Abstract
We report a computational study of the bending deformation of two-dimensional nanoribbons by classical molecular dynamics methods. Two-dimensional double transition metal carbides, together with monometallic ones, belong to the family of novel nanomaterials, so-called MXenes. Recently, it was reported that within molecular dynamics simulations, Ti4C3 MXene nanoribbons demonstrated higher resistance to bending deformation than thinner Ti2C MXene and other two-dimensional materials, such as graphene and molybdenum disulfide. Here, we apply a similar method to that used in a previous study to investigate the behavior of Mo2Ti2C3 nanoribbon under bending deformation, in comparison to the Ti4C3 sample that has a similar structure. Our calculations show that Mo2Ti2C3 is characterized by higher bending rigidity at eV than monometallic Ti4C3 nanoribbon at eV, which has a similar thickness. Moreover, approximately the same magnitude of critical central deflection of the nanoribbon before fracture was observed for both Mo2Ti2C3 and Ti4C3 samples, nm, while Mo2Ti2C3 MXene is characterized by almost two times higher critical value of related external force.
1. Introduction
Two-dimensional (2D) crystals are a relatively new class of structure that includes several families of various materials. The most popular and well-known among 2D crystals is graphene—an atomically thin layer of carbon atoms with hexagonal structure and covalent bonds, discovered in 2004 [1]. Other popular representatives of 2D crystals are 2D boron nitride [2], 2D molybdenum disulfide MoS2 [3], 2D transition metal dichalcogenides (2D TMDs), and other materials [4]. Since their discovery, two-dimensional materials have opened new possibilities to significantly improve nanoelectronics, due to their ability to be integrated into devices with atomic-scale precision, leading to the development of smaller, faster, and more efficient electronic components. Thus, each of the 2D materials has unique physical properties that define their potential application. For instance, graphene has exceptional electron mobility, mechanical strength, and thermal conductivity. Its application in nanoelectronics includes the development of high-speed transistors, flexible transparent conductive films, and advanced sensors [5,6]. Graphene-based transistors, for instance, can operate at higher frequencies compared to traditional silicon-based transistors, enabling faster data processing and communication [7,8].
In 2011, the family of 2D materials became even bigger, as a new group of crystals—transition metals carbides and nitrides (as well as carbonitrides) with general chemical formula Mn+1Xn (M is an early transition metal, and X is carbon C and/or nitrogen N) called MXenes—were discovered [9]. MXenes are synthesized from bulk precursors, so-called MAX-phases (nanolaminated materials with general chemical formula Mn+1AXn, where M, as above, is an early transition metal, A is an element from group IIIA or IVA (or group 13 or 14), and X is carbon C and/or nitrogen N, and n = 1, 2, or 3) by etching a layer of element A atoms from MAX phases [10,11].
Together with unique electrochemical properties [12,13,14], MXenes exhibit exceptional mechanical properties [15,16], including high strength, flexibility, and tunability, making them highly attractive for applications in nanoelectronics. Their ability to withstand mechanical deformation while maintaining electronic functionality opens up new possibilities for the development of flexible, durable, and high-performance electronic devices [17,18].
Additionally, the layered structure of MXenes contributes to their mechanical stability and flexibility. The layers can slide relative to each other, providing an intrinsic mechanism for accommodating strain and preventing fracture. This characteristic is particularly beneficial for creating multilayered or composite structures that combine MXenes with other materials, resulting in devices that are both mechanically robust and electrically superior. The integration of MXenes into nanoelectromechanical systems (NEMSs), leverages their mechanical properties to enhance device performance and longevity [17,18].
The flexibility and mechanical robustness of MXenes are further enhanced by their unique surface chemistry. MXenes can be functionalized with various surface groups, such as hydroxyl, oxygen, and fluorine, which not only modify their electronic properties but also influence their mechanical behavior. This tunability allows researchers to design MXenes with tailored mechanical properties to meet specific application requirements. For instance, by adjusting the surface chemistry, MXenes can achieve an optimal balance between flexibility and strength, making them ideal for applications in nanoelectronics where mechanical reliability is paramount.
One of the important properties of nanomaterials that defines their use in nanoelectromechanical system devices is bending or flexural rigidity [19,20]. It characterizes the ability of a particular sample to resist bending deformation and maintain its flexibility and elasticity. However, the experimental measurement of the mechanical properties of nanomaterials requires special facilities and represents a serious challenge due to the small sizes of the samples (see for example [21,22,23]). Therefore, mechanical properties of nanomaterials are also studied by computational [15,16,17,18,19,20] and theoretical [24,25,26] methods. Thus, one of the illustrative examples of such study is the calculation of bending rigidity of graphene nanoribbons (GNRs) by classical molecular dynamics simulations [20]. The developed approach also allowed the authors to propose a model for an ultrasensitive pressure sensor based on GNRs, and also suggested further applications of graphene nanoribbons in NEMSs devices in general [27].
