginosa. Proc. Natl. Acad. Sci. USA 1981, 78, 4256–4260. [Google Scholar] [CrossRef] [PubMed]
- Albrecht-Gary, A.-M.; Blanc, S.; Rochel, N.; Ocaktan, A.Z.; Abdallah, M.A. Bacterial Iron Transport: Coordination Properties of Pyoverdin PaA, a Peptidic Siderophore of Pseudomonas Aeruginosa. Inorg. Chem. 1994, 33, 6391–6402. [Google Scholar] [CrossRef]
- Brandel, J.; Humbert, N.; Elhabiri, M.; Schalk, I.J.; Mislin, G.L.A.; Albrecht-Gary, A.M. Pyochelin, a Siderophore of Pseudomonas Aeruginosa: Physicochemical Characterization of the Iron(III), Copper(II) and Zinc(II) Complexes. Dalton Trans. 2012, 41, 2820–2834. [Google Scholar] [CrossRef] [PubMed]
- Visca, P.; Imperi, F.; Lamont, I.L. Pyoverdine Siderophores: From Biogenesis to Biosignificance. Trends Microbiol. 2007, 15, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Demange, P.; Wendenbaum, S.; Linget, C.; Mertz, C.; Cung, M.T.; Dell, A.; Abdallah, M.A. Bacterial Siderophores: Structure and NMR Assignment of Pyoverdins Pa, Siderophores of Pseudomonas Aeruginosa ATCC 15692. Biol. Metals 1990, 3, 155–170. [Google Scholar] [CrossRef]
- Wasielewski, E.; Atkinson, R.A.; Abdallah, M.A.; Kieffer, B. The Three-Dimensional Structure of the Gallium Complex of Azoverdin, a Siderophore of Azomonas Macrocytogenes ATCC 12334, Determined by NMR Using Residual Dipolar Coupling Constants. Biochemistry 2002, 41, 12488–12497. [Google Scholar] [CrossRef] [PubMed]
- Budzikiewicz, H.; Schäfer, M.; Fernández, D.U.; Matthijs, S.; Cornelis, P. Characterization of the Chromophores of Pyoverdins and Related Siderophores by Electrospray Tandem Mass Spectrometry. BioMetals 2007, 20, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Wirth, C.; Meyer-Klaucke, W.; Pattus, F.; Cobessi, D. From the Periplasmic Signaling Domain to the Extracellular Face of an Outer Membrane Signal Transducer of Pseudomonas Aeruginosa: Crystal Structure of the Ferric Pyoverdine Outer Membrane Receptor. J. Mol. Biol. 2007, 368, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Poole, K.; Neshat, S.; Heinrichs, D. Pyoverdine-Mediated Iron Transport in Pseudomonas Aeruginosa: Involvement of a High-Molecular-Mass Outer Membrane Protein. FEMS Microbiol. Lett. 1991, 78, 1–6. [Google Scholar] [CrossRef]
- Ghysels, B.; Dieu, B.T.M.; Beatson, S.A.; Pirnay, J.P.; Ochsner, U.A.; Vasil, M.L.; Cornelis, P. FpvB, an Alternative Type I Ferripyoverdine Receptor of Pseudomonas Aeruginosa. Microbiology 2004, 150, 1671–1680. [Google Scholar] [CrossRef]
- Greenwald, J.; Nader, M.; Celia, H.; Gruffaz, C.; Geoffroy, V.; Meyer, J.M.; Schalk, I.J.; Pattus, F. FpvA Bound to Non-Cognate Pyoverdines: Molecular Basis of Siderophore Recognition by an Iron Transporter. Mol. Microbiol. 2009, 72, 1246–1259. [Google Scholar] [CrossRef]
- Meyer, J.-M.; Stintzi, A.; Poole, K. The Ferripyoverdine Receptor FpvA of Pseudomonas Aeruginosa PAO1 Recognizes the Ferripyoverdines of P. Aeruginosa PAO1 and P. Fluorescens ATCC 13525. FEMS Microbiol. Lett. 1999, 170, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.; Schäfer, M.; Geoffroy, V.; Meyer, J.-M. Siderotyping-A Powerful Tool for the Characterization of Pyoverdines. Curr. Top. Med. Chem. 2001, 1, 31–57. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.K.; Burrows, L.L. Pseudomonas Aeruginosa FpvB Is a High-Affinity Transporter for Xenosiderophores Ferrichrome and Ferrioxamine B. mBio 2023, 14, e0314922. [Google Scholar] [CrossRef] [PubMed]
- Brillet, K.; Ruffenach, F.; Adams, H.; Journet, L.; Gasser, V.; Hoegy, F.; Guillon, L.; Hannauer, M.; Page, A.; Schalk, I.J. An ABC Transporter with Two Periplasmic Binding Proteins Involved in Iron Acquisition in Pseudomonas Aeruginosa. ACS Chem. Biol. 2012, 7, 2036–2045. [Google Scholar] [CrossRef]
- Yeterian, E.; Martin, L.W.; Lamont, I.L.; Schalk, I.J. An Efflux Pump Is Required for Siderophore Recycling by Pseudomonas Aeruginosa. Environ. Microbiol. Rep. 2010, 2, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Hannauer, M.; Yeterian, E.; Martin, L.W.; Lamont, I.L.; Schalk, I.J. An Efflux Pump Is Involved in Secretion of Newly Synthesized Siderophore by Pseudomonas Aeruginosa. FEBS Lett. 2010, 584, 4751–4755. [Google Scholar] [CrossRef]
- Tseng, C.F.; Burger, A.; Mislin, G.L.A.; Schalk, I.J.; Yu, S.S.F.; Chan, S.I.; Abdallah, M.A. Bacterial Siderophores: The Solution Stoichiometry and Coordination of the Fe(III) Complexes of Pyochelin and Related Compounds. J. Biol. Inorg. Chem. 2006, 11, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Namiranian, S.; Richardson, D.J.; Russell, D.A.; Sodeau, J.R. Excited State Properties of the Siderophore Pyochelin and Its Complex with Zinc Ions. Photochem. Photobiol. 1997, 65, 777–782. [Google Scholar] [CrossRef]
- Cobessi, D.; Celia, H.; Pattus, F. Crystal Structure at High Resolution of Ferric-Pyochelin and Its Membrane Receptor FptA from Pseudomonas Aeruginosa. J. Mol. Biol. 2005, 352, 893–904. [Google Scholar] [CrossRef]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.L.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete Genome Sequence of Pseudomonas Aeruginosa PAO1, an Opportunistic Pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Michel, L.; Bachelard, A.; Reimmann, C. Ferripyochelin Uptake Genes Are Involved in Pyochelin-Mediated Signalling in Pseudomonas Aeruginosa. Microbiology 2007, 153, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Cuív, P.Ó.; Clarke, P.; Lynch, D.; O’Connell, M. Identification of RhtX and FptX, Novel Genes Encoding Proteins That Show Homology and Function in the Utilization of the Siderophores Rhizobactin 1021 by Sinorhizobium Meliloti and Pyochelin by Pseudomonas Aeruginosa, Respectively. J. Bacteriol. 2004, 186, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, J.; Cheng, J.; Zeng, J.; Ma, X.; Lin, J. PQS and Pyochelin in Pseudomonas Aeruginosa Share Inner Membrane Transporters to Mediate Iron Uptake. Microbiol. Spectr. 2024, 12, e03256-23. [Google Scholar] [CrossRef] [PubMed]
- Roche, B.; Garcia-Rivera, M.A.; Normant, V.; Kuhn, L.; Hammann, P.; Brönstrup, M.; Mislin, G.L.A.; Schalk, I.J. A Role for PchHI as the ABC Transporter in Iron Acquisition by the Siderophore Pyochelin in Pseudomonas Aeruginosa. Environ. Microbiol. 2022, 24, 866–877. [Google Scholar] [CrossRef]
- Dean, C.R.; Neshat, S.; Poole, K. PfeR, an Enterobactin-Responsive Activator of Ferric Enterobactin Receptor Gene Expression in Pseudomonas Aeruginosa. J. Bacteriol. 1996, 178, 5361–5369. [Google Scholar] [CrossRef] [PubMed]
- Ghysels, B.; Ochsner, U.; Möllman, U.; Heinisch, L.; Vasil, M.; Cornelis, P.; Matthijs, S. The Pseudomonas Aeruginosa Pir A Gene Encodes a Second Receptor for Ferrienterobactin and Synthetic Catecholate Analogues. FEMS Microbiol. Lett. 2005, 246, 167–174. [Google Scholar] [CrossRef]
- Moynié, L.; Milenkovic, S.; Mislin, G.L.A.; Gasser, V.; Malloci, G.; Baco, E.; McCaughan, R.P.; Page, M.G.P.; Schalk, I.J.; Ceccarelli, M.; et al. The Complex of Ferric-Enterobactin with Its Transporter from Pseudomonas Aeruginosa Suggests a Two-Site Model. Nat. Commun. 2019, 10, 3673. [Google Scholar] [CrossRef] [PubMed]
- Gasser, V.; Baco, E.; Cunrath, O.; August, P.S.; Perraud, Q.; Zill, N.; Schleberger, C.; Schmidt, A.; Paulen, A.; Bumann, D.; et al. Catechol Siderophores Repress the Pyochelin Pathway and Activate the Enterobactin Pathway in Pseudomonas Aeruginosa: An Opportunity for Siderophore-Antibiotic Conjugates Development. Environ. Microbiol. 2016, 18, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Cornish, A.S.; Page, W.J. The Catecholate Siderophores of Azotobacter Vinelandii: Their Affinity for Iron and Role in Oxygen Stress Management. Microbiology 1998, 144, 1747–1748. [Google Scholar] [CrossRef]
- Miller, M.J.; Walz, A.J.; Zhu, H.; Wu, C.; Moraski, G.; Möllmann, U.; Tristani, E.M.; Crumbliss, A.L.; Ferdig, M.T.; Checkley, L.; et al. Design, Synthesis, and Study of a Mycobactin-Artemisinin Conjugate That Has Selective and Potent Activity against Tuberculosis and Malaria. J. Am. Chem. Soc. 2011, 133, 2076–2079. [Google Scholar] [CrossRef]
- Faurant, C. From Bark to Weed: The History of Artemisinin. Parasite 2011, 18, 215–218. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Malaria Report 2023. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2023 (accessed on 17 July 2024).
