Abstract
The stem bark of black locust (Robinia pseudoacacia L.) was extracted, and nine antioxidant compounds (R1–R9) were detected by high-performance thin-layer chromatography combined with the radical scavenging 2,2-diphenyl-1-picrylhydrazyl (DPPH•) assay, multi-detection, and heated electrospray high-resolution mass spectrometry. For structure elucidation, the methanolic crude extract was fractionated by solid-phase extraction, and the compounds were isolated by reversed-phase high-performance liquid chromatography with diode array detection. The structures of isolated compounds were elucidated by nuclear magnetic resonance and attenuated total reflectance Fourier-transform infrared spectroscopy as well as gas chromatography-mass spectrometry to determine the double bond position. 3-O-Caffeoyl oleanolic acid (R1), oleyl (R2), octadecyl (R3), gadoleyl (R4), eicosanyl (R5), (Z)-9-docosenyl (R6), docosyl (R7), tetracosyl (R8), and hexacosanyl (R9) caffeates were identified. While R1 has been reported in R. pseudoacacia stem bark, the known R3, R5, R7, R8, and R9 are described for the first time in this species, and the R2, R4, and R6 are new natural compounds. All nine caffeates demonstrated antioxidant activity. The antioxidant effects of the isolated compounds R1–R8 were quantified by a microplate DPPH• assay, with values ranging from 0.29 to 1.20 mol of caffeic acid equivalents per mole of isolate.
Keywords:
black locust (Robinia pseudoacacia L.); phenolic esters of fatty alcohols; high-performance thin-layer chromatography—effect-directed analysis (HPTLC–EDA); antioxidant assay; heated electrospray high-resolution mass spectrometry (HESI-HRMS); bioassay-guided isolation; solid-phase extraction (SPE); reversed-phase high-performance liquid chromatography with diode array detection (RP-HPLC–DAD); nuclear magnetic resonance (NMR) spectroscopy; attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy; gas chromatography-mass spectrometry (GC–MS) 1. Introduction
The North American black locust (Robinia pseudoacacia L., family Fabaceae) has been widely planted all over the world, initially as an ornamental tree and later for soil and water conversation, like in Europe since the 17th century [1,2]. It is a fast-growing tree that reproduces both sexually and vegetatively; therefore, it has become one of the most aggressively invasive woody plants with a high biomass worldwide [2]. Due to its allelopathic potential, it often overgrows the indigenous plants, and as a nitrogen-fixing species, it can alter the native vegetation [3]. Despite its environmental drawbacks, it offers economic benefits, particularly in the honey and wood industries [4]. Furthermore, black locust has been used in traditional folk medicine, especially in Europe and Asia, due to its astringent, cholagogue, diuretic, anti-inflammatory, purgative, spasmolytic, and sedative properties, as well as Cherokee treated toothache with it [5,6]. Its beneficial effects [7,8], such as antibacterial, antifungal, antioxidant, anti-inflammatory, and cytotoxic properties, are primarily attributed to the high content of diverse phenoloids [9]. Flowers and leaves are rich sources of phenolic acids (e.g., caffeoylquinic acids, caffeic and coumaric acids and their hexosides, coumaroylquinic acids, ellagic acid hexoside, gallic acid, and p-hydroxybenzoic acid), flavonoids (apigenin, catechin, procyanidin dimers and trimers, quercetin and kaempferol derivatives, and vescalagins), and tannins [9,10], all of which exhibit various biological activities. The diversity of flavonoid aglycones and hydroxycinnamic acid derivatives originated from propolis and nectar-derived kaempferol glycosides enables the floral authentication of black locust honey [11]. However, black locust also contains toxic glycoproteins, lectins, and the homo-monoterpene robinlin that can possess pharmacological activities beyond cytotoxicity [12].
The wood of R. pseudoacacia is predominantly composed of structural polysaccharides (e.g., cellulose, hemicellulose, and lignin) that conceal a wide range of valuable compounds [13], and it can be the source of biofuels [14]. The heartwood contains several phenolic acids (e.g., caffeic acid, chlorogenic acid, ellagic acid, ferulic acid, p-coumaric acid, gallic acid, ellagic acid, p-hydroxybenzoic acid, and protocatechuic acid), flavonoids (e.g., di-O-methylquercetin B, quercetin, epigallocatechin, fustin, catechin, kaempferol, myricetin, procyanidin dimer, robinetin, and dihydrorobinetin), along with stilbenes (resveratrol and piceatannol) [15,16,17]. Interestingly, the bark lacks robinetin, but dihydrorobinetin and phenolic acids like catechin, epicatechin, caffeic acid, and ferulic acid have been detected as defensive compounds in the bark [18,19].