A similar approach was recently adopted to study the bending properties of the Tin+1Cn MXene nanoribbons [19]. The performed calculations confirmed the assumption that Tin+1Cn samples are characterized by higher bending rigidity than graphene, due to the higher thickness of MXene nanoribbons, as well as the increasing of bending rigidity with the growth of the thickness of the samples. Moreover, recent discoveries of the ordered double transition metal carbides [28,29] opened a new way of controlling and tuning the electronic properties of MXenes by altering the outer metal layer in Mn+1Xn sheets. Thus, after the synthesis of double transition metals, MXene Mo2Ti2C3 was reported [29], which naturally opened a question of how the changing of two outer layers of titanium in Ti4C3 to molybdenum affects the flexural properties of MXene nanoribbons. In this paper, we report the comparative analysis of the bending properties of Mo2Ti2C3 and Ti4C3 by classical molecular dynamics simulations that were performed using a previously developed computational scheme of numerical experiments on bending of Tin+1Cn MXene nanoribbons [19]. As no experiments on bending rigidity of pristine Mo2Ti2C3 and Ti4C3 nanoribbons have been reported by now, our study may provide the first insights on how mechanical properties of MXenes can be affected by replacing monometallic Ti sample by (Mo, Ti) double transition metal carbide.
2. Model
We followed a previous study on the bending rigidity of Tin+1Cn MXene nanoribbons [19], where preliminary results on the bending of Ti4C3 nanoribbon were reported. In our simulations, we consider two different MXene nanoribbons, Mo2Ti2C3 and Ti4C3. The chemical composition of the considered samples was chosen due to the already developed MD approach for the simulation of Tin+1Cn samples [15] and similar structures of Mo2Ti2C3 and Ti4C3 MXenes [28,29]. As it is reported in [29], the double transition metal Mo2Ti2C3 is characterized by almost equivalent interatomic distances between Mo-C and Ti-C atoms, similar lattice parameters, and the same sample thickness as Ti4C3 MXene. The structures of both Mo2Ti2C3 and Ti4C3 MXenes and the general view of the studied nanoribbons are shown in Figure 1 and Figure 2, respectively (all snapshots of atomistic configurations of the samples were prepared with visual molecular dynamics software version 1.9 [30]).
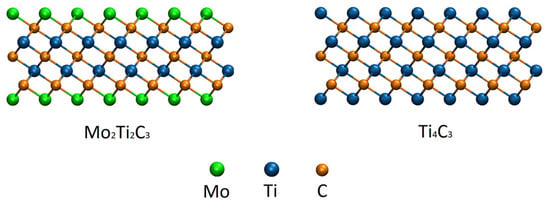
Figure 1.
Atomistic configurations of the studied Mo2Ti2C3 (left panel) and Ti4C3 (right panel) samples. Related colors of the atoms are shown in the figure.
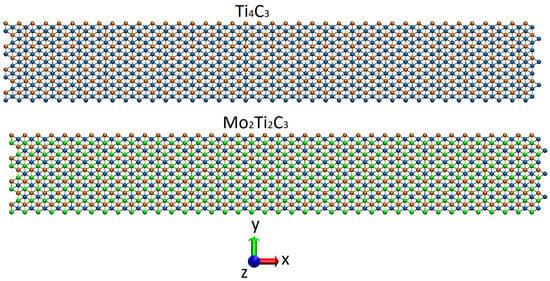
Figure 2.
Snapshots (top view in Cartesian coordinate system XYZ) of the initial configuration of the studied Ti4C3 (top panel) and Mo2Ti2C3 (bottom panel) nanoribbons. The colors of atoms are the same as in Figure 1.
Moreover, with the assumption that bending rigidity depends on the thickness h of the sample being [19], simulations of the thickest (seven atomic layers) M4X3 samples under bending deformation may provide the approximal value of maximal bending rigidity that can be expected among (Mo,Ti)n+1Cn and Tin+1Cn MXenes.
