

Microwave-Promoted Total Synthesis of Puniceloid D for Modulating the Liver X Receptor
 , , and
, , and
Abstract
:
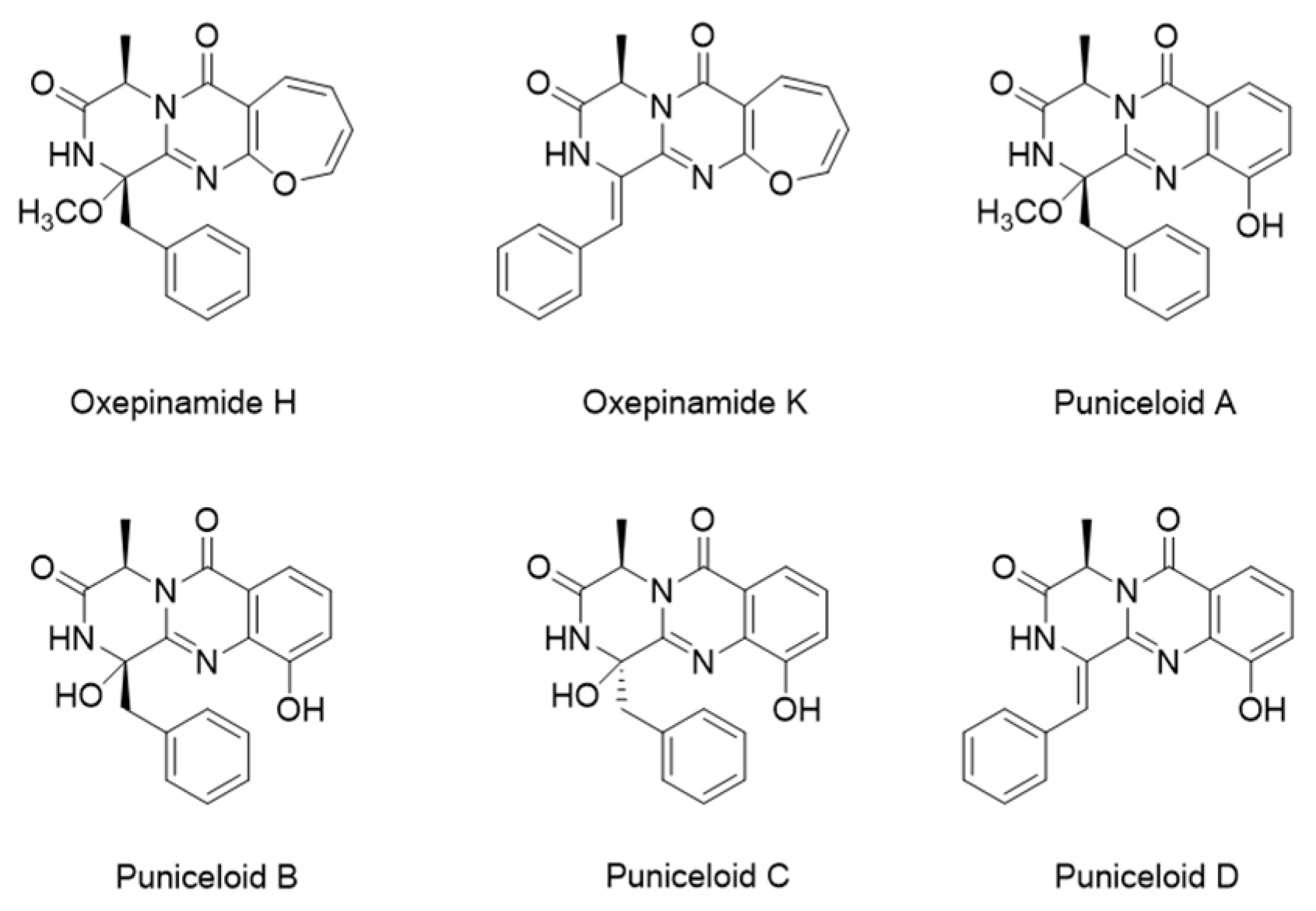
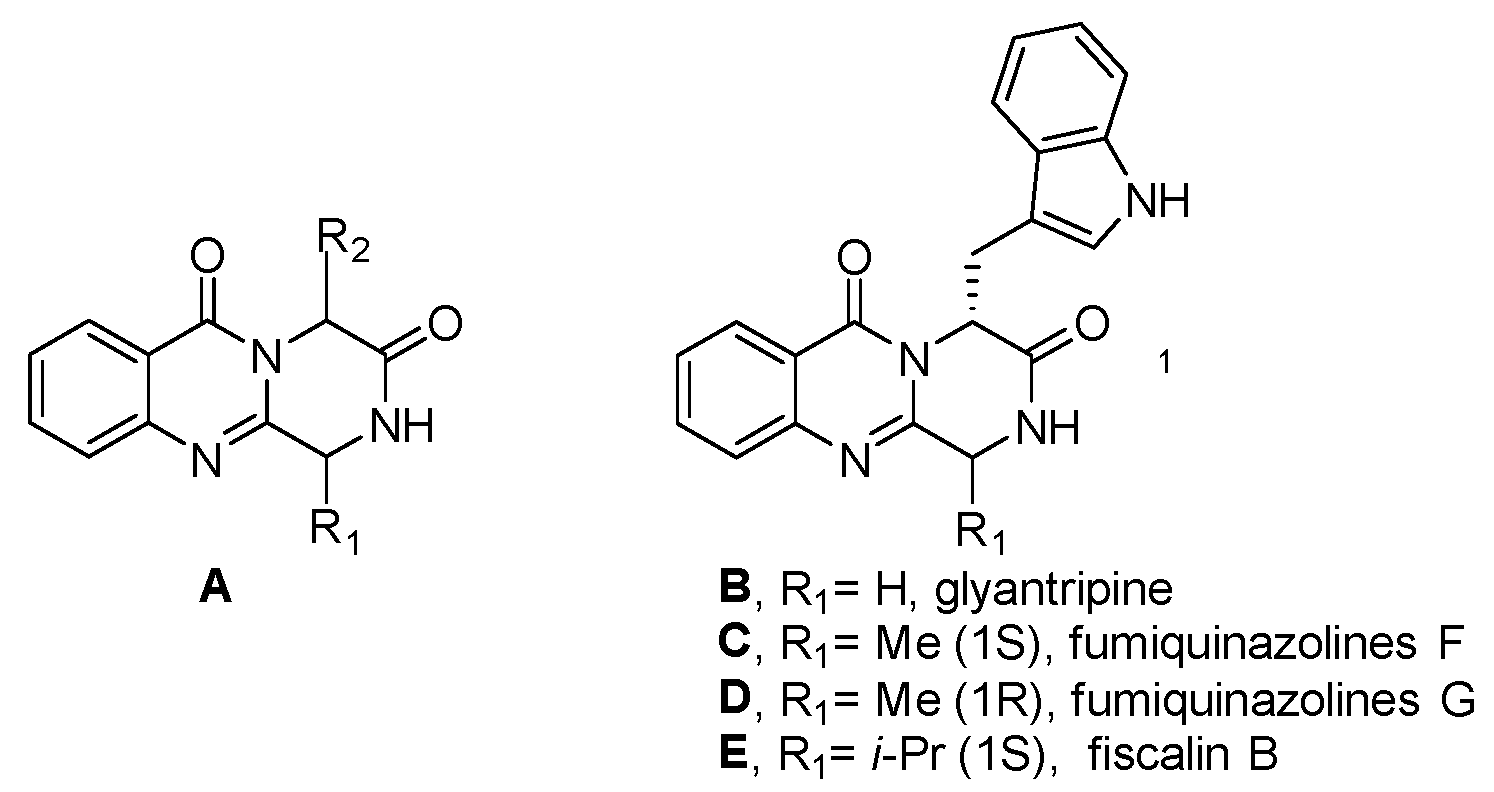
1. Introduction
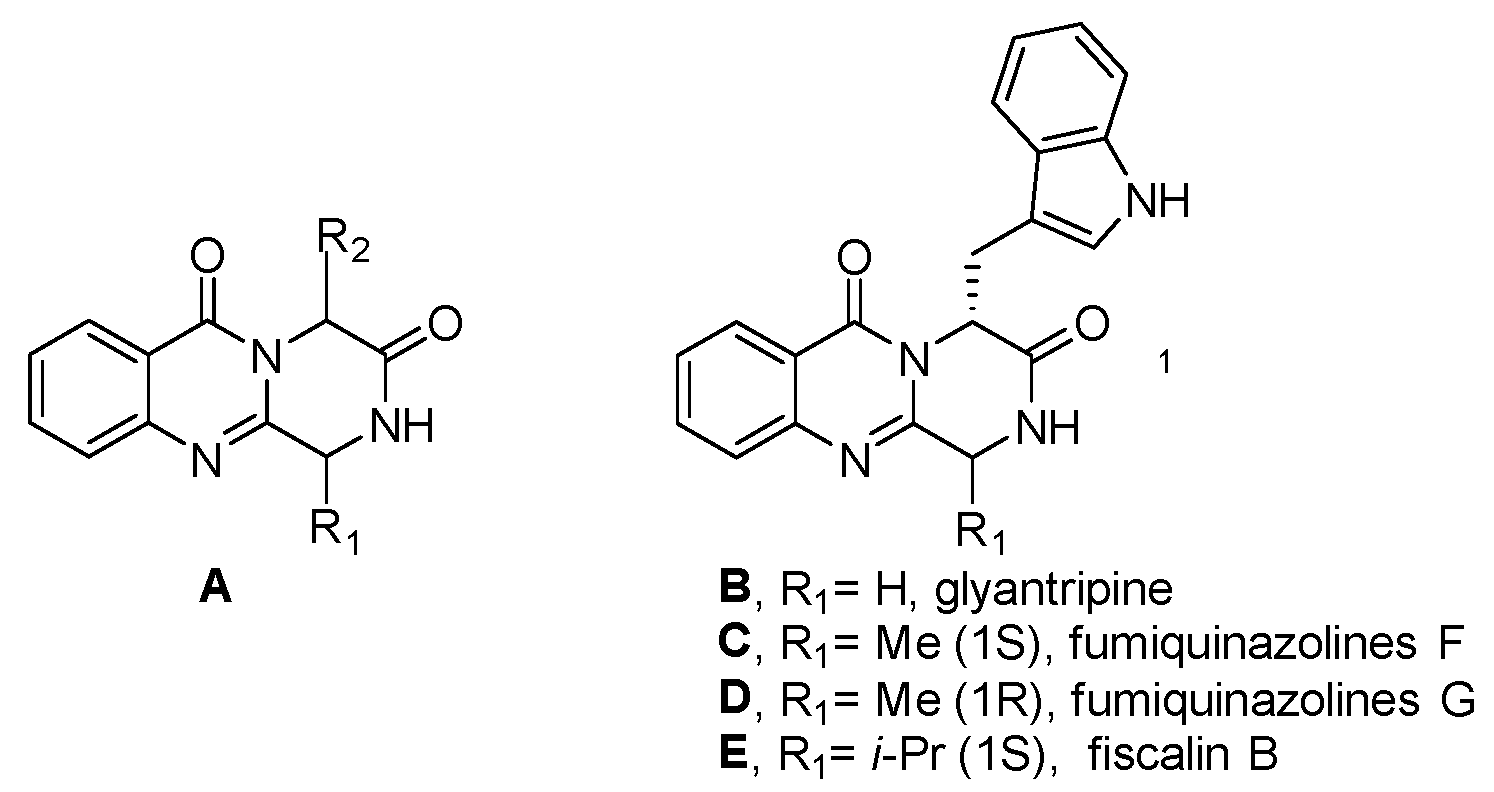
2. Results
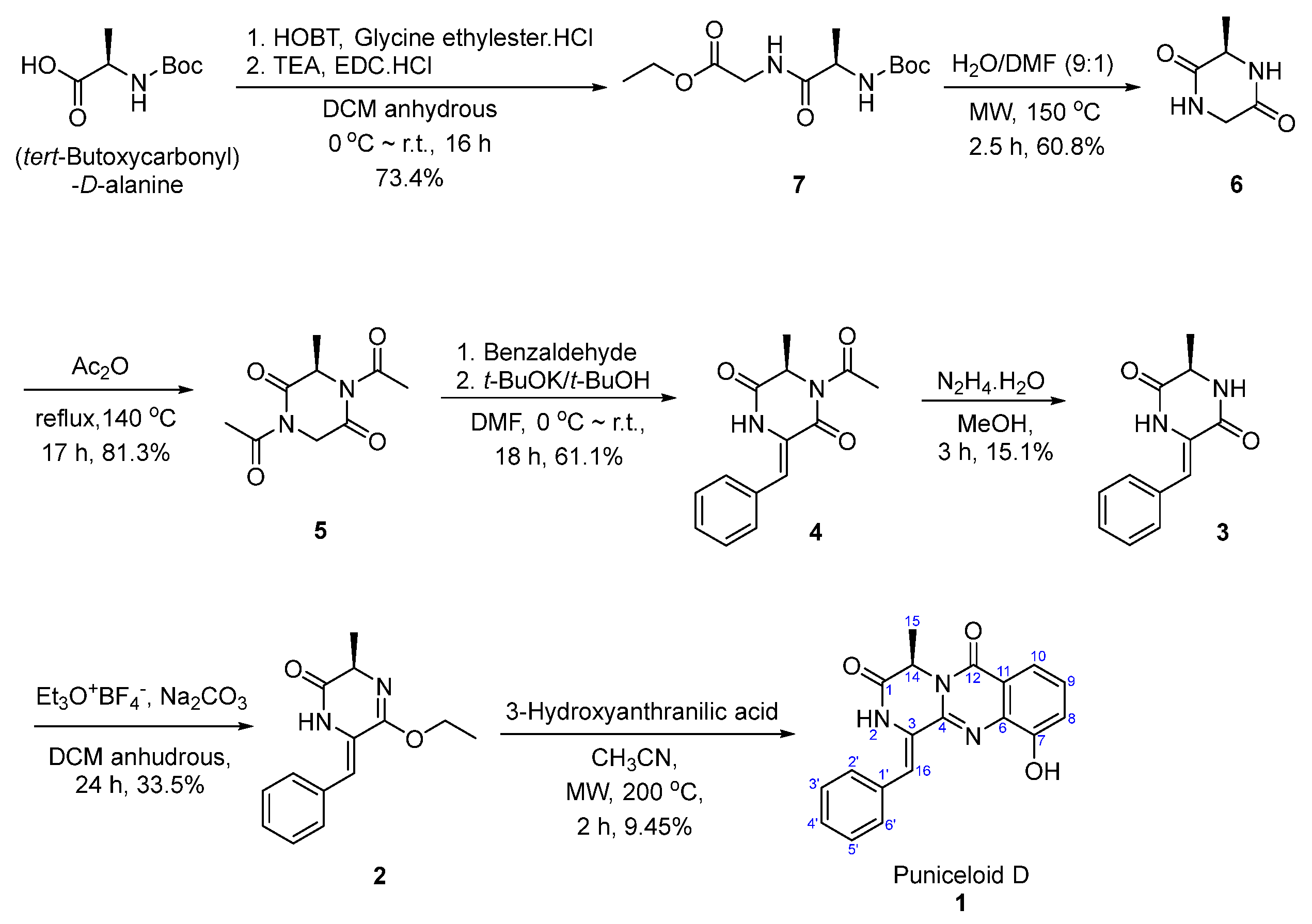
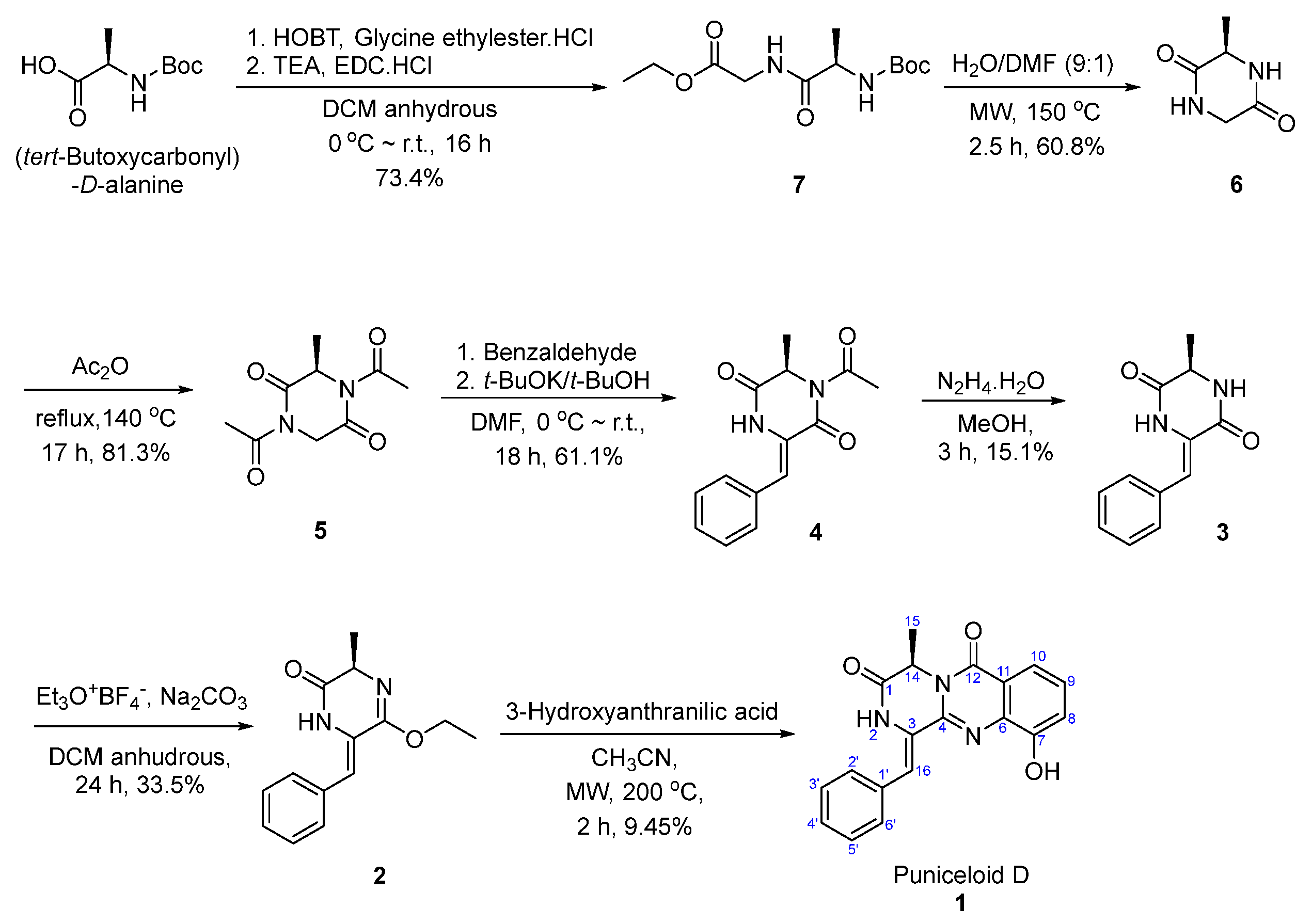
Total Synthesis of Puniceloid D
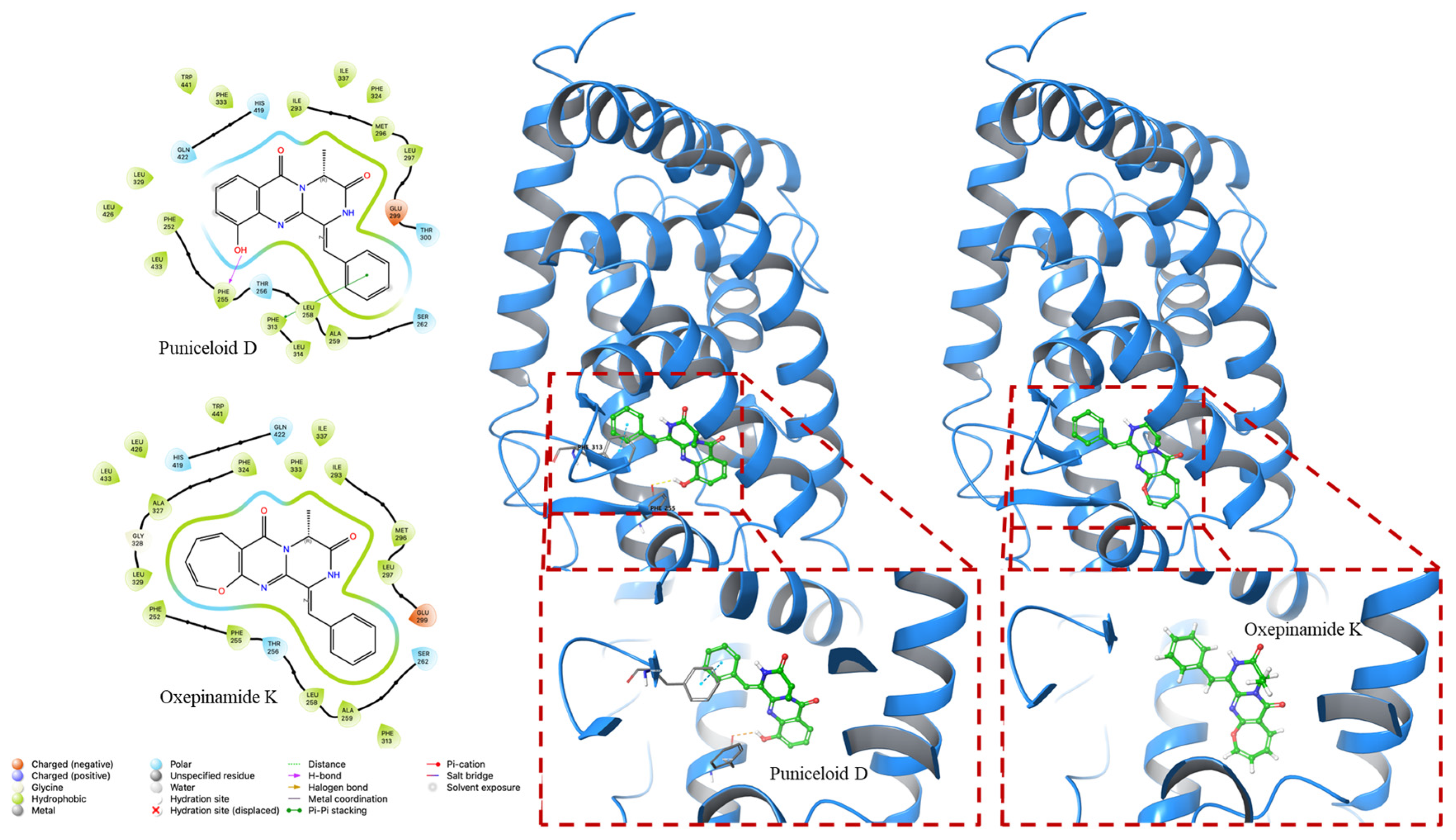
3. Discussion
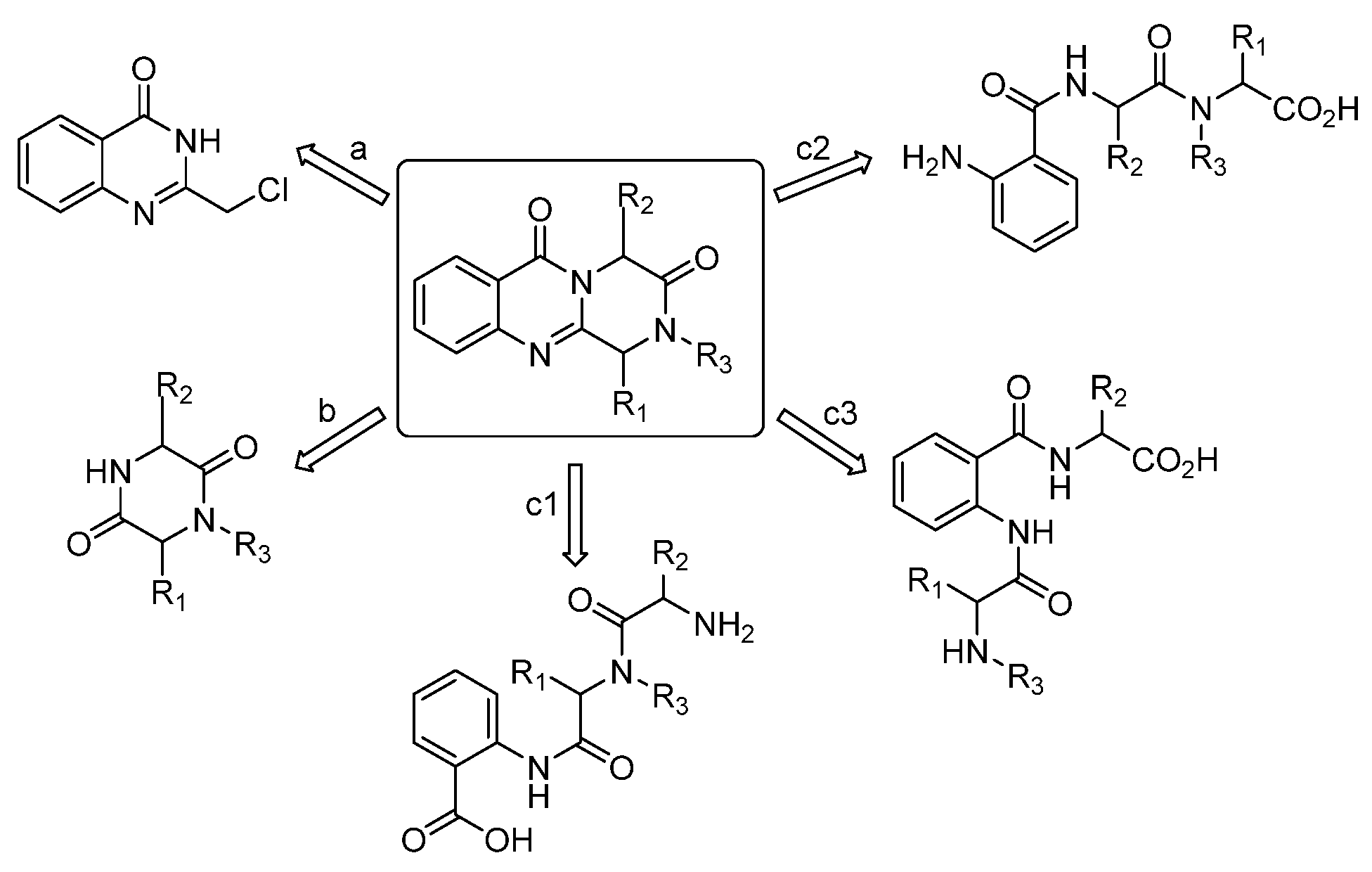
4. Materials and Methods
4.1. Materials and Synthetic Methods
4.1.1. Synthesis of Ethyl (tert-Butoxycarbonyl)-D-alanylglycinate (7)
4.1.2. Synthesis of (R)-3-methylpiperazine-2,5-dione (6)
4.1.3. Synthesis of (R)-1,4-diacetyl-3-methylpiperazine-2,5-dione (5)
4.1.4. Synthesis of (R,Z)-1-acetyl-3-benzylidene-6-methylpiperazine-2,5-dione (4)
4.1.5. Synthesis of (R,Z)-3-benzylidene-6-methylpiperazine-2,5-dione (3)
4.1.6. Synthesis of (R,Z)-6-benzylidene-5-ethoxy-3-methyl-3,6-dihydropyrazin-2(1H)-one (2)
4.1.7. Synthesis of Puniceloid D (1)
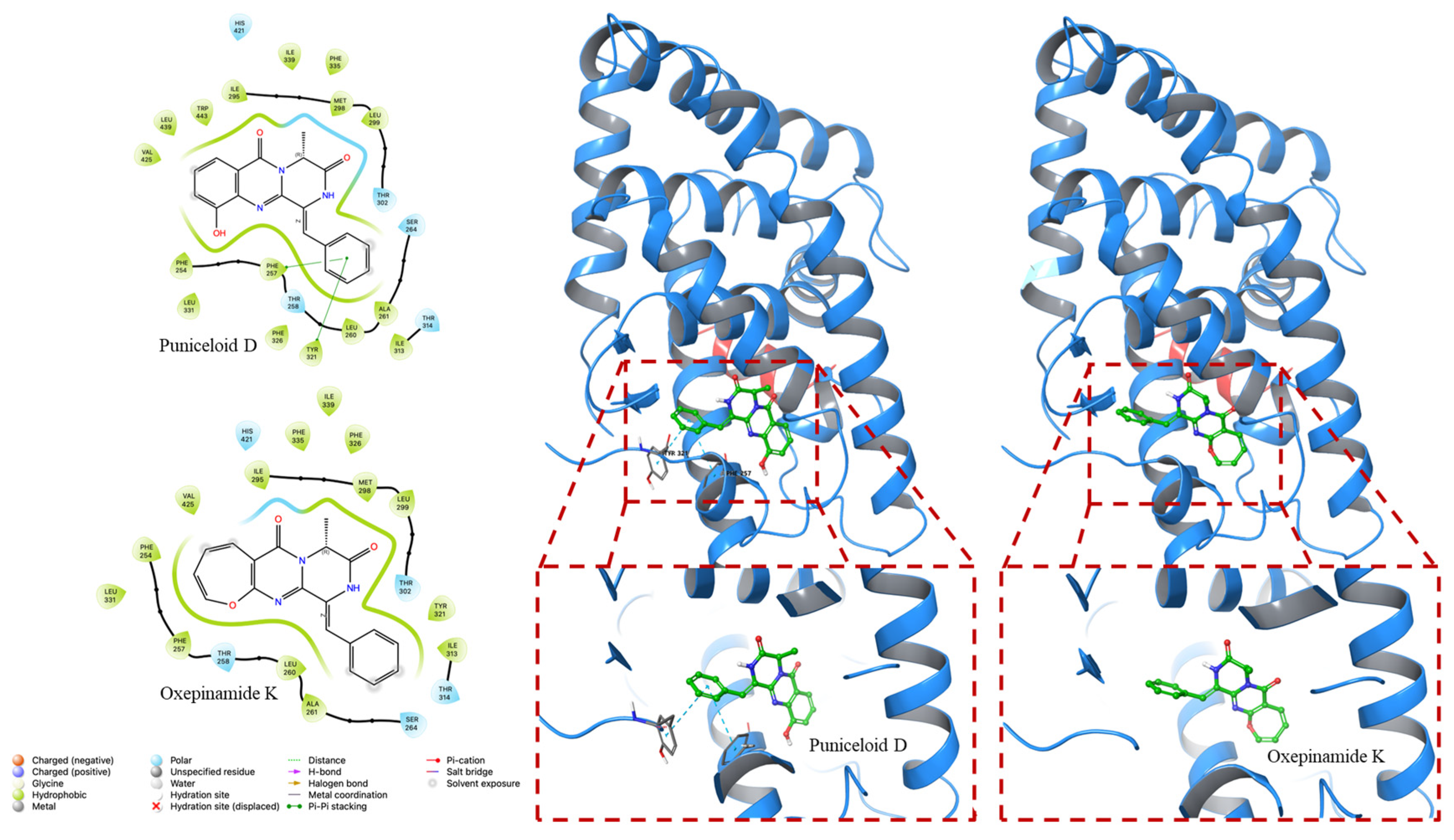
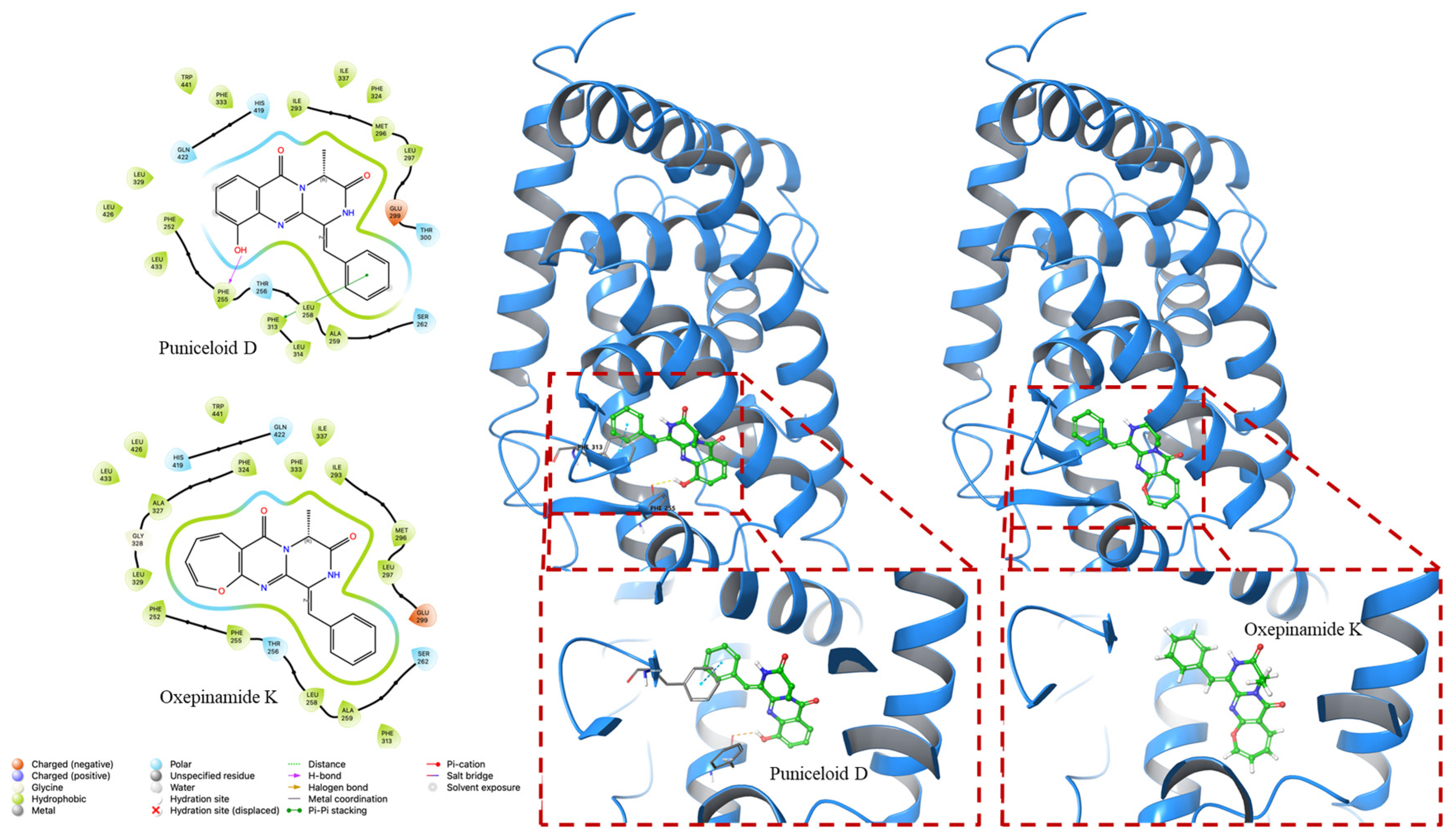
4.2. Molecular Docking Experiment
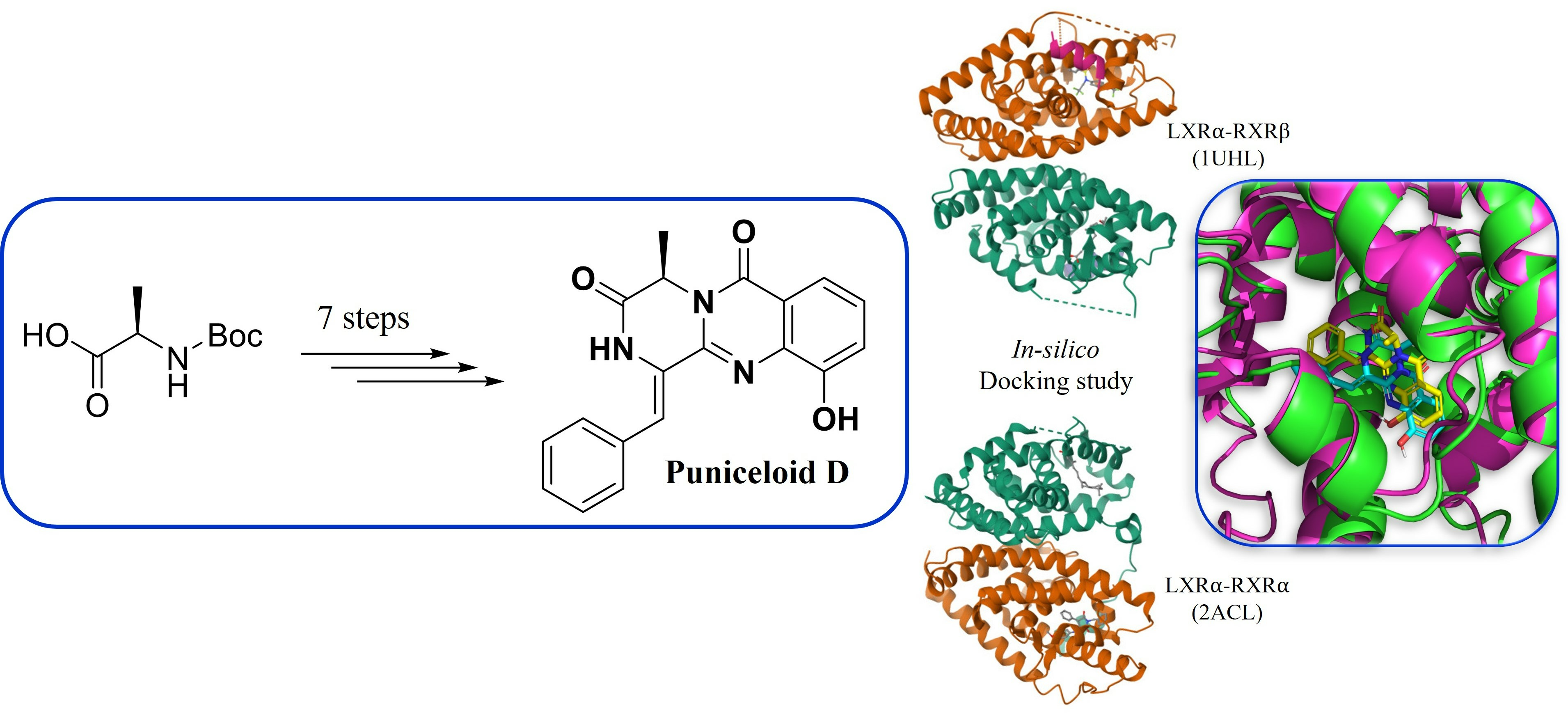
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hiebl, V.; Ladurner, A.; Latkolik, S.; Dirsch, V.M. Natural Products as Modulators of the Nuclear Receptors and Metabolic Sensors LXR, FXR and RXR. Biotechnol. Adv. 2018, 36, 1657–1698. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Park, Y.J. Metabolic Regulation of Nuclear Receptors. J. Korean Endocr. Soc. 2008, 23, 155–164. [Google Scholar] [CrossRef]