Plant phenolics play a pivotal role in plant growth, development, and defense by displaying antioxidant, antimicrobial, allelopathic, and UV-blocking effects [20]. Reactive oxygen species (ROS) and free radicals are essential for cell signaling and other vital physiological processes. However, during various diseases, including inflammatory and infectious conditions, their overproduction can lead to potential cellular damage [20,21,22]. Natural antioxidants, such as phenolic compounds, can diminish this unfavorable effect by scavenging the free radicals and converting them into stable forms [23]. High-performance thin-layer chromatography combined with multi-detection (HPTLC–UV/VIS/FLD), the 2,2-diphenyl-1-picrylhydrazyl (DPPH•) assay, and derivatization via the Natural Product reagent A ensure an efficient, high-throughput screening for identifying antioxidant compounds in complex matrices, such as plant extracts [24,25,26]. It is an effective monitoring tool in bioactivity-guided compound isolation [27].
The study aimed at screening, characterization, isolation, and identification of antioxidant compounds from the methanolic bark extracts of the black locust. Various analytical techniques were utilized, including reversed-phase (RP)-HPTLC–DPPH• assay, RP-HPTLC–UV/VIS/FLD–densitometry, RP-HPTLC–heated electrospray high-resolution mass spectrometry (HESI-HRMS), reversed-phase high-performance liquid chromatography diode array detection (RP-HPLC–DAD), attenuated total reflectance Fourier-transform infrared (ATR–FTIR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and gas chromatography–mass spectrometry (GC–MS). The antioxidant activity of the isolated compounds was assessed by a DPPH• microplate assay.
2. Results and Discussion
2.1. RP-HPTLC–DPPH• Assay Screening and Assignment by RP-HPTLC–HESI-HRMS
Antioxidant compounds of the methanolic crude extract obtained from the black locust bark were separated on RP18 HPTLC plates using acetonitrile—ethanol 3:2 V/V as a mobile phase and detected via fluorescence detection (FLD) after derivatization with the Natural Product reagent A and via white light illumination (Vis) after the radical scavenging DPPH• assay (Figure 1d–g). Nine antioxidant compound zones at hRF 16 (R9), 22 (R8), 28 (R7), 33 (R5), 37 (R6), 42 (R3), 46 (R4), 54 (R2), and 68 (R1) were revealed. Via the derivatization with the Natural Product reagent A, the natively weak blue fluorescence of the zones R1–R9 was enhanced, indicating that the compounds responsible for the antioxidant effect belong to the group of phenolics.

Figure 1.
Flowers (a), stem with leaves (b), and stem bark (c) of Robinia pseudoacacia along with HPTLC chromatograms of bark crude extract (3 µL) separated on RP18 plates with acetonitrile-ethanol 3:2 V/V and detected at 254 nm (d), 365 nm (e), and after derivatization with natural product reagent A at 365 nm (f) as well as after the DPPH• assay under white light illumination (g) revealing the antioxidant compounds R1–R9.
This hypothesis (regarding the presence of phenolics) was confirmed by their densitometrically recorded RP-HPTLC–UV spectra showing characteristic absorption bands between 300 and 350 nm (Figure S1). The antioxidant compounds were further characterized by RP-HPTLC–HESI-HRMS. In the positive ionization mode, the intensity of signals corresponding to sodium adducts ([M+Na]+) was low, whereas in the negative ionization mode, signals of deprotonated molecules ([M−H]−) were intense (Table 1).

Table 1.
Antioxidant compounds (R1–R9) isolated from the bark extract of black locust detected by RP-HPTLC–DPPH•–Vis and characterized by RP-HPTLC–HRMS.