- Kong, K.F.; Schneper, L.; Mathee, K. Beta-Lactam Antibiotics: From Antibiosis to Resistance and Bacteriology. APMIS 2010, 118, 1–36. [Google Scholar] [CrossRef]
- Drlica, K.; Hiasa, H.; Kerns, R.; Malik, M.; Mustaev, A.; Zhao, X. Quinolones: Action and Resistance Updated. Curr. Top. Med. Chem. 2009, 9, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-Mediated Bacterial Death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mechanisms of Action of Antimicrobials: Focus on Fluoroquinolones inhibition of cell wall synthesis. Mech. Fluoroquinolone Action CID 2001, 32, 9–15. [Google Scholar]
- Anderson, V.E.; Osheroff, N. Type II Topoisomerases as Targets for Quinolone Antibacterials: Turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 2001, 7, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, J.K.; Miller, K.; O’Neill, A.J.; Chopra, I. Consequences of Daptomycin-Mediated Membrane Damage in Staphylococcus Aureus. J. Antimicrob. Chemother. 2008, 62, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.D.; Palmer, M. The Action Mechanism of Daptomycin. Bioorg. Med. Chem. 2016, 24, 6253–6268. [Google Scholar] [CrossRef] [PubMed]
- Foti, C.; Piperno, A.; Scala, A.; Giuffrè, O. Oxazolidinone Antibiotics: Chemical, Biological and Analytical Aspects. Molecules 2021, 26, 4280. [Google Scholar] [CrossRef]
- Maraskolhe, D.L.; Deotale, V.S.; Mendiratta, D.K.; Narang, P. Comparision of Three Laboratory Tests for Detection of Ampc β Lactamases in Klebsiella Species and E. coli. J. Clin. Diagn. Res. 2014, 8, DC05–DC08. [Google Scholar] [CrossRef]
- Andrew, B.; Kagirita, A.; Bazira, J. Prevalence of Extended-Spectrum Beta-Lactamases-Producing Microorganisms in Patients Admitted at KRRH, Southwestern Uganda. Int. J. Microbiol. 2017, 2017, 3183076. [Google Scholar] [CrossRef] [PubMed]
- Andriole, V.T. The Quinolones: Past, Present, and Future. Clin. Infect. Dis. 2005, 41, S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A. Global Fluoroquinolone Resistance Epidemiology and Implictions for Clinical Use. Interdiscip. Perspect. Infect. Dis. 2012, 2012, 976273. [Google Scholar] [CrossRef]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural Basis of Quinolone Inhibition of Type IIA Topoisomerases and Target-Mediated Resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef]