It is worth to note that several approaches for MD simulations of MXenes were reported in the literature so far (see for example [15,16,31,32]). The most precise and yet most complicated approach employs ReaxFF interatomic potential [33]. Such an approach allowed us to obtain important data on chemical reactions and phase transitions within MXenes, and to study their mechanical properties as well [16,33]. However, ReaxFF can be straightforwardly used for simulations of certain MXenes only, and its application for Mo2Ti2C3 requires additional parametrization, which is a rather hard challenge [33]. Therefore, in our simulations, we adopted a previously developed approach that is based on combinations of interatomic potentials [15]. The full description of the developed model is provided in the original paper; therefore, here we focused on the adjustments that were introduced in the model to adapt for simulations of Mo2Ti2C3 MXene. Following the assumption that interactions between titanium atoms in Tin+1Cn MXenes can be described within embedded atom method (EAM) [15], we suppose that EAM potential can also be used to describe interactions between molybdenum atoms in Mo2Ti2C3 samples in a similar manner, as EAM potential is already parametrized for molybdenum [34]. Moreover, with this assumption, interactions between Ti and Mo atoms in Mo2Ti2C3 MXene sample can be described with an EAM alloy model for Ti-Mo [34]. As in Mo2Ti2C3, Mo-C and Ti-C bonds have almost similar lengths [29], interatomic forces between both metals and carbon atoms are calculated in the same way as forces between titanium and carbon in Ti4C3 [19]. This is a rough approximation, which makes our model rather qualitative; nevertheless, it makes it possible to obtain preliminary data on flexural properties of double metals Mo2Ti2C3 MXene, without the complicated parametrization of other interatomic potentials.
In our experiments on bending, we followed an already existing methodic that was proposed for studying the flexural properties of graphene nanoribbons [20], and which was further adopted for the calculation of the bending rigidity of Tin+1Cn MXenes [19]. We consider Mo2Ti2C3 and Ti4C3 nanoribbons with sizes of 1.7 nm × 12.0 nm located in a Cartesian coordinate box with periodic boundary conditions in X and Y direction, and free boundary condition in Z, as shown by schematics in Figure 3. For the same reason, as in [19], we applied similar constraints to the boundary atoms across long edges of the nanoribbons, not allowing them to relax in the XY plane during bending.
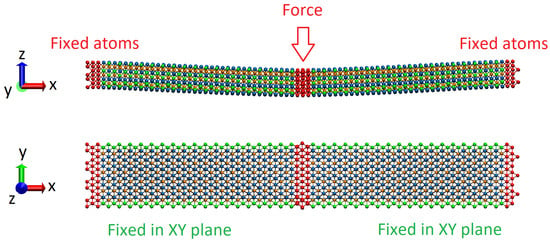
Figure 3.
Schematics of the experiments on the bending of Mo2Ti2C3 and Ti4C3 nanoribbons in Cartesian coordinate system XYZ. Fixed atoms, on the edges of the sample and central atoms to which external bending load is applied, are shown in red color. Constrained atoms across long edges are denoted in green.
Thus, by applying the external force F, as is shown in Figure 3, and measuring the corresponding central deflection of nanoribbon , it is possible to calculate the bending rigidity D of the sample, from the well-known equation [19,20]:
where L is the length of the nanoribbon and β is the coefficient in the range from 0.0056 to 0.00725 for a nanoribbon length-to-width ratio from 1 to , respectively.
In our simulation, we adopted an in-house code, developed for the study of mechanical properties and thermal stability of Tin+1Cn MXenes [15,19,35,36]. The code was implemented on a graphics processing unit (GPU) for parallel calculations. The calculations were performed on an NVIDIA Tesla P100 GPU (NVIDIA Corporation, Santa Clara, CA, USA). Other details of the simulation setup can be found in [19]. The obtained results are described in the following section.
3. Results
As described above, the simulations of the bending deformation of MXene nanoribbons were performed by applying external force in the –Z direction to the central part of the sample (see Figure 3). Applications of the external load results in a deflection of the nanoribbon from the initial configuration and its bending. Similar to the simulations described in [19,20], we performed the simulation by gradually increasing the force applied to the sample and analyzing its behavior. During the experiments, the time dependencies of the immediate position of the nanoribbon center and other data needed for calculations of bending parameters were recorded. The typical behavior of the samples under an external bending load is shown in Figure 4.
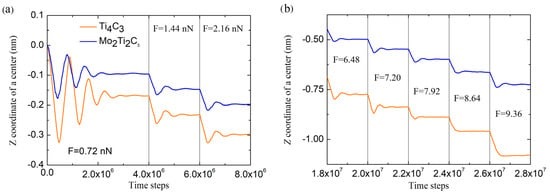
Figure 4.