- Degirolamo, C.; Sabbà, C.; Moschetta, A. Intestinal Nuclear Receptors in HDL Cholesterol Metabolism. J. Lipid Res. 2015, 56, 1262–1270. [Google Scholar] [CrossRef]
- Gronemeyer, H.; Gustafsson, J.-Å.; Laudet, V. Principles for Modulation of the Nuclear Receptor Superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, C.; Gödtke, L.; Reichstein, A.; Keiser, M.; Busch, D.; Drozdzik, M.; Oswald, S. Gene Expression and Protein Abundance of Nuclear Receptors in Human Intestine and Liver: A New Application for Mass Spectrometry-Based Targeted Proteomics. Molecules 2022, 27, 4629. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Evans, R.M. The RXR Heterodimers and Orphan Receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An Oxysterol Signalling Pathway Mediated by the Nuclear Receptor LXRα. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Jamroz-Wiśniewska, A.; Wójcicka, G.; Horoszewicz, K.; Bełtowski, J. Liver X Receptors (LXRs). Part I: Structure, Function, Regulation of Activity, and Role in Lipid Metabolism. Adv. Hyg. Exp. Med. 2007, 61, 736–759. [Google Scholar]
- Li, N.; Wang, X.; Zhang, J.; Liu, C.; Li, Y.; Feng, T.; Xu, Y.; Si, S. Identification of a Novel Partial Agonist of Liver X Receptor α (LXRα) via Screening. Biochem. Pharmacol. 2014, 92, 438–447. [Google Scholar] [CrossRef]