- Aldred, K.J.; McPherson, S.A.; Wang, P.; Kerns, R.J.; Graves, D.E.; Turnbough, C.L.; Osheroff, N. Drug Interactions with Bacillus Anthracis Topoisomerase IV: Biochemical Basis for Quinolone Action and Resistance. Biochemistry 2012, 51, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; McPherson, S.A.; Turnbough, C.L.; Kerns, R.J.; Osheroff, N. Topoisomerase IV-Quinolone Interactions Are Mediated through a Water-Metal Ion Bridge: Mechanistic Basis of Quinolone Resistance. Nucleic Acids Res. 2013, 41, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Braun, V.; Gunthner, K.; Hantke, K.; Zimmermann, L. Intracellular Activation of Albomycin in Escherichia coli and Salmonella Typhimurium. J. Bacteriol. 1983, 156, 308–315. [Google Scholar] [CrossRef]
- Ji, C.; Miller, M.J. Siderophore-Fluoroquinolone Conjugates Containing Potential Reduction-Triggered Linkers for Drug Release: Synthesis and Antibacterial Activity. BioMetals 2015, 28, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Ghosh, M.; Niu, C.; Malouin, F.; Moellmann, U.; Miller, M.J. Iron Transport-Mediated Drug Delivery Using Mixed-Ligand Siderophore-Beta-Lactam Conjugates. Chem. Biol. 1996, 3, 1011–1019. [Google Scholar] [CrossRef]
- Wencewicz, T.A.; Miller, M.J. Biscatecholate-Monohydroxamate Mixed Ligand Siderophore-Carbacephalosporin Conjugates Are Selective Sideromycin Antibiotics That Target Acinetobacter Baumannii. J. Med. Chem. 2013, 56, 4044–4052. [Google Scholar] [CrossRef]
- Ghosh, M.; Miller, P.A.; Möllmann, U.; Claypool, W.D.; Schroeder, V.A.; Wolter, W.R.; Suckow, M.; Yu, H.; Li, S.; Huang, W.; et al. Targeted Antibiotic Delivery: Selective Siderophore Conjugation with Daptomycin Confers Potent Activity against Multidrug Resistant Acinetobacter Baumannii Both In Vitro and In Vivo. J. Med. Chem. 2017, 60, 4577–4583. [Google Scholar] [CrossRef] [PubMed]
- Randall, C.P.; Mariner, K.R.; Chopra, I.; O’Neill, A.J. The Target of Daptomycin Is Absent from Escherichia coli and Other Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2013, 57, 637–639. [Google Scholar] [CrossRef]
- Zheng, T.; Nolan, E.M. Enterobactin-Mediated Delivery of ß-Lactam Antibiotics Enhances Antibacterial Activity against Pathogenic Escherichia coli. J. Am. Chem. Soc. 2014, 136, 9677–9691. [Google Scholar] [CrossRef]
- Troxell, B.; Hassan, H.M. Transcriptional Regulation by Ferric Uptake Regulator (Fur) in Pathogenic Bacteria. Front. Cell. Infect. Microbiol. 2013, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Bullock, J.L.; Nolan, E.M. Siderophore-Mediated Cargo Delivery to the Cytoplasm of Escherichia coli and Pseudomonas Aeruginosa: Syntheses of Monofunctionalized Enterobactin Scaffolds and Evaluation of Enterobactin-Cargo Conjugate Uptake. J. Am. Chem. Soc. 2012, 134, 18388–18400. [Google Scholar] [CrossRef] [PubMed]