Time dependencies of the Z coordinate of centers of the samples. Panel (a) shows the behavior of the samples in the initial stage of the experiment for the first three steps of loading, while panel (b) demonstrates the behavior in the middle of simulations.
Presented in Figure 4, data show that at the beginning of experiments, the application of the external force of the smallest magnitude F0 (equivalent to 0.72 nN) initiates damping oscillations of both nanoribbons (see opening part of the dependencies in Figure 4a), after which the sample deflects from the original position. After applying forces of larger magnitudes (with an increment ), central deflection was measured after slight damping oscillations when samples reached the stationary configuration. In our experiments, samples were allowed to reach a stationary position for 2 × 106 time steps (4 × 106 time steps for initial indentation, as shown in Figure 4a). The central deflection was measured by time averaging of the Z coordinate of the central part of the sample, to which the external force is applied (see Figure 3) over the last 5 × 105 time steps during each force increment. Figure 4b shows that each force increment resulted in approximately constant changes in each sample’s increment of central deflection , which suggests that in this part of experiment, nanoribbons are bending in an almost linear regime.
To illustrate the behavior of the samples in elastic regimes of bending deformation, we performed additional indentation of the Ti4C3 sample with the doubled force increment , as well as the gradual relaxation of Mo2Ti2C3 nanoribbon. Obtained data are shown in Figure 5.
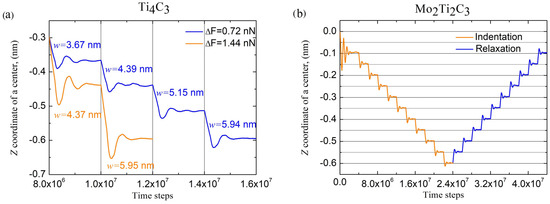
Figure 5.
(a) Time dependencies of Z coordinate of Ti4C3 nanoribbon during bending with different force increments, and (magnitudes of central deflection and force increments are shown in the figure). Time dependencies of Z coordinate of Mo2Ti2C3 nanoribbons during indentation and relaxation (b).
As it follows from the data presented in Figure 5a, the application of a two-times larger force increment to Ti4C3 sample results in approximately two times larger increments of related central deflection compared to the initial conditions of the experiments. The presented dependencies are also characterized by a larger amplitude of damping oscillations after each increase in the bending force. Figure 5b shows the behavior of the Mo2Ti2C3 sample during the first 10 steps of indentation (increasing of the applied force with constant increment ) and following relaxation (decreasing of with constant increment ) to the previous position. As it follows in the figure, consecutive relaxation of the applied force results in decreases in the central deflection by almost the same increment as during the indentation. Therefore, the presented dependence illustrates the reversibility of the performed experiments within the elastic mode of the bending deformation of nanoribbons.
Further in our experiments, the application of the increasing external force continued until the complete fracture of the nanoribbon. The total time dependencies of the Z coordinate of nanoribbon centers and the load curves for both samples are shown in Figure 6a and Figure 6b, respectively.
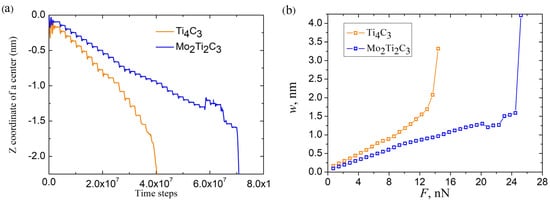
Figure 6.
(a) Time dependencies of Z coordinate of Ti4C3 and Mo2Ti2C3 nanoribbons during the whole experiment. (b) Corresponding load curves for Ti4C3 and Mo2Ti2C3 nanoribbons. The final points on the dependencies plotted in panel (b) do not relate to corresponding magnitude of central deflection of nanoribbons; these points were measured after the fracture of nanoribbons in the certain time moments, according to computational algorithm and plotted in the figure to illustrate the fracturing of the samples.
The fracture of nanoribbon is indicated by the sharp drop in Z(t) dependencies to large negative values of Z (see Figure 6a). As it follows from the figure, both samples are characterized by a close magnitude of critical deflection nm before the destruction of the nanoribbon. At the same time, as Figure 6b shows, the fracturing of Mo2Ti2C3 nanoribbon was observed at an almost two times larger magnitude of the critical external force (note that there were significantly more indentation steps and correspondingly more time steps in simulations for Mo2Ti2C3 nanoribbon before fracturing, as shown in Figure 6a).