- Smith, T.K.T.; Kahiel, Z.; LeBlond, N.D.; Ghorbani, P.; Farah, E.; Al-Awosi, R.; Cote, M.; Gadde, S.; Fullerton, M.D. Characterization of Redox-Responsive LXR-Activating Nanoparticle Formulations in Primary Mouse Macrophages. Molecules 2019, 24, 3751. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yang, F.; Kang, J.; Gan, H.; Yang, X.; Lai, X.; Gao, Y. Identfication of Potent LXRβ-Selective Agonists without LXRα Activation by in Silico Approaches. Molecules 2018, 23, 1349. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Mangelsdorf, D.J. The Role of Orphan Nuclear Receptors in the Regulation of Cholesterol Homeostasis. Annu. Rev. Cell Dev. Biol. 2000, 16, 459–481. [Google Scholar] [CrossRef]
- Apfel, R.; Benbrook, D.; Lernhardt, E.; Ortiz, M.A.; Salbert, G.; Pfahl, M. A Novel Orphan Receptor Specific for a Subset of Thyroid Hormone-Responsive Elements and Its Interaction with the Retinoid/Thyroid Hormone Receptor Subfamily. Mol. Cell. Biol. 1994, 14, 7025–7035. [Google Scholar] [PubMed]
- Ni, M.; Zhang, B.; Zhao, J.; Feng, Q.; Peng, J.; Hu, Y.; Zhao, Y. Biological Mechanisms and Related Natural Modulators of Liver X Receptor in Nonalcoholic Fatty Liver Disease. Biomed. Pharmacother. 2019, 113, 108778. [Google Scholar] [CrossRef]
- Maqdasy, S.; Trousson, A.; Tauveron, I.; Volle, D.H.; Baron, S.; Lobaccaro, J.-M.A. Once and for All, LXRα and LXRβ Are Gatekeepers of the Endocrine System. Mol. Aspects Med. 2016, 49, 31–46. [Google Scholar] [CrossRef]
- El-Gendy, B.E.-D.M.; Goher, S.S.; Hegazy, L.S.; Arief, M.M.H.; Burris, T.P. Recent Advances in the Medicinal Chemistry of Liver X Receptors: Miniperspective. J. Med. Chem. 2018, 61, 10935–10956. [Google Scholar] [CrossRef]