- Neumann, W.; Sassone-Corsi, M.; Raffatellu, M.; Nolan, E.M. Esterase-Catalyzed Siderophore Hydrolysis Activates an Enterobactin-Ciprofloxacin Conjugate and Confers Targeted Antibacterial Activity. J. Am. Chem. Soc. 2018, 140, 5193–5201. [Google Scholar] [CrossRef]
- Wiles, T.J.; Kulesus, R.R.; Mulvey, M.A. Origins and Virulence Mechanisms of Uropathogenic Escherichia coli. Exp. Mol. Pathol. 2008, 85, 11–19. [Google Scholar] [CrossRef]
- Henderson, J.P.; Crowley, J.R.; Pinkner, J.S.; Walker, J.N.; Tsukayama, P.; Stamm, W.E.; Hooton, T.M.; Hultgren, S.J. Quantitative Metabolomics Reveals an Epigenetic Blueprint for Iron Acquisition in Uropathogenic Escherichia coli. PLoS Pathog. 2009, 5, e1000305. [Google Scholar] [CrossRef]
- Garcia, E.C.; Brumbaugh, A.R.; Mobley, H.L.T. Redundancy and Specificity of Escherichia coli Iron Acquisition Systems during Urinary Tract Infection. Infect. Immun. 2011, 79, 1225–1235. [Google Scholar] [CrossRef]
- Ji, C.; Miller, P.A.; Miller, M.J. Iron Transport-Mediated Drug Delivery: Practical Syntheses and In Vitro Antibacterial Studies of Tris-Catecholate Siderophore-Aminopenicillin Conjugates Reveals Selectively Potent Antipseudomonal Activity. J. Am. Chem. Soc. 2012, 134, 9898–9901. [Google Scholar] [CrossRef]
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An Archetype for Microbial Iron Transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef]
- Ghosh, M.; Lin, Y.M.; Miller, P.A.; Möllmann, U.; Boggess, W.C.; Miller, M.J. Siderophore Conjugates of Daptomycin Are Potent Inhibitors of Carbapenem Resistant Strains of Acinetobacter Baumannii. ACS Infect. Dis. 2018, 4, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, L.; Wittmann, S.; Stoiber, T.; Berg, A.; Ankel-Fuchs, D.; Möllmann, U. Highly Antibacterial Active Aminoacyl Penicillin Conjugates with Acylated Bis-Catecholate Siderophores Based on Secondary Diamino Acids and Related Compounds. J. Med. Chem. 2002, 10, 1659–1670. [Google Scholar] [CrossRef]
- Möllmann, U.; Heinisch, L.; Bauernfeind, A.; Köhler, T.; Ankel-Fuchs, D. Siderophores as Drug Delivery Agents: Application of the “Trojan Horse” Strategy. BioMetals 2009, 22, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Oki, N.; Aoki, B.; Kuroki, T.; Matsumoto, M.; Kojima, K.; Nehashi, T. Semisynthetic Beta-Lactam Antibiotics. III. Effect on Antibacterial Activity and Comt-Susceptibility of Chlorine-Introduction into the Catechol Nucleus of 6-[(R)-2-[3-(3,4-Dihydroxybenzoyl)-3-(3-Hydroxypropyl)-1-Ureido]-2-Phenylacetamido]penicillanic Acid. J. Antibiot. 1987, 40, 22–28. [Google Scholar] [CrossRef]
- Jungheim, L.N.; Shepherd, T.A. Design of Antitumor Prodrugs: Substrates for Antibody Targeted Enzymes. Chem. Rev. 1994, 94, 1553–1566. [Google Scholar] [CrossRef]