It is important to note that nonlinear modes of bending, related to plastic deformation and partial destruction of the samples, are also clearly visible from the data presented in Figure 6. The mentioned modes of bending relate to the part of the dependencies where the growth of the external force results in increases in the corresponding increments of central deflection or in decreases in the current magnitude of central deflection presented in Figure 6b. The latter case relates to the plastic deformation and partial destruction of the sample, which may lead to the local rearrangement of atoms and relaxation of mechanical stresses. Moreover, it is worth noting that the nonlinear dependence of central deflection on the applied load may also be caused by the axial extension effect [37]. An example of atomistic configuration of Mo2Ti2C3 nanoribbon with clearly visible plastic deformation and partial destruction of the central part is shown in Figure 7.
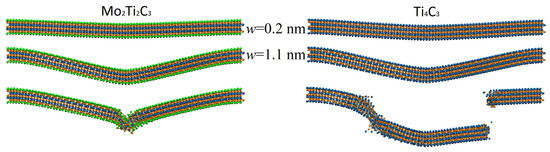
Figure 7.
Examples of typical atomistic configuration of Mo2Ti2C3 (left panel) and Ti4C3 (right panel) nanoribbons at different magnitudes of central deflection. The bottom panels show the plastic deformation of the Mo2Ti2C3 sample and the fracture of the Ti4C3 nanoribbon.
Figure 7 shows a typical atomistic configuration of studied samples in different modes of bending deformation. The top panels show configurations of the samples at central deflection nm, which relate to the elastic mode of bending, corresponding to linear parts of the dependencies of central deflection on external force , as shown in Figure 6b. The middle panels of Figure 7 depict the configuration of the samples at larger central deflection nm. At these conditions, force increment no longer leads to constant increments of central deflection ; however, no plastic deformation or fracture of the samples is observed in atomistic configurations, and samples can be relaxed to initial configuration. The bottom panels of Figure 7 show the fracture of the samples. Notably, samples are characterized by different fracture dynamics. Thus, Mo2Ti2C3 nanoribbon is characterized by severe plastic deformation in the central area where the external force is applied, following the destruction of the MXene crystal structure and the fracture of the nanoribbon. At the same time, fracture dynamics of Ti4C3 nanoribbon are more typical for brittle materials, with the destruction of the sample in several areas, and following the formation of fragments that preserve the two-dimensional structure of Ti4C3 MXene. Such behavior of Ti4C3 MXenes was already observed in our earlier MD simulations [19]. The bending and fracture dynamics of both nanoribbons are also shown in Supplementary Videos S1 and S2.
Performing simulations and recorded data allowed us to calculate elastic parameters that characterize the bending properties of MXene nanoribbons. However, the straightforward installation of obtained dependence (shown in Figure 6b) in Equation (1) leads to the bending rigidity D of the samples, as presented in Figure 8.
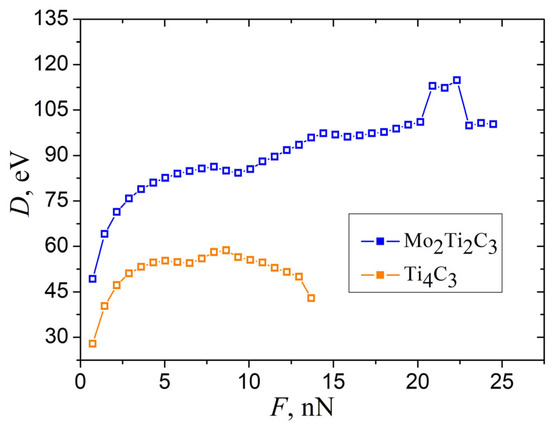
Figure 8.
The dependence of bending rigidity D of Mo2Ti2C3 and Ti4C3 samples, calculated from the data shown in Figure 6b, according to Equation (1).
As it can be seen from the figure, the calculated D strongly depends on applied force even in the region of small magnitudes of central deflections. This makes it hard to obtain the exact value of the elastic parameters that characterize the bending properties of MXene nanoribbons. However, this feature can be resolved by analyzing the dependencies shown in Figure 4a and Figure 6.