- Komati, R.; Spadoni, D.; Zheng, S.; Sridhar, J.; Riley, K.E.; Wang, G. Ligands of Therapeutic Utility for the Liver X Receptors. Molecules 2017, 22, 88. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, X.; Lu, X.; Zheng, Z.; Ma, X.; Qi, S. Diketopiperazine-Type Alkaloids from a Deep-Sea-Derived Aspergillus Puniceus Fungus and Their Effects on Liver X Receptor α. J. Nat. Prod. 2019, 82, 1558–1564. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Chen, J.; Wu, X.-F. Recent Advances in 4 (3 H)-Quinazolinone Syntheses. RSC Adv. 2014, 4, 12065–12077. [Google Scholar] [CrossRef]
- Mhaske, S.B.; Argade, N.P. The Chemistry of Recently Isolated Naturally Occurring Quinazolinone Alkaloids. Tetrahedron 2006, 62, 9787–9826. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2005, 22, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Avendano, C.; Menendez, J. Chemistry of Pyrazino [2, 1-b] Quinazoline-3, 6-Diones. Curr. Org. Chem. 2003, 7, 149–173. [Google Scholar] [CrossRef]
- Park, J.; Nurkolis, F.; Won, H.; Yang, J.; Oh, D.; Jo, H.; Choi, J.; Chung, S.; Kurniawan, R.; Kim, B. Could Natural Products Help in the Control of Obesity? Current Insights and Future Perspectives. Molecules 2023, 28, 6604. [Google Scholar] [CrossRef] [PubMed]
- Han, D.-G.; Cho, S.-S.; Kwak, J.-H.; Yoon, I.-S. Medicinal Plants and Phytochemicals for Diabetes Mellitus: Pharmacokinetic Characteristics and Herb-Drug Interactions. J. Pharm. Investig. 2019, 49, 603–612. [Google Scholar] [CrossRef]