- Liu, R.; Miller, P.A.; Vakulenko, S.B.; Stewart, N.K.; Boggess, W.C.; Miller, M.J. A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria to Commit Suicide with a Gram-Positive Antibiotic. J. Med. Chem. 2018, 61, 3845–3854. [Google Scholar] [CrossRef]
- Rivault, F.; Liébert, C.; Burger, A.; Hoegy, F.; Abdallah, M.A.; Schalk, I.J.; Mislin, G.L.A. Synthesis of Pyochelin-Norfloxacin Conjugates. Bioorg. Med. Chem. Lett. 2007, 17, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Noël, S.; Gasser, V.; Pesset, B.; Hoegy, F.; Rognan, D.; Schalk, I.J.; Mislin, G.L.A. Synthesis and Biological Properties of Conjugates between Fluoroquinolones and a N3′′-Functionalized Pyochelin. Org. Biomol. Chem. 2011, 9, 8288–8300. [Google Scholar] [CrossRef]
- Paulen, A.; Hoegy, F.; Roche, B.; Schalk, I.J.; Mislin, G.L.A. Synthesis of Conjugates between Oxazolidinone Antibiotics and a Pyochelin Analogue. Bioorg. Med. Chem. Lett. 2017, 27, 4867–4870. [Google Scholar] [CrossRef]
- Ghosh, M.; Miller, P.A.; Miller, M.J. Antibiotic Repurposing: Bis-Catechol- and Mixed Ligand (Bis-Catechol-Mono-Hydroxamate)-Teicoplanin Conjugates Are Active against Multidrug Resistant Acinetobacter Baumannii. J. Antibiot. 2020, 73, 152–157. [Google Scholar] [CrossRef]
- Kahne, D.; Leimkuhler, C.; Lu, W.; Walsh, C. Glycopeptide and Lipoglycopeptide Antibiotics. Chem. Rev. 2005, 105, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Domingues, S.; Lima, T.; Saavedra, M.J.; Da Silva, G.J. An Overview of Cefiderocol’s Therapeutic Potential and Underlying Resistance Mechanisms. Life 2023, 13, 1427. [Google Scholar] [CrossRef]
- Christensen, S.B. Drugs That Changed Society: History and Current Status of the Early Antibiotics: Salvarsan, Sulfonamides, and β-Lactams. Molecules 2021, 26, 6057. [Google Scholar] [CrossRef]
- Katsu, K.; Kitoh, K.; Inoue, M.; Mitsuhashi, S. In Vitro Antibacterial Activity of E-0702, a New Semisynthetic Cephalosporin. Antimicrob. Agents Chemother. 1982, 22, 181–185. [Google Scholar] [CrossRef]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin. Infect. Dis. 2019, 69, S538–S543. [Google Scholar] [CrossRef]
- Ito-Horiyama, T.; Ishii, Y.; Ito, A.; Sato, T.; Nakamura, R.; Fukuhara, N.; Tsuji, M.; Yamano, Y.; Yamaguchi, K.; Tateda, K. Stability of Novel Siderophore Cephalosporin S-649266 against Clinically Relevant Carbapenemases. Antimicrob. Agents Chemother. 2016, 60, 4384–4386. [Google Scholar] [CrossRef]