As can be seen in Figure 4a, the first increment of a central deflection in the opening part of dependencies has a larger magnitude than the following deflection increments for both samples. Moreover, the system requires more simulation time to reach the stationary configuration, and related damped oscillations of the nanoribbon last longer, compared to the next steps of indentation. This may be explained by the presence of initial deflection of the samples and related initial stresses (pre-tension) due to the applied boundary conditions in the XY plane. It is worth to mention that the pre-tension of the samples is often observed during the calculation of mechanical parameters in MD simulations [16]. The pre-tension of the samples can be taken into account by introducing the correction to measure deflection for each sample correspondingly. The magnitude of can be estimated as a residual part of the first increment after subtracting the deflection increment averaged over the next few steps of indentation, =. Thus, magnitudes of initial deflection, calculated according to this assumption, equal nm and 0.103 nm for Mo2Ti2C3, and Ti4C3 nanoribbons, respectively. Dependencies of bending rigidity on external force, calculated from Equation (1) after introducing of such correction, are shown in Figure 9.
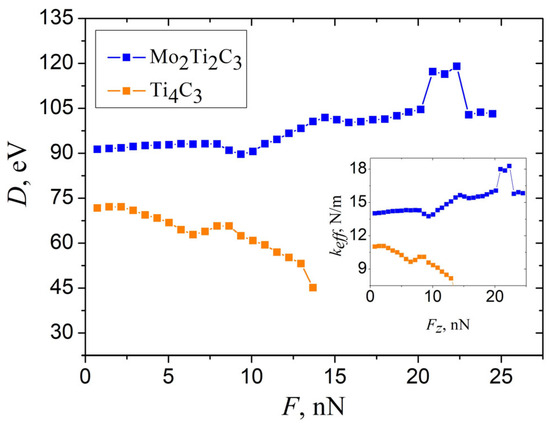
Figure 9.
The dependence of bending rigidity D of Mo2Ti2C3 and Ti4C3 samples, calculated from the data shown in Figure 6b, according to Equation (1), taking into account of the pre-tension of the sample. Inset shows related dependencies of effective spring constant .
As it follows from the figure, dependence calculated by taking into account of the initial deflection of Mo2Ti2C3 sample almost independently of applied force in the elastic region of bending deformation nN and has an almost constant value of approximately eV. At the same time, Ti4C3 nanoribbon is characterized by a shorter linear mode compared to Mo2Ti2C3; however, it is possible to estimate the approximate value of bending rigidity eV from the initial part of related dependence.
4. Discussion
We reported the results of classical molecular dynamics simulations of bending deformation of Mo2Ti2C3 and Ti4C3 MXene nanoribbons. Our experiments revealed that bending rigidity obtained from the linear part of load curve for double transition metal carbide Mo2Ti2C3 eV is notably higher than the related parameter of Ti4C3 MXene eV. Moreover, a complete fracture of Mo2Ti2C3 sample was observed at almost twice the larger magnitude of external force nN, compared to nN for Ti4C3. Also, linear mode of bending of Mo2Ti2C3 sample was observed within a wider range of magnitudes of external force, compared to Ti4C3. This situation may be caused mainly by a higher mass of the molybdenum, compared to titanium, which makes Mo2Ti2C3 sample more massive than Ti4C3, and it requires a larger force magnitude for acceleration. Another reason for such behavior may be the different interactions between metal atoms within Mo layers compared to Ti layers, as within our model, bonds between Mo and C have the same strength and length as bonds between Ti and C. The latter are features of the developed approach; however, may be the reason for almost the same magnitude of critical central deflection before fracture nm for both samples.
It is important to note that in our calculations, we used empirically chosen parameters to describe interatomic forces between atoms within Mo2Ti2C3 sample; therefore, obtained parameters need further validation by experimental measurements or more accurate computations by MD or DFT methods. Nevertheless, our study provides qualitative insights into the processes that occur at atomistic levels during the bending deformation of 2D nanoribbons. Also, the obtained data may become useful in further theoretical and experimental studies of mechanical properties of 2D materials and their possible applications in the design of NEMSs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29194668/s1, Video S1: Animation of the bending deformation and fracture dynamics of Ti4C3 nanoribbon. Video S2: Animation of the bending deformation and fracture dynamics of Mo2Ti2C3 nanoribbon.
Author Contributions
Supervision, project administration, writing—review and editing, V.L.P.; hardware, visualization, writing—review and editing, I.A.L.; conceptualization, methodology, software, simulation, data analysis, visualization, writing—original draft preparation, V.B. All authors have read and agreed to the published version of the manuscript.