- Hosseini Nasab, N.; Shah, F.H.; Kim, S.J. Pharmacological Role of Ostericum Koreanum: A Short Viewpoint. Nat. Prod. Commun. 2021, 16, 1934578X211050790. [Google Scholar] [CrossRef]
- Cledera, P.; Avendaño, C.; Menéndez, J.C. Comparative Study of Synthetic Approaches to 1-Arylmethylenepyrazino [2, 1-b] Quinazoline-3, 6-Diones. Tetrahedron 1998, 54, 12349–12360. [Google Scholar] [CrossRef]
- Kim, J.; Movassaghi, M. Concise Total Synthesis and Stereochemical Revision of (+)-Naseseazines A and B: Regioselective Arylative Dimerization of Diketopiperazine Alkaloids. J. Am. Chem. Soc. 2011, 133, 14940–14943. [Google Scholar] [CrossRef]
- Smeenk, J.M.; Ayres, L.; Stunnenberg, H.G.; van Hest, J.C.M. Polymer Protein Hybrids. Macromol. Symp. 2005, 225, 1–8. [Google Scholar] [CrossRef]
- Mollica, A.; Davis, P.; Ma, S.-W.; Lai, J.; Porreca, F.; Hruby, V.J. Synthesis and Biological Evaluation of New Biphalin Analogues with Non-Hydrazine Linkers. Bioorg. Med. Chem. Lett. 2005, 15, 2471–2475. [Google Scholar] [CrossRef]
- Coursindel, T.; Restouin, A.; Dewynter, G.; Martinez, J.; Collette, Y.; Parrot, I. Stereoselective Ring Contraction of 2, 5-Diketopiperazines: An Innovative Approach to the Synthesis of Promising Bioactive 5-Membered Scaffolds. Bioorg. Chem. 2010, 38, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Qin, X.; Li, D.; Tu, Z.; Li, J.; Zhou, X.; Wang, J.; Yang, B.; Lin, X.; Liu, J. Design and Synthesis of Novel Soluble 2, 5-Diketopiperazine Derivatives as Potential Anticancer Agents. Eur. J. Med. Chem. 2014, 83, 236–244. [Google Scholar] [CrossRef]