- Poirel, L.; Kieffer, N.; Nordmann, P. Stability of Cefiderocol against Clinically Significant Broad-Spectrum Oxacillinases. Int. J. Antimicrob. Agents 2018, 52, 866–867. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Yoshizawa, H.; Yamawaki, K.; Yokoo, K.; Sato, J.; Hisakawa, S.; Hasegawa, Y.; Kusano, H.; Sano, M.; Sugimoto, H.; et al. Cefiderocol (S-649266), A New Siderophore Cephalosporin Exhibiting Potent Activities against Pseudomonas Aeruginosa and Other Gram-Negative Pathogens Including Multi-Drug Resistant Bacteria: Structure Activity Relationship. Eur. J. Med. Chem. 2018, 155, 847–868. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Nishikawa, T.; Matsumoto, S.; Yoshizawa, H.; Sato, T.; Nakamura, R.; Tsuji, M.; Yamano, Y. Siderophore Cephalosporin Cefiderocol Utilizes Ferric Iron Transporter Systems for Antibacterial Activity against Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 7396–7401. [Google Scholar] [CrossRef]
- Ito, A.; Sato, T.; Ota, M.; Takemura, M.; Nishikawa, T.; Toba, S.; Kohira, N.; Miyagawa, S.; Ishibashi, N.; Matsumoto, S.; et al. In Vitro Antibacterial Properties of Cefiderocol, a Novel Siderophore Cephalosporin, against Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2017, 62, e01454-17. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Kaminski, M.; Landman, D.; Quale, J. Cefiderocol Resistance in Acinetobacter baumannii: Roles of-Lactamases, Siderophore Receptors, and Penicillin Binding Protein 3. Antimicrob. Agents Chemother. 2020, 64, e01221-20. [Google Scholar] [CrossRef] [PubMed]



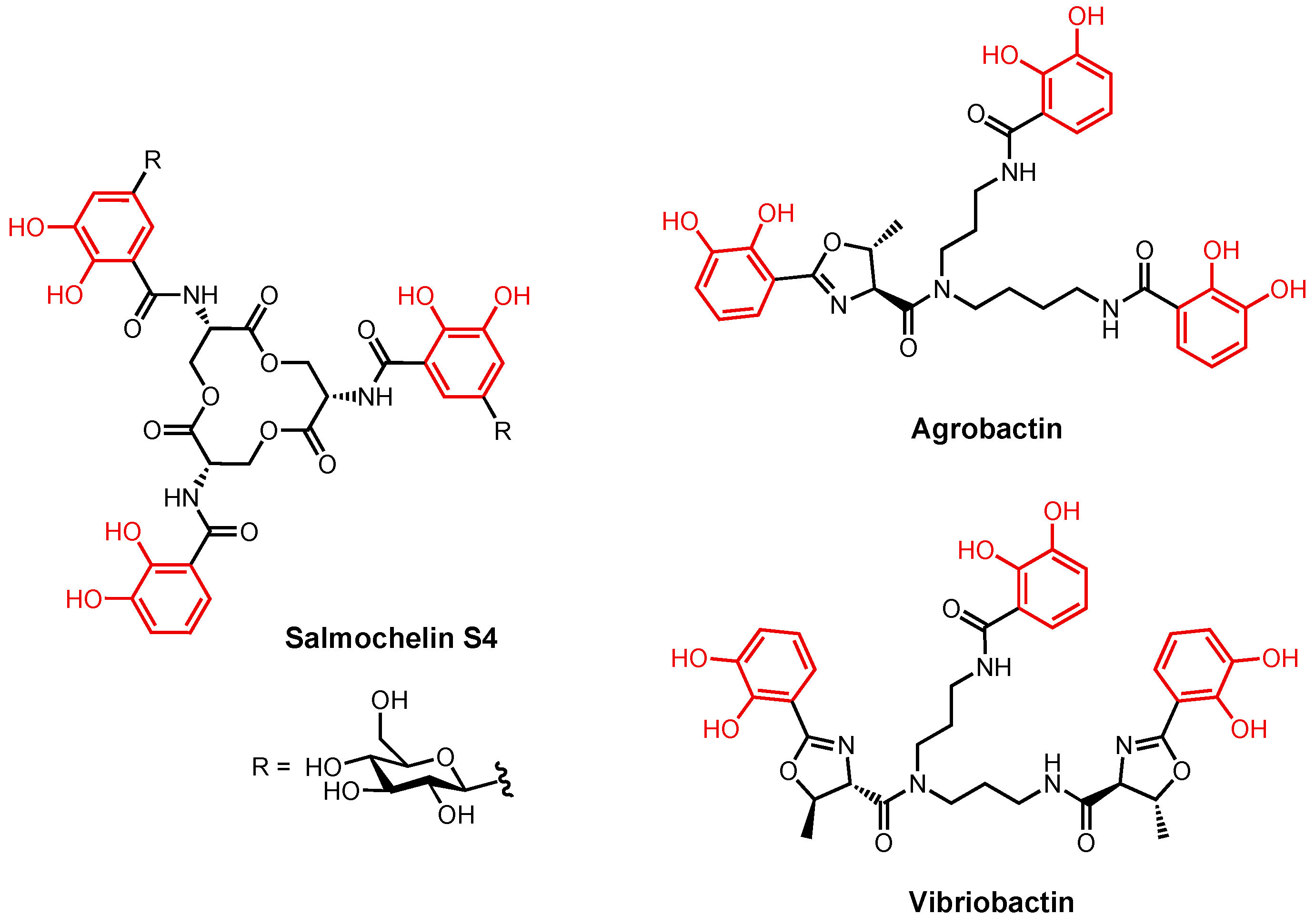
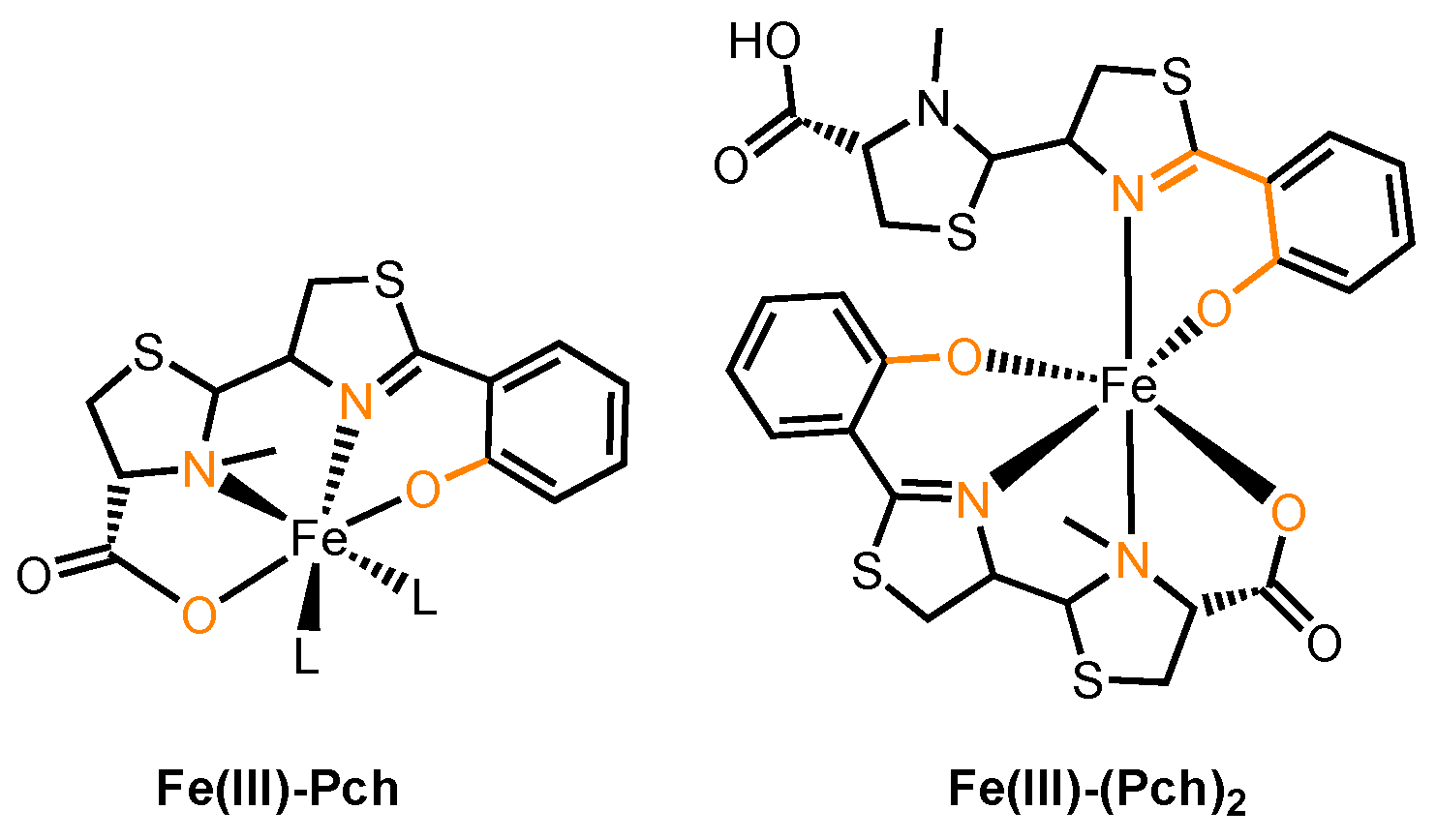
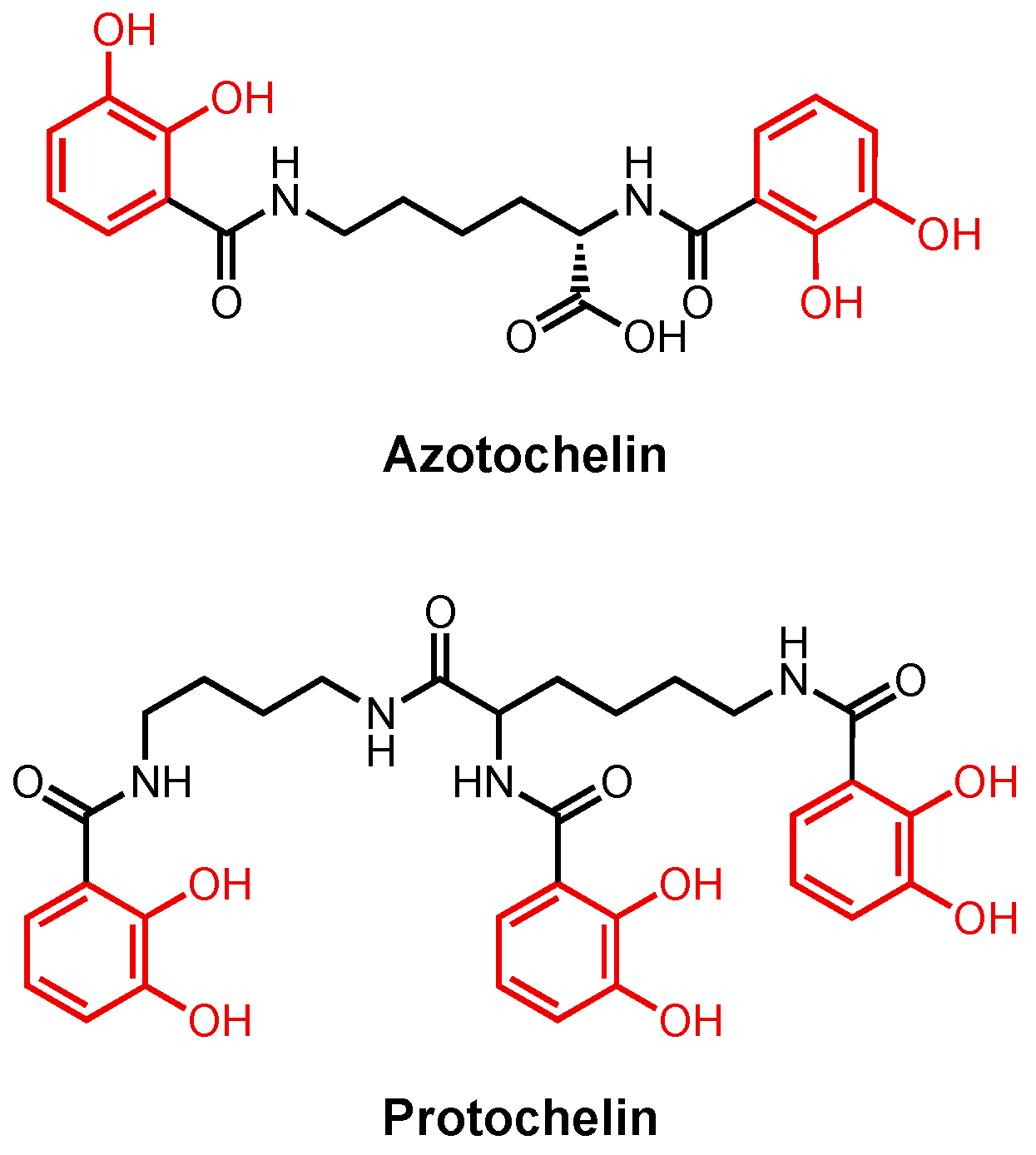
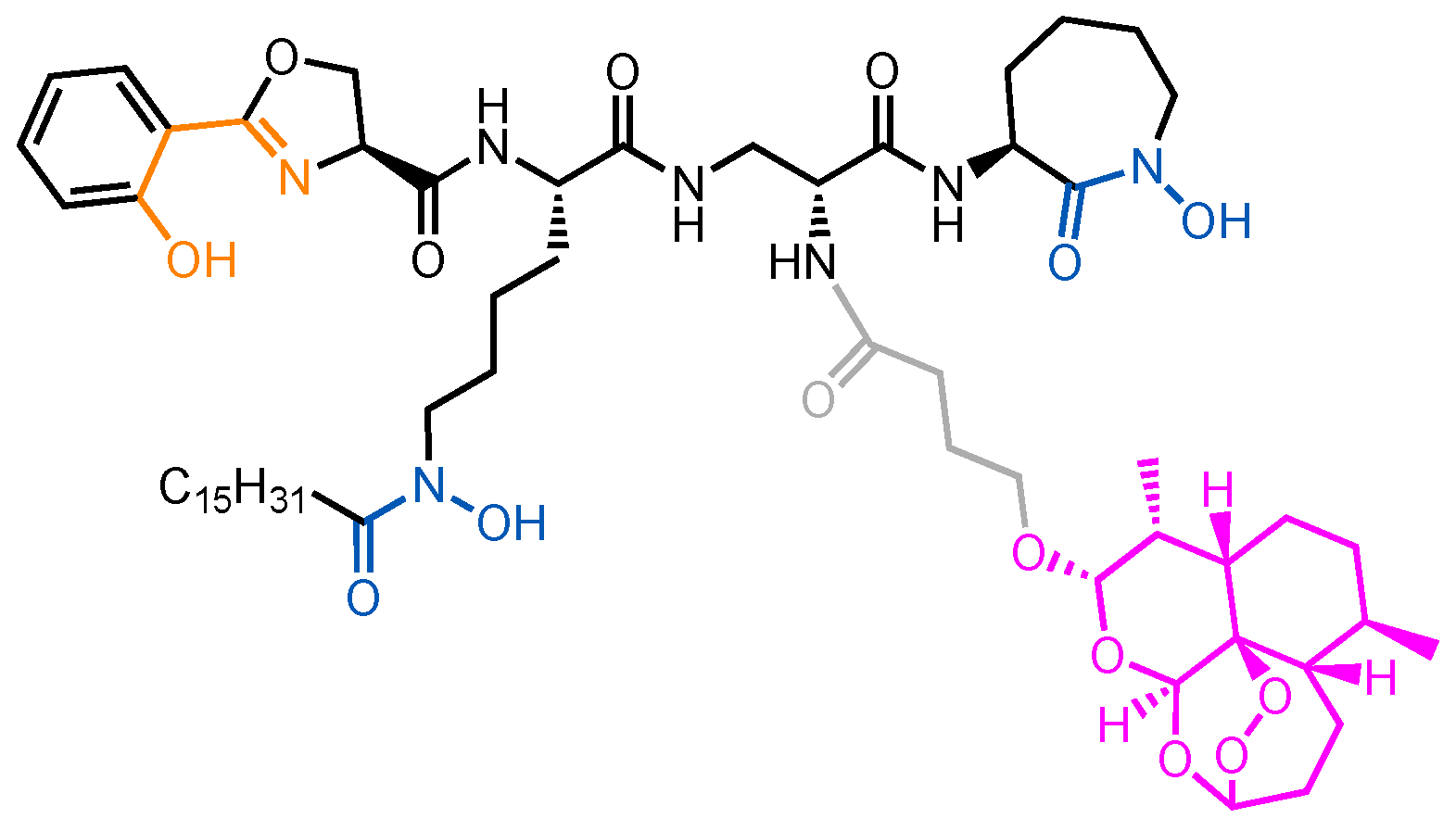
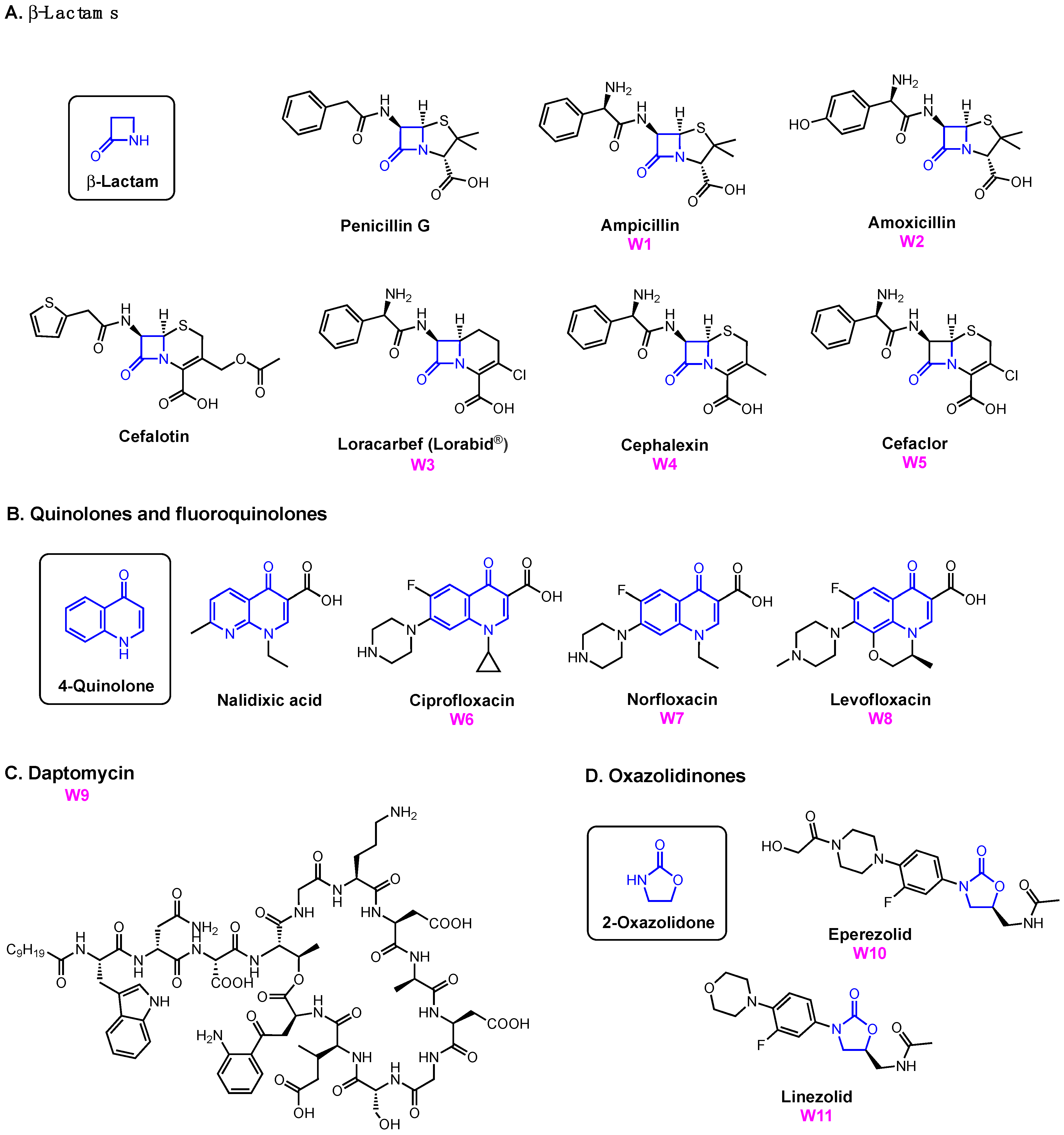
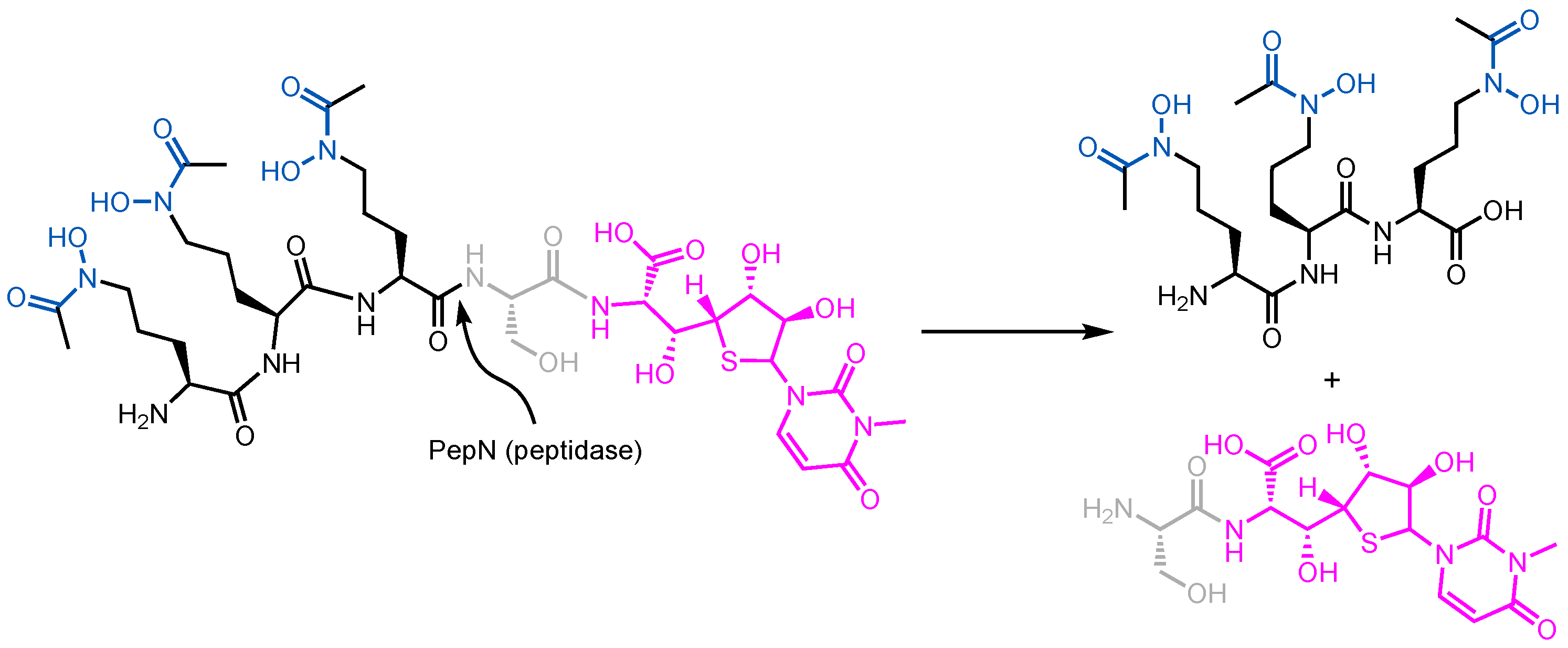


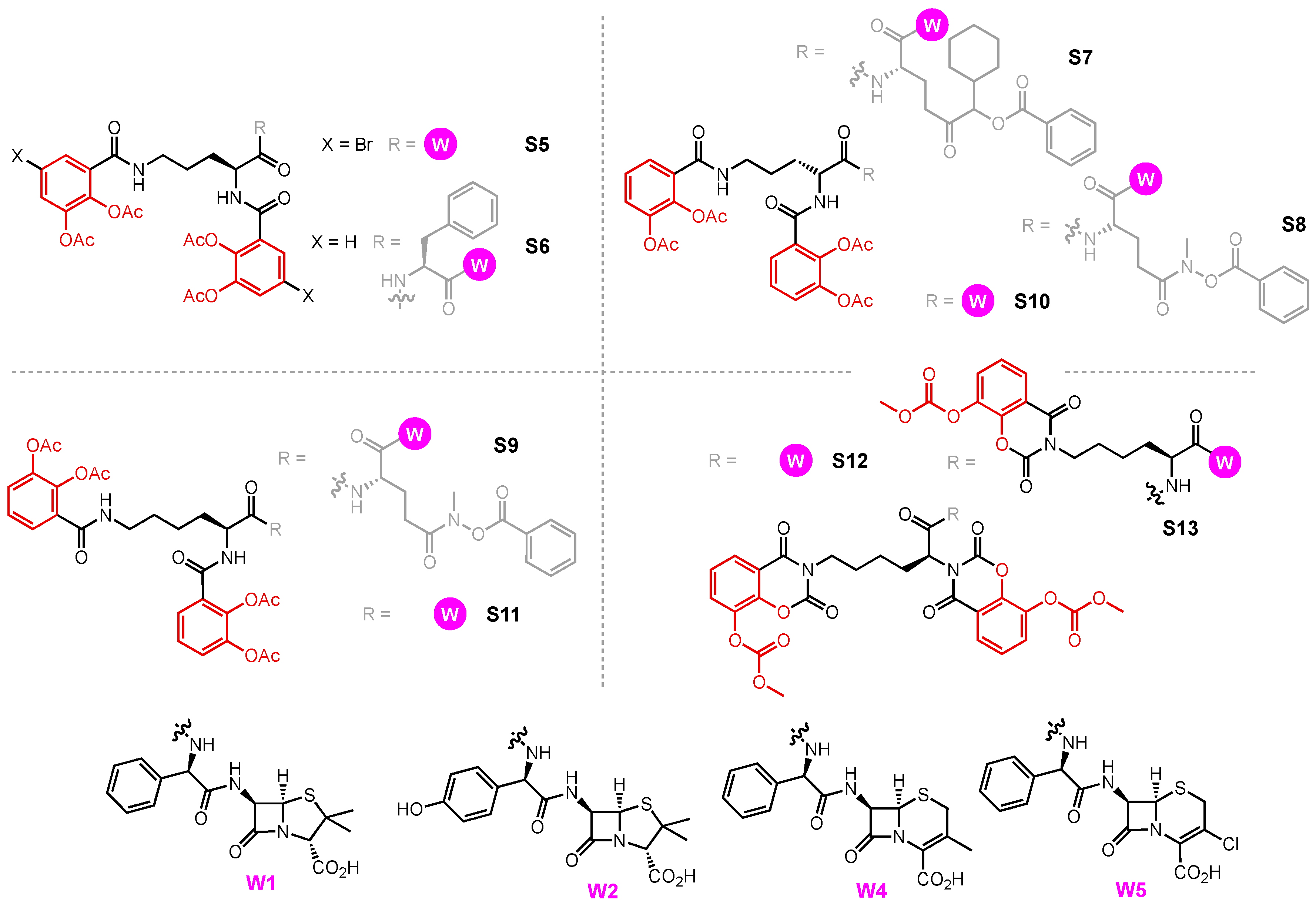
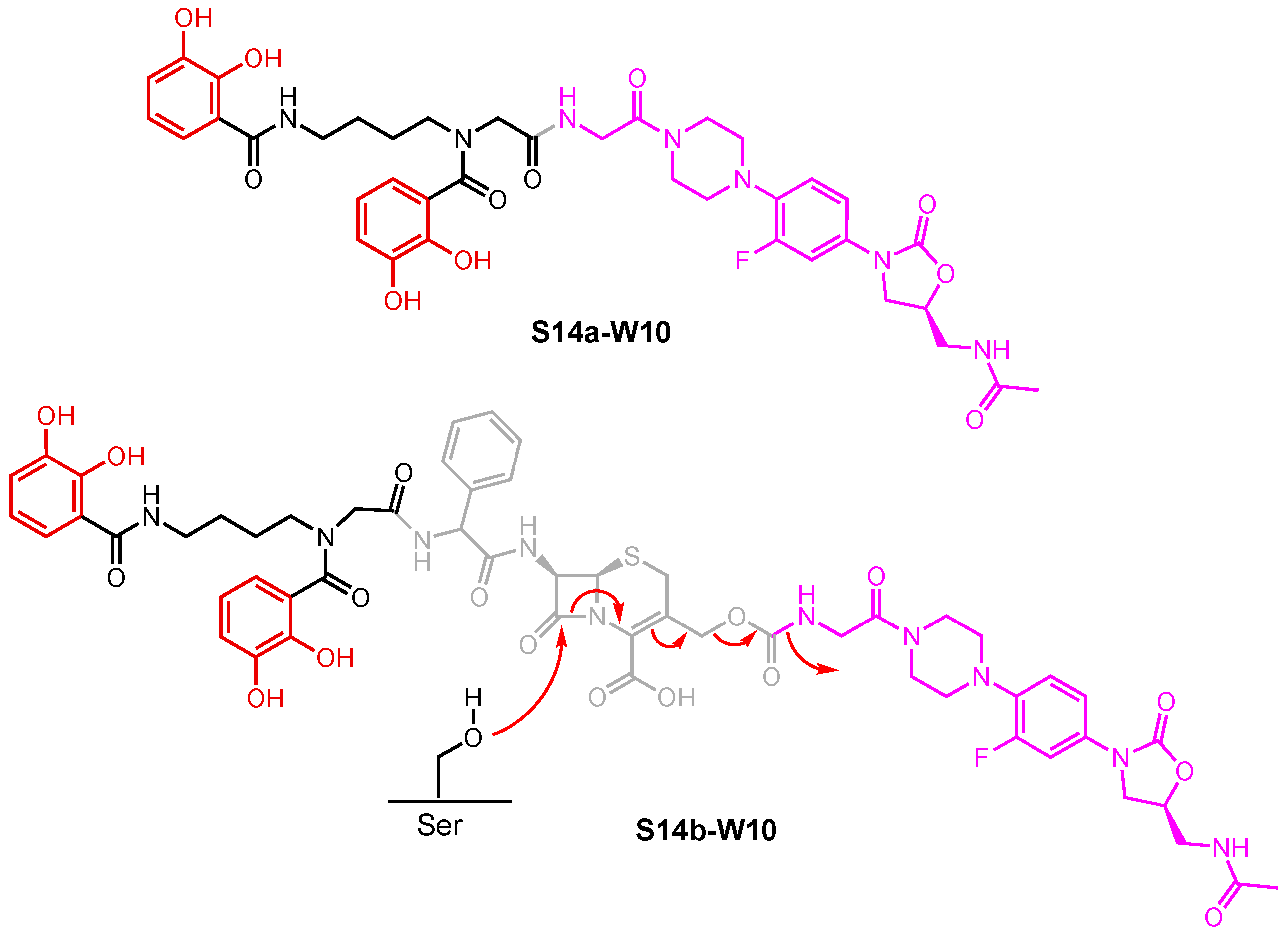
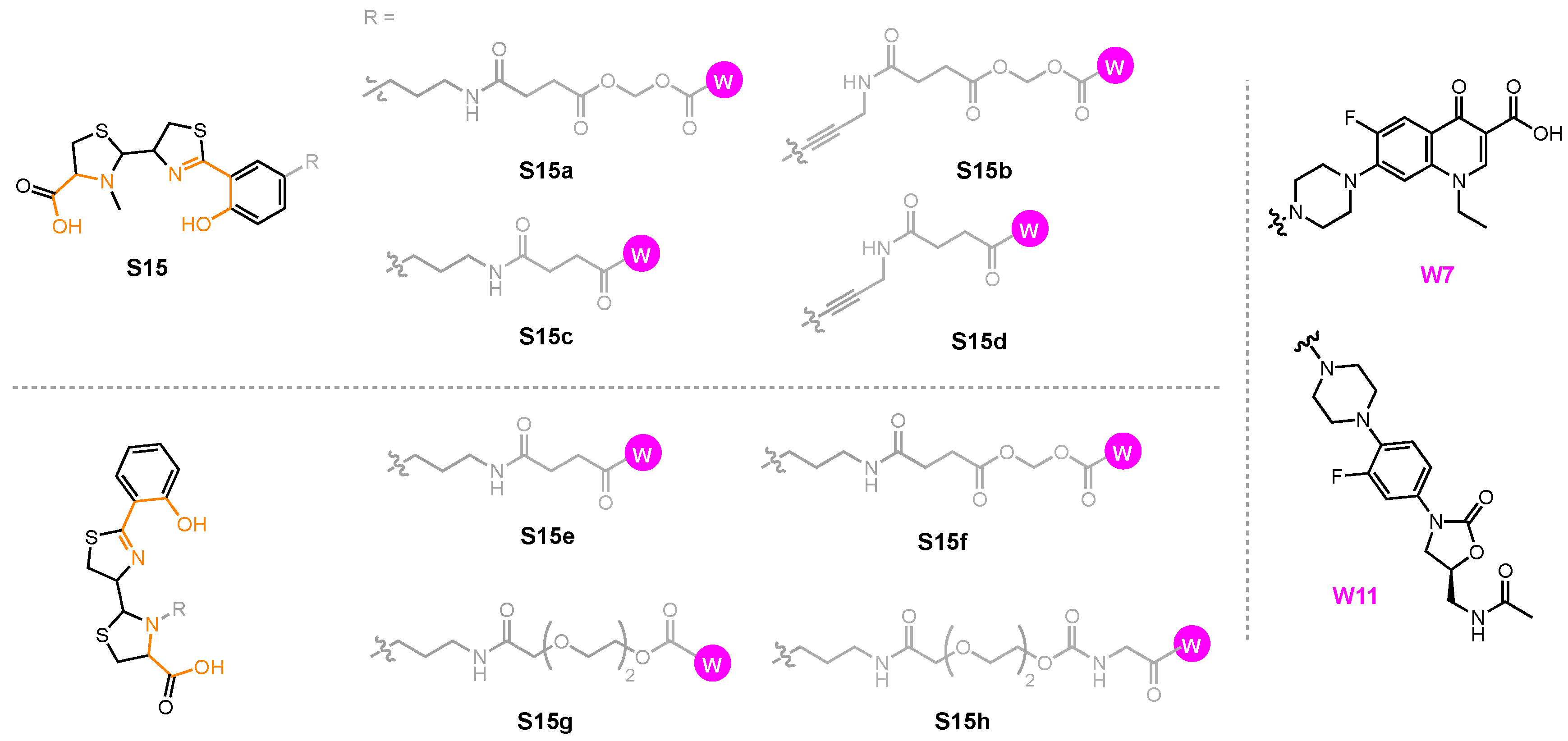
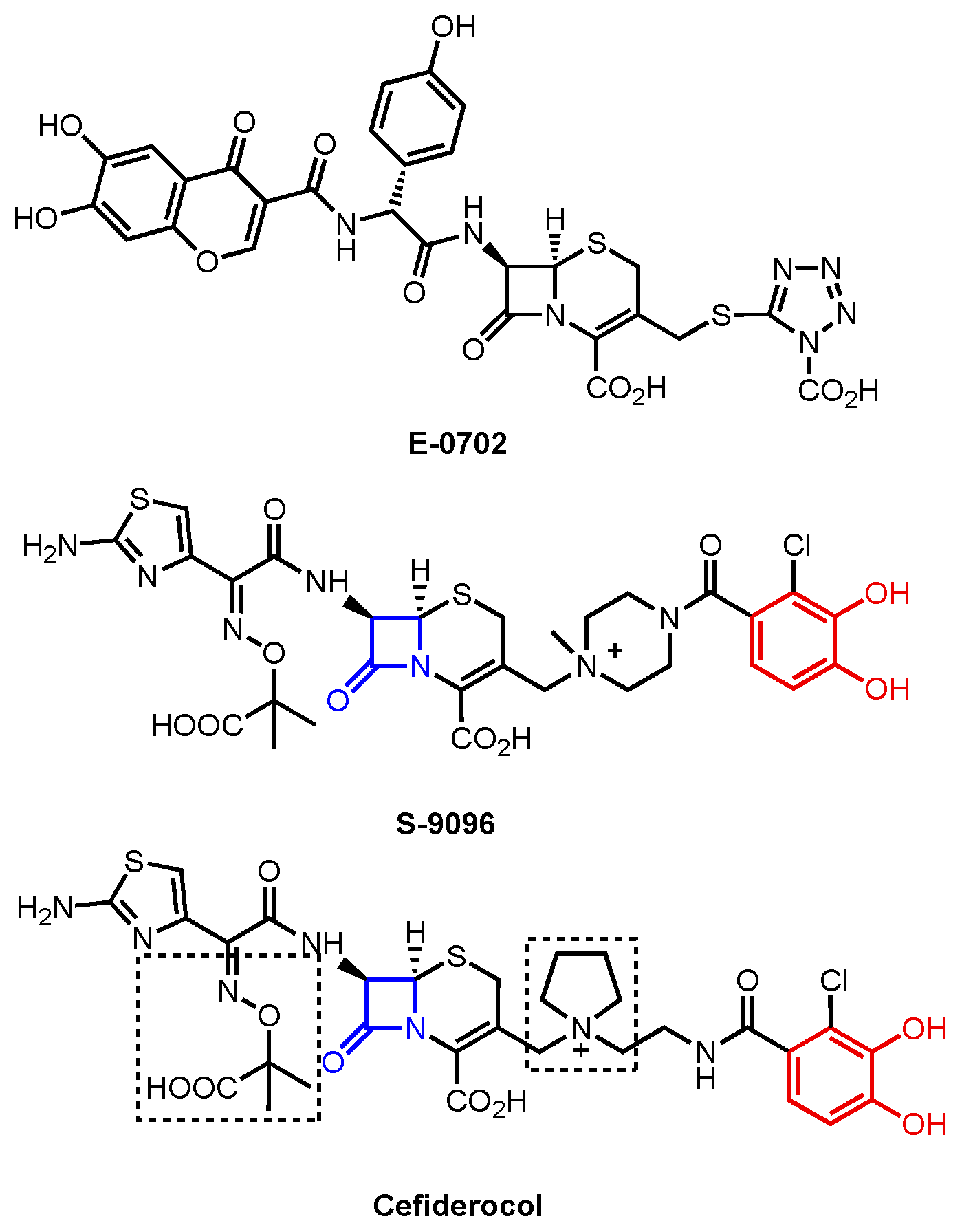

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. baumannii | E. coli | P. aeruginosa | ||||
|---|---|---|---|---|---|---|
| (Pre)acinetobactin (Pre)Acb | Ferrichrome Fch | Enterobactin Ent | Catecholate Siderophores | Pyochelin Pch | Enterobactin Ent | |
| Outer membrane (TBDT) | BauA | FhuA | FepA IroN | Fiu CirA | FptA | PfeA PirA |
| Periplasm (PBP) | BauB | FhuD | FepB | Unknown ? | FepB ? | FepB ? |
| Inner membrane (ABC) | BauCDE | FhuCB | FepDGC | Unknown ? | FptX FepDGC PchHI | FepDGC ? |
| Cytoplasm | BauF * | FhuF * | Fes # YgjH * | Unknown ? | Unknown ? | Unknown ? |
| Entry | Strain | W6 | S1a-W6 | S1b-W6 | S2a-W6 | S2b-W6 |
|---|---|---|---|---|---|---|
| 1 a | A. baumannii ATCC 17961 | 15 b | - | - | 18 | 20 |
| 2 | E. coli X580 | 31 c | 34 | 32 | 21 | 27 |