Funding
V.B. was supported by the scholarship of the Verkhovna Rada of Ukraine for young scientists—Doctors of Science for 2023. This work was supported in parts by the German Academic Exchange Service (DAAD).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Pacilé, D.; Meyer, J.C.; Girit, Ç.Ö.; Zettl, A. The two-dimensional phase of boron nitride: Few-atomic-layer sheets and suspended membranes. Appl. Phys. Lett. 2008, 92, 133107. [Google Scholar] [CrossRef]
- Lee, C.; Yan, H.; Brus, L.E.; Heinz, T.F.; Hone, J.; Ryu, S. Anomalous lattice vibrations of single- and few-layer MoS2. ACS Nano. 2010, 4, 2695–2700. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.G.; Rack, P.D.; Jariwala, D. Emerging nanofabrication and quantum confinement techniques for 2D materials beyond graphene. Npj 2D Mater. Appl. 2018, 2, 20. [Google Scholar] [CrossRef]
- Rizzato, S.; Monteduro, A.G.; Leo, A.; Todaro, M.T.; Maruccio, G. From ion-sensitive field-effect transistor to 2D materials field-effect-transistor biosensors. Electrochem. Sci. Adv. 2023, 3, e2200006. [Google Scholar] [CrossRef]
- Schwierz, F. Graphene transistors. Nat. Nanotechnol. 2010, 5, 487–496. [Google Scholar] [CrossRef]
- Cai, Q.; Ye, J.; Jahannia, B.; Wang, H.; Patil, C.; Al Foysal Redoy, R.; Sidam, A.; Sameer, S.; Aljohani, S.; Umer, M.; et al. Comprehensive Study and Design of Graphene Transistor. Micromachines 2024, 15, 406. [Google Scholar] [CrossRef]
- Caridad, J.M.; Castelló, Ó.; López Baptista, S.M.; Taniguchi, T.; Watanabe, K.; Roskos, H.G.; Delgado-Notario, J.A. Room-Temperature Plasmon-Assisted Resonant THz Detection in Single-Layer Graphene Transistors. Nano Lett. 2024, 24, 935–942. [Google Scholar] [CrossRef]
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-dimensional nanocrystals produced by exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248–4253. [Google Scholar] [CrossRef]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th anniversary article: MXenes: A new family of two-dimensional materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef]
- Barsoum, M.W. MAX Phases: Properties of Machinable Ternary Carbides and Nitrides; John Wiley & Sons: Weinheim, Germany, 2013. [Google Scholar] [CrossRef]
- Naguib, M.; Halim, J.; Lu, J.; Cook, K.M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. New two-dimensional niobium and vanadium carbides as promising materials for Li-ion batteries. J. Am. Chem. Soc. 2013, 135, 15966–15969. [Google Scholar] [CrossRef] [PubMed]
- Kurtoglu, M.; Naguib, M.; Gogotsi, Y.; Barsoum, M. First Principles Study of Two-dimensional Early Transition Metal Carbides. MRS Commun. 2012, 2, 133–137. [Google Scholar] [CrossRef]
- Khazaei, M.; Ranjbar, A.; Arai, M.; Sasaki, T.; Yunoki, S. Electronic properties and applications of MXenes: A theoretical review. J. Mater. Chem. C 2017, 5, 2488–2503. [Google Scholar] [CrossRef]
- Borysiuk, V.N.; Mochalin, V.N.; Gogotsi, Y. Molecular dynamic study of the mechanical properties of two-dimensional titanium carbides Tin+1Cn (MXenes). Nanotechnology 2015, 26, 265705. [Google Scholar] [CrossRef]
- Plummer, G.; Anasori, B.; Gogotsi, Y.; Tucker, G.J. Nanoindentation of monolayer Tin+1CnTx MXenes via atomistic simulations: The role of composition and defects on strength. Comput. Mater. Sci. 2019, 157, 168–174. [Google Scholar] [CrossRef]
- Ibrahim, Y.; Mohamed, A.; Abdelgawad, A.M.; Eid, K.; Abdullah, A.M.; Elzatahry, A. The Recent Advances in the Mechanical Properties of Self-Standing Two-Dimensional MXene-Based Nanostructures: Deep Insights into the Supercapacitor. Nanomaterials 2020, 10, 1916. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Li, Y.; He, H.; Wang, Q.; Lan, L.; Song, W.; Qiu, T.; Lu, W. Flexible two-dimensional MXene-based antennas. Nanoscale Horiz. 2023, 8, 309–319. [Google Scholar] [CrossRef]