2.2. Fractionation by Solid-Phase Extraction and Isolation by RP-HPLC–DAD
The methanolic crude extract was fractionated by reversed-phase solid-phase extraction (SPE). The separation and peak identification of the compounds were achieved by RP-HPLC–DAD–ESI-MS. The isolation from the ethanol eluate was carried out by RP-HPLC–DAD (Figure 2). The yields of compounds R1–R9 ranged from 0.9 to 2.3 mg (Table 1), which were used for subsequent structure elucidation.
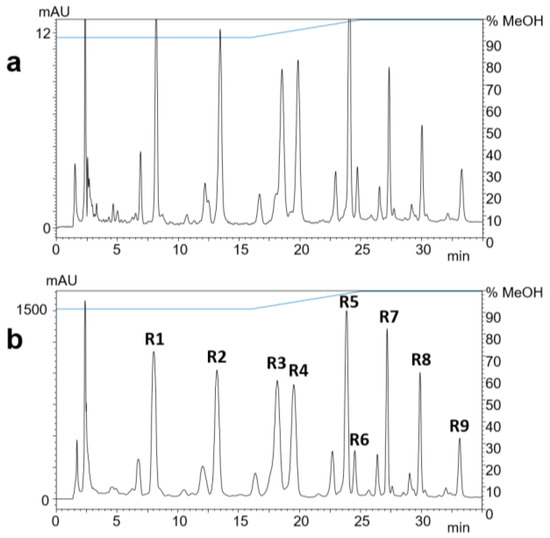
Figure 2.
RP-HPLC-UV chromatograms at 323 nm of 1 µL (a) and 100 µL (b) of black locust bark extract after SPE. Compounds R1–R9 were identified by ESI-MS.
2.3. Results of NMR and ATR-FTIR Spectra Recording
The NMR (Figures S3–S35) and ATR-FTIR spectra (Figures S36–S41) were recorded, and the data were compiled and listed as follows for 3-O-caffeoyl oleanolic acid (R1): IR (ATR) νmax 3191, 2941, 2927, 2854, 1696, 1600, 1524, 1463, 1389, 1365, 1266, 1170, 1146, 1117, 1019 cm–1; 1H and 13C NMR data (Table 2).
Table 2.
1H and 13C NMR (CD3OD, 600/151 MHz) resonance assignments of 3-O-caffeoyl oleanolic acid (R1).
- Oleyl caffeate (R2): IR (ATR) νmax 2924, 2854, 1712, 1593, 1509, 1460, 1370, 1263, 1167, 1121, 1091, 1050, 1018 cm–1; 1H NMR (CD3OD, 600 MHz) δ 7.53 (d, J = 16.0 Hz, 1H, H-7′), 7.04 (d, J = 2.1 Hz, 1H, H-1′), 6.94 (dd, J = 8.3, 2.1 Hz, 1H, H-5′), 6.78 (d, J = 8.1 Hz, 1H, H-4′), 6.25 (d, J = 15.9 Hz, 1H, H-8′), 5.34 (m, 2H, H-9, H-10), 4.17 (t, J = 6.6 Hz, 2H, H-1), 2.03 (m, 4H, H-8, H-11), 1.69 (p, J = 7.0 Hz, 2H, H-2), 1.41 (m, 2H, H-3), 1.29 (br s, 20H, H-4–H-7, H-12–H-17), 0.91 (m, 3H, H-18); 13C NMR (CD3OD, 151 MHz) δ (2D HSQC, HMBC) 169.5 (C, C-9′), 149.7 (C, C-3′), 146.9 (C, C-2′), 146.8 (CH, C-7′), 130.8 (CH, C-9), 130.8 (CH, C-10), 127.8 (C, C-6′), 122.9 (CH, C-5′), 116.6 (CH, C-4′), 115.3 (CH, C-8′), 115.1 (CH, C-1′), 65.6 (CH2, C-1), 33.2 (CH2, C-16), 30.9–30.2 (CH2, C-4–C-7, C-12–C-15), 29.8 (CH2, C-2), 28.0 (CH2, C-8, C-11), 27.1 (CH2, C-3), 23.4 (CH2, C-17), 14.4 (CH3, C-18)