- Mollica, A.; Costante, R.; Fiorito, S.; Genovese, S.; Stefanucci, A.; Mathieu, V.; Kiss, R.; Epifano, F. Synthesis and Anti-Cancer Activity of Naturally Occurring 2, 5-Diketopiperazines. Fitoterapia 2014, 98, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Fan, W.; Szajda, M.; Li, Q.; Caster, K.C. Conjugated Systems Derived from Piperazine-2, 5-dione. J. Heterocycl. Chem. 1988, 25, 591–597. [Google Scholar] [CrossRef]
- Wang, Z. Comprehensive Organic Name Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; Volume 2. [Google Scholar]
- Cledera, P.; Sanchez, J.D.; Caballero, E.; Yates, T.; Ramirez, E.G.; Avendano, C.; Ramos, M.T.; Menendez, J.C. Microwave-Assisted, Solvent-Free Synthesis of Several Quinazoline Alkaloid Frameworks. Synthesis 2007, 21, 3390–3398. [Google Scholar]
- Argyrakis, W.; Köppl, C.; Werner, H.; Frey, W.; Baro, A.; Laschat, S. A Combined Quantum Mechanical and Experimental Approach towards Chiral Diketopiperazine Hydroperoxides. J. Phys. Org. Chem. 2011, 24, 682–692. [Google Scholar] [CrossRef]
- Albers, M.; Blume, B.; Schlueter, T.; Wright, M.B.; Kober, I.; Kremoser, C.; Deuschle, U.; Koegl, M. A Novel Principle for Partial Agonism of Liver X Receptor Ligands: Competitive Recruitment of Activators and Repressors. J. Biol. Chem. 2006, 281, 4920–4930. [Google Scholar] [CrossRef]
- Senbonmatsu, Y.; Kimura, S.; Akiba, M.; Ando, S.; Saito, N. Preparation of Chiral Right-Half Models of Antitumor Bistetrahydroisoquinolinequinone Natural Products. Heterocycles 2018, 97, 1050–1067. [Google Scholar]