- Borysiuk, V.N.; Mochalin, V.N.; Gogotsi, Y. Bending rigidity of two-dimensional titanium carbide (MXene) nanoribbons: A molecular dynamics study. Comput. Mater. Sci. 2018, 143, 418–424. [Google Scholar] [CrossRef]
- Kang, J.W.; Lee, S. Molecular dynamics study on the bending rigidity of graphene nanoribbons. Comput. Mater. Sci. 2013, 74, 107–113. [Google Scholar] [CrossRef]
- Lipatov, A.; Lu, H.; Alhabeb, M.; Anasori, B.; Gruverman, A.; Gogotsi, Y.; Sinitskii, A. Elastic properties of 2D Ti3C2Tx MXene monolayers and bilayers. Sci. Adv. 2018, 4, eaat0491. [Google Scholar] [CrossRef]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Chernov, S.V.; Makukha, Z.M.; Protsenko, I.Y.; Nepijko, S.A.; Elmers, H.J.; Schönhense, G. Test object for emission electron microscope. Appl. Phys. A 2014, 114, 1383–1385. [Google Scholar] [CrossRef]
- Watanabe, T.; Hatanaka, S.; Nakano, K.; Popov, V.L. Sliding friction in contacts with one- and two-dimensional viscoelastic foundations and viscoelastic half-space. Facta Univ. Ser. Mech. Eng. 2024, 22, 529–545. [Google Scholar]
- Willert, E. Self-consistency conditions in static three-body elastic tangential contact. Facta Univ. Ser. Mech. Eng. 2024, 22, 449–457. [Google Scholar]
- Lyashenko, I.A.; Khomenko, A.V.; Metlov, L.S. Nonlinear thermodynamic model of boundary friction. J. Frict. Wear 2011, 32, 113–123. [Google Scholar] [CrossRef]
- Kwon, O.K.; Lee, J.H.; Kim, K.S.; Kang, J.W. Developing ultrasensitive pressure sensor based on graphene nanoribbon: Molecular dynamics simulation. Phys. E. Low Dimens. Syst. Nanostruct. 2013, 47, 6–11. [Google Scholar] [CrossRef]
- Anasori, B.; Xie, Y.; Beidaghi, M.; Lu, J.; Hosler, B.C.; Hultman, L.; Kent, P.R.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional, Ordered, Double Transition Metals Carbides (MXenes). ACS Nano 2015, 9, 9507–9516. [Google Scholar] [CrossRef]
- Anasori, B.; Shi, C.; Moon, E.J.; Xie, Y.; Voigt, C.A.; Kent, P.R.; May, S.J.; Billinge, S.J.; Barsoum, M.W.; Gogotsi, Y. Control of electronic properties of 2D carbides (MXenes) by manipulating their transition metal layers. Nanoscale Horiz. 2016, 13, 227–234. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zhang, D.; Ashton, M.; Ostadhossein, A.; Van Duin, A.C.T.; Hennig, R.G.; Sinnott, S.B. Computational study of low interlayer friction in Tin+1Cn (n = 1, 2, and 3) MXene. ACS Appl. Mater. Interfaces 2017, 9, 34467–34479. [Google Scholar] [CrossRef]
- Branco, D.d.C.; Cheng, G.J. Employing Hybrid Lennard-Jones and Axilrod-Teller Potentials to Parametrize Force Fields for the Simulation of Materials’ Properties. Materials 2021, 14, 6352. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, R.; Naguib, M.; Yilmaz, D.E.; Nanda, J.; Duin, A.C. A comparative study on the oxidation of two-dimensional Ti3C2 MXene structures in different environments. J. Mater. Chem. A 2018, 6, 12733–12743. [Google Scholar] [CrossRef]
- Zhou, X.W.; Wadley, H.N.G.; Johnson, R.A.; Larson, D.J.; Tabat, N.; Cerezo, A.; Petford-Long, A.K.; Smith, G.D.W.; Clifton, P.H.; Martens, R.L.; et al. Atomic scale structure of sputtered metal multilayers. Acta Mater. 2001, 49, 4005–4015. [Google Scholar] [CrossRef]
- Borysiuk, V.; Mochalin, V.N. Thermal stability of two-dimensional titanium carbides Tin+1Cn (MXenes) from classical molecular dynamics simulations. MRS Commun. 2019, 9, 203–208. [Google Scholar] [CrossRef]
- Borysiuk, V.; Lyashenko, I.A.; Popov, V.L. Molecular Dynamics Study of Friction between Ag Nanoparticle and Two-Dimensional Titanium Carbide Ti2C (MXene). Crystals 2024, 14, 272. [Google Scholar] [CrossRef]
- Jiang, S.; Sun, L.; Zhan, H.; Zheng, Z.; Peng, X.; Lü, C. Bending behavior of diamane and twisted bilayer graphene: Insights from four-point bending deformation. Thin Walled Struct. 2024, 195, 111415. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).