- Octadecyl caffeate (R3): IR (ATR) νmax 3222, 2918, 2851, 1710, 1593, 1520, 1466, 1382, 1269, 1165, 1119, 1077, 1049 cm–1; 1H NMR (CD3OD, 600 MHz) δ 7.53 (d, J = 15.9 Hz, 1H, H-7′), 7.04 (d, J = 2.1 Hz, 1H, H-1′), 6.94 (dd, J = 8.1, 2.1 Hz, 1H, H-5′), 6.78 (d, J = 8.2 Hz, 1H, H-4′), 6.25 (d, J = 15.9 Hz, 1H, H-8′), 4.17 (t, J = 6.6 Hz, 2H, H-1), 1.70 (p, J = 6.8 Hz, 2H, H-2), 1.41 (m, 2H, H-3), 1.29 (br s, 28H, H-4–H-17), 0.90 (t, J = 7.0 Hz, 3H, H-18); 13C NMR (CD3OD, 151 MHz) δ 169.4 (C, C-9′), 149.6 (C, C-3′), 146.8 (C, C-2′), 146.8 (CH, C-7′), 127.7 (C, C-6′), 122.9 (CH, C-5′), 116.5 (CH, C-4′), 115.2 (CH, C-8′), 115.1 (CH, C-1′), 65.6 (CH2, C-1), 33.1 (CH2, C-16), 30.8–30.6 (CH2, C-6–C-15), 30.5 (CH2, C-4), 30.3 (CH2, C-5), 29.8 (CH2, C-2), 27.1 (CH2, C-3), 23.7 (CH2, C-17), 14.4 (CH3, C-18)
- Gadoleyl caffeate (R4): IR (ATR) νmax 2925, 2854, 1713, 1683, 1648, 1592, 1540, 1459, 1347, 1266, 1165, 1122, 1050, 1013 cm–1; 1H NMR (CD3OD, 600 MHz) δ 7.53 (d, J = 16.0 Hz, 1H, H-7′), 7.04 (d, J = 2.1 Hz, 1H, H-1′), 6.94 (dd, J = 8.1, 2.0 Hz, 1H, H-5′), 6.78 (d, J = 8.1 Hz, 1H, H-4′), 6.25 (d, J = 15.9 Hz, 1H, H-8′), 5.34 (m, 2H, H-9, H-10), 4.17 (t, J = 6.6 Hz, 2H, H-1), 2.03 (m, 4H, H-8, H-11), 1.69 (p, J = 7.1 Hz, 2H, H-2), 1.41 (m, 2H, H-3), 1.29 (br s, 24H, H-4–H-7, H-12–H-19), 0.91 (m, 3H, H-20); 13C NMR (CD3OD, 151 MHz) δ (2D HSQC, HMBC) 169.5 (C, C-9′), 149.6 (C, C-3′), 146.9 (C, C-2′), 146.8 (CH, C-7′), 130.8 (CH, C-9), 130.8 (CH, C-10), 127.8 (C, C-6′), 123.0 (CH, C-5′), 116.6 (CH, C-4′), 115.3 (CH, C-8′), 115.1 (CH, C-1′), 65.6 (CH2, C-1), 33.1 (CH2, C-18), 30.9–30.2 (CH2, C-4–C-7, C-12–C-17), 29.8 (CH2, C-2), 28.0 (CH2, C-8, C-11), 27.1 (CH2, C-3), 23.4 (CH2, C-19), 14.4 (CH3, C-20)
- Eicosanyl caffeate (R5): IR (ATR) νmax 3327, 2918, 2851, 1713, 1599, 1523, 1468, 1380, 1265, 1165, 1118, 1089, 1048 cm–1; 1H NMR (CD3OD, 600 MHz) δ 7.53 (d, J = 15.9 Hz, 1H, H-7′), 7.04 (d, J = 2.0 Hz, 1H, H-1′), 6.94 (dd, J = 8.3, 2.1 Hz, 1H, H-5′), 6.78 (d, J = 8.2 Hz, 1H, H-4′), 6.25 (d, J = 15.9 Hz, 1H, H-8′), 4.17 (t, J = 6.6 Hz, 2H, H-1), 1.70 (p, J = 6.9 Hz, 2H, H-2), 1.41 (m, 2H, H-3), 1.29 (br s, 32H, H-4–H-19), 0.90 (t, J = 7.0 Hz, 3H, H-20); 13C NMR (CD3OD, 151 MHz) δ (2D HSQC, HMBC) 169.5 (C, C-9′), 149.7 (C, C-3′), 146.9 (C, C-2′), 146.8 (CH, C-7′), 127.8 (C, C-6′), 122.9 (CH, C-5′), 116.5 (CH, C-4′), 115.2 (CH, C-8′), 115.1 (CH, C-1′), 65.6 (CH2, C-1), 33.1 (CH2, C-18), 30.8–30.0 (CH2, C-4–C-17), 29.8 (CH2, C-2), 27.1 (CH2, C-3), 23.7 (CH2, C-19), 14.4 (CH3, C-20)
- (Z)-9-docosenyl caffeate (R6): 1H NMR (CD3OD, 600 MHz) δ 7.53 (d, J = 16.0 Hz, 1H, H-7′), 7.04 (d, J = 2.0 Hz, 1H, H-1′), 6.94 (dd, J = 8.0, 1.7 Hz, 1H, H-5′), 6.78 (d, J = 8.1 Hz, 1H, H-4′), 6.25 (d, J = 15.9 Hz, 1H, H-8′), 5.34 (m, 2H, H-9, H-10), 4.17 (t, J = 6.5 Hz, 2H, H-1), 2.03 (m, 4H, H-8, H-11), 1.70 (m, 2H, H-2), 1.41 (m, 2H, H-3), 1.29 (br s, 28H, H-4–H-7, H-12–H-21), 0.90 (t, J = 6.7 Hz, 3H, H-22)