| 3 | P. aeruginosa K799/WT | 21 d | 27 | 18 | 14 | 19 |
| 4 | P. aeruginosa K799/61 | 24 d | 31 | 32 | 0 | 19 |
| Entry | Strain | W3 | S2a-W3 |
|---|---|---|---|
| 1 | A. baumannii ATCC 17961 | >128 | 0.125 |
| 2 | E. coli ATCC 25922 | 2 | 8 |
| 3 | P. aeruginosa ATCC 27853 | >128 | >128 |
| Entry | Strain | W9 | S2a-W9 | S4-W9 |
|---|---|---|---|---|
| 1 | A. baumannii ATCC 17961 | >100 | 0.4 | 0.2 |
| 2 | A. baumannii ATCC 17978 | - | - | 0.8 |
| 3 | A. baumannii BAA 1710 | >100 | 0.8 | - |
| 4 | A. baumannii BAA 1793 | >100 | 0.8 | - |
| 5 | A. baumannii BAA 1797 | >100 | 0.8 | - |
| 6 | A. baumannii BAA 1800 | >100 | 0.8 | - |
| 7 | A. baumannii ARC 3484 | >100 | 0.4 | 3 |
| 8 | A. baumannii ARC 3486 | >100 | 0.4 | 3 |
| 9 | A. baumannii ARC 5079 | >100 | 0.8 | 12.5 |
| 10 | A. baumannii ARC 5081 | >100 | 0.4 | 12.5 |
| 11 | A. baumannii ATCC 19606 | - | 0.8 | - |
| 12 | E. coli DCO | >100 | >100 | >50 |
| 13 | P. aeruginosa PAO1 | >100 | >100 | - |
| 14 | P. aeruginosa KW799/WT | >50 | - | >50 |
| 15 | P. aeruginosa ARC 3502 | >50 | - | >50 |
| Entry | Strain | W1 | W2 | S3a-W1 | S3a-W2 | S4-W1 | S4-W2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | ||
| 1 | P. aeruginosa PAO1 | n.a. | n.a. | 10 | 10 | 10 | 10 | 50 | 0.39 | 50 | 0.39 | ||
| 2 | E. coli ATCC 25922 | 16.7 | 12.5 | 4.17 | 4.17 | 10 | 0.1 | 10 | 0.1 | 150 | 1.56 | 100 | 6.15 |
| 3 | E. coli UTI89 | 10 | 10 | 10 | 10 | 1 | 0.1 | 10 | 0.1 | - | - | - | - |
| 4 | E. coli CFT073 | 10 | 10 | 10 | 10 | 0.1 | 0.01 | 0.1 | 0.01 | - | - | - | - |
| 5 | E. coli H9049 | 10 | 10 | 10 | 10 | 10 | 0.1 | 10 | 0.1 | - | - | - | - |
| 6 | E. coli 35401 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | - | - | - | - |
| 7 | E. coli 43895 | 10 | 10 | 10 | 10 | 10 | 1 | 10 | 1 | - | - | - | - |
| 8 | E. coli K-12 | 10 | 10 | 10 | 10 | 10 | 0.1 | 10 | 0.1 | - | - | - | - |
| 9 | E. coli K-12 fepA- | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | - | - | - | - |
| 10 | E. coli K-12 fepC- | 10 | 10 | 10 | 10 | 1 | 1 | 10 | 0.1 | - | - | - | - |
| 11 | E. coli K-12 fes- | 10 | 10 | 10 | 10 | 1 | 0.1 | 1 | 0.1 | - | - | - | - |
| 12 | P. aeruginosa K799/WT | n.a. | n.a. | - | - | - | - | 33 | 0.05 | 25 | 0.05 | ||
| 13 | P. aeruginosa K799/61 | 0.52 | 0.78 | 0.46 | 0.39 | - | - | - | - | 12.5 | 0.067 | 12.5 | 0.083 |
| 14 | P. aeruginosa Pa4 | n.a. | n.a. | - | - | - | - | 25 | 0.39 | 25 | 0.21 | ||
| 15 | P. aeruginosa Pa6 | n.a. | n.a. | - | - | - | - | n.a. | n.a. | n.a. | n.a. | ||
| Entry | Strain | W6 (Cipro) | S3b-W6 | S3c-W6 * | |
|---|---|---|---|---|---|
| +Fe | −Fe | ||||