- Kanzaki, H.; Imura, D.; Nitoda, T.; Kawazu, K. Enzymatic Conversion of Cyclic Dipeptides to Dehydro Derivatives That Inhibit Cell Division. J. Biosci. Bioeng. 2000, 90, 86–89. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, S.; Chen, Y.; Tian, X.; Zhang, H.; Zhang, G.; Zhu, Y.; Zhang, S.; Zhang, W.; Zhang, C. New Diketopiperazine Derivatives from a Deep-Sea-Derived Nocardiopsis Alba SCSIO 03039. J. Antibiot. 2013, 66, 31–36. [Google Scholar] [CrossRef]
- Nishioka, T.; Endo-Umeda, K.; Ito, Y.; Shimoda, A.; Takeuchi, A.; Tode, C.; Hirota, Y.; Osakabe, N.; Makishima, M.; Suhara, Y. Synthesis and in Vitro Evaluation of Novel Liver X Receptor Agonists Based on Naphthoquinone Derivatives. Molecules 2019, 24, 4316. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Synthesized | Reported [19] | ||
|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 166.3 | data | 166.6, C | |
| 2-NH | 10.56, s | 10.52, s | ||
| 3 | 125.9 | 126.0, C | ||
| 4 | 143.3 | 144.1, C | ||
| 6 | 135.6 | 136.1, C | ||
| 7 | 151.7 | 152.8, C | ||
| 7-OH | 9.76, s | 9.75, s | ||
| 8 | 117.8 | 7.26, dd (7.9, 1.4) | 118.4, CH | 7.26, dd (8.0, 1.0) |
| 9 | 128.6 | 7.37, t (7.9) | 128.1, CH | 7.35, t (8.0) |
| 10 | 116.3 | 7.58, dd (7.9, 1.4) | 116.0, CH | 7.58, dd (8.0, 1.0) |
| 11 | 120.4 | 120.4, C | ||
| 12 | 160.0 | 159.5, C | ||
| 14 | 52.0 | 5.22, q (6.8) | 51.2, CH | 5.22, q (7.0) |
| 15 | 19.5 | 1.49, d (7.0) | 18.2, CH3 | 1.50, d (7.0) |