- Docosyl caffeate (R7): IR (ATR) νmax 2917, 2850, 1716, 1583, 1512, 1467, 1433, 1373, 1259, 1168, 1120, 1056 cm–1; 1H NMR (CD3OD, 600 MHz) δ 7.53 (d, J = 15.9 Hz, 1H, H-7′), 7.04 (d, J = 2.1 Hz, 1H, H-1′), 6.94 (dd, J = 8.2, 2.1 Hz, 1H, H-5′), 6.78 (d, J = 8.2 Hz, 1H, H-4′), 6.25 (d, J = 15.8 Hz, 1H, H-8′), 4.17 (t, J = 6.6 Hz, 2H, H-1), 1.70 (p, J = 6.8 Hz, 2H, H-2), 1.40 (m, 2H, H-3), 1.29 (br s, 36H, H-4–H-21), 0.90 (t, J = 7.1 Hz, 3H, H-22); 13C NMR (CD3OD, 151 MHz) δ (2D HSQC, HMBC) 169.5 (C, C-9′), 149.7 (C, C-3′), 146.8 (C, C-2′), 146.6 (CH, C-7′), 127.8 (C, C-6′), 122.9 (CH, C-5′), 116.6 (CH, C-4′), 115.3 (CH, C-8′), 115.1 (CH, C-1′), 65.6 (CH2, C-1), 33.2 (CH2, C-20), 30.9–30.0 (CH2, C-4–C-19), 29.9 (CH2, C-2), 27.1 (CH2, C-3), 23.7 (CH2, C-21), 14.4 (CH3, C-22)
- Tetracosyl caffeate (R8): 1H NMR (CD3OD, 600 MHz) δ 7.53 (d, J = 15.8 Hz, 1H, H-7′), 7.04 (d, J = 2.1 Hz, 1H, H-1′), 6.94 (dd, J = 8.0, 1.8 Hz, 1H, H-5′), 6.78 (d, J = 8.1 Hz, 1H, H-4′), 6.25 (d, J = 15.9 Hz, 1H, H-8′), 4.17 (t, J = 6.7 Hz, 2H, H-1), 1.70 (p, J = 6.9 Hz, 2H, H-2), 1.40 (m, 2H, H-3), 1.29 (br s, 40H, H-4–H-23), 0.90 (t, J = 6.8 Hz, 3H, H-24)
- Hexacosanyl caffeate (R9): 1H NMR (CD3OD, 600 MHz) for O-caffeoyl moiety δ 7.53 (d, J = 15.7 Hz, 1H, H-7′), 7.04 (d, J = 2.1 Hz, 1H, H-1′), 6.94 (dd, J = 8.0, 1.8 Hz, 1H, H-5′), 6.78 (d, J = 8.2 Hz, 1H, H-4′), 6.25 (d, J = 15.8 Hz, 1H, H-8′)
2.4. Structure Elucidation of the Isolated Compounds
The NMR and ATR-FTIR spectroscopy data were consistent with HESI-HRMS data and in good agreement with the literature cited. The downfield region of the 1H NMR spectra of the isolated compounds R1–R9 highly resembled each other with a set of resonances at δH 7.04–7.03 (d, J ≈ 2 Hz, 1H, H-1′), 6.94 (dd, J ≈ 8 and 2 Hz, 1H, H-5′), and 6.78 (d, J ≈ 8 Hz, 1H, H-4′) ppm suggesting the presence of a 1,2,4-trisubstituted aromatic ring. Moreover, 1H signals at δH 7.53–7.52 (d, J ≈ 16 Hz, 1H, H-7′) and 6.25–6.24 (d, J ≈ 16 Hz, 1H, H-8′) ppm implied a trans-oriented, disubstituted carbon-carbon double bond. The 13C or the 1H–13C HMBC spectra of compounds R1–R8 revealed one ester carbonyl carbon at δC 169.5–169.2 located at the C-9′ position, as