| 1 | E. coli K-12 | 0.1 | n.a. | n.a. | n.a. |
| 2 | E. coli B | 0.1 | n.a. | n.a. | n.a. |
| 3 | E. coli UTI89 | 0.1 | n.a. | 0.1 | 0.1 |
| 4 | E. coli CFT073 | 0.1 | n.a. | 1 | 0.1 |
| 5 | E. coli CFT073 fepA- | - | - | 0.1 | |
| 6 | E. coli CFT073 iroN- | - | - | 0.1 | |
| 7 | E. coli CFT073 fepA- iroN- | - | - | n.a. | |
| 8 | E. coli CFT073 fepC- | - | - | n.a. | |
| 9 | E. coli CFT073 fepDG- | - | - | n.a. | |
| 10 | E. coli CFT073 fes- | - | - | 1 | |
| 11 | E. coli CFT073 iroD- | - | - | n.a. | |
| Entry | Strain | W1 | W5 | S5-W1 | S6-W1 | S7-W1 | S8-W1 | S9-W1 | S10-W4 | S11-W4 | S12-W1 | S13-W1 | S13-W2 | S13-W5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | E. coli ATCC 25922 | 6.26 | 12.5 | 0.78 | 0.4 | 0.2 | 0.4 | 0.1 | n.a. | 100 | 50 | 6.25 | 3.12 | 1.56 |
| 2 | P. aeruginosa ATCC 27853 | n.a. | 100 | 0.4 | 0.4 | 0.78 | 0.4 | 0.2 | 3.12 | 25 | 6.25 | 0.78 | 6.25 | 50 |
| 3 | P. aeruginosa SG 137 | n.a. | 100 | <0.05 | <0.05 | 0.05 | <0.05 | 0.01 | <0.05 | 12.5 | 0.4 | 0.2 | 0.78 | 50 |
| Entry | Strain | W10 | S14a-W10 | S14b-W10 |
|---|---|---|---|---|
| 1 | A. baumannii ATCC 17961 | n.a. | n.a. | 0.8 |
| 2 | A. baumannii ATCC BAA 1793 | n.a. | n.a. | 0.8–0.16 |
| 3 | A. baumannii ATCC BAA 1797 | n.a. | n.a. | 6.25 |
| 4 | A. baumannii ATCC BAA 1800 | n.a. | n.a. | 0.8 |
| 5 | E. coli DC0 | n.a. | n.a. | <0.025 |
| 6 | P. aeruginosa KW799/WT | n.a. | n.a. | 0.2–0.4 |
| Entry | Strain | W6 | W7 | W8 | S15f-W6 | S15f-W7 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | +Fe | −Fe | ||
| 1 | P. aeruginosa PAO1 | 0.060 | 0.040 | 0.200 | 0.110 | 0.350 | 0.200 | 0.700 | 0.600 | 1.000 | n.a. |
| 2 | P. aeruginosa PAD07 (pvd- pch-) | 0.045 | 0.060 | 0.190 | 0.200 | 0.360 | 0.350 | 0.600 | 0.700 | 1.000 | 1.000 |
| 3 | P. aeruginosa PAD14 (tonB-) | 0.040 | 0.035 | 0.120 | 0.120 | 0.210 | 0.180 | 0.200 | 0.170 | 0.550 | 0.450 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, V.C.; Peczuh, M.W. Location, Location, Location: Establishing Design Principles for New Antibacterials from Ferric Siderophore Transport Systems. Molecules 2024, 29, 3889. https://doi.org/10.3390/molecules29163889
Luo VC, Peczuh MW. Location, Location, Location: Establishing Design Principles for New Antibacterials from Ferric Siderophore Transport Systems. Molecules. 2024; 29(16):3889. https://doi.org/10.3390/molecules29163889
Chicago/Turabian StyleLuo, Vivien Canran, and Mark W. Peczuh. 2024. "Location, Location, Location: Establishing Design Principles for New Antibacterials from Ferric Siderophore Transport Systems" Molecules 29, no. 16: 3889. https://doi.org/10.3390/molecules29163889