| 16 | 117.7 | 7.71, s | 117.2, CH | 7.74, s |
| 1′ | 133.0 | 134.0, C | ||
| 2′,6′ | 129.2 | 7.66, d (7.71) | 129.4, CH | 7.65, d (7.5) |
| 3′,5′ | 128.7 | 7.47, t (7.62) | 128.7, CH | 7.47, t (7.5) |
| 4′ | 129.7 | 7.37, m | 127.6, CH | 7.37, m |
| PDB ID | Compounds | Docking Score (Kcal/mol) | G Score (Kcal/mol) |
|---|---|---|---|
| 1UHL | Puniceloid D | −8.535 | −8.841 |
| Oxepinamide K | −8.113 | −8.113 | |
| 2ACL | Puniceloid D | −9.783 | −10.089 |
| Oxepinamide K | −8.252 | −8.252 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.J.; Hosseininasab, N.; Park, J.; Hyun, S.; Jung, J.-K.; Kwak, J.-H. Microwave-Promoted Total Synthesis of Puniceloid D for Modulating the Liver X Receptor. Molecules 2024, 29, 416. https://doi.org/10.3390/molecules29020416
Jung YJ, Hosseininasab N, Park J, Hyun S, Jung J-K, Kwak J-H. Microwave-Promoted Total Synthesis of Puniceloid D for Modulating the Liver X Receptor. Molecules. 2024; 29(2):416. https://doi.org/10.3390/molecules29020416
Chicago/Turabian StyleJung, Young Jin, Narges Hosseininasab, Jungjin Park, Soonsil Hyun, Jae-Kyung Jung, and Jae-Hwan Kwak. 2024. "Microwave-Promoted Total Synthesis of Puniceloid D for Modulating the Liver X Receptor" Molecules 29, no. 2: 416. https://doi.org/10.3390/molecules29020416
APA StyleJung, Y. J., Hosseininasab, N., Park, J., Hyun, S., Jung, J.-K., & Kwak, J.-H. (2024). Microwave-Promoted Total Synthesis of Puniceloid D for Modulating the Liver X Receptor. Molecules, 29(2), 416. https://doi.org/10.3390/molecules29020416