evidenced by HMBC correlations H-7′/C-9′ and H-8′/C-9′. Besides, they exhibited typical carbon signals at δC 149.7–149.6 (C, C-3′), 146.9–146.7 (C, C-2′), 146.8–146.6 (CH, C-7′), 127.8–127.7 (C, C-6′), 123.0–122.9 (CH, C-5′), 116.6–116.5 (CH, C-4′), 115.6–115.2 (CH, C-8′), and 115.1 (CH, C-1′) ppm, indicating that their structure contains an O-caffeoyl basic skeleton [28,29], which were corroborated by the ATR-FTIR absorption bands at around 3350–3200 cm–1 (O–H stretch) and at 1716–1710 cm–1 (α,β-unsaturated ester C=O stretch). The connectivity between the aromatic ring and the double bond was confirmed by long-range HMBC correlations H-8′/C-6′, H-7′/C-1′, H-7′/C-5′, and H-7′/C-6′. The 13C chemical shifts could not be determined for compounds R6 and R8 due to their low isolated quantity of below 1 mg (Table 1).
The HESI-HRMS spectrum of R1 showed a deprotonated molecule peak at m/z 617.3848 [M−H]− establishing its molecular formula as C39H54O6 (Table 1), which corresponded to 13 double bond equivalents. Its 1H NMR spectrum (Table 2) indicated the presence of seven isolated methyl groups at δH 1.19 (s, 3H, H-27), 1.01 (s, 3H, H-25), 0.97 (s, 3H, H-24), 0.95 (s, 3H, H-30), 0.91 (s, 3H, H-23), 0.91 (s, 3H, H-29), and 0.84 (s, 3H, H-26) ppm, an olefinic proton at δH 5.26 (t, J = 3.6 Hz, 1H, H-12) ppm, and an oxymethine proton at δH 4.57 (dd, J = 11.5, 4.3 Hz, 1H, H-3) ppm. The 13C NMR spectrum (Table 2) revealed 30 carbon resonances excluding the O-caffeoyl moiety, including seven methyl carbons at δC 33.6 (C-29), 28.7 (C-23), 26.4 (C-27), 24.0 (C-30), 17.7 (C-26), 17.3 (C-24), 15.9 (C-25) ppm, two olefinic carbons at δC 145.3 (C-13), 123.5 (C-12) ppm, one oxygenated carbon at δC 82.3 (C-3) ppm, as well as one carboxylic carbon at δC 182.1 (C-28) ppm. The caffeate and carboxylic moiety and the carbon-carbon double bond account for eight double bond equivalents, implying a pentacyclic triterpene skeleton with seven angular methyl groups. Based on the spectral data along with the 2D homo- and heteronuclear correlations and by comparing the 1H, 13C NMR, and ATR-FTIR data with those reported in the literature [30,31], the triterpene was identified as oleanolic acid. The point of attachment between the O-caffeoyl moiety and oleanolic acid was determined by the downfield chemical shifts of H-3 at δH 4.57 ppm and C-3 at δC 82.3 ppm, and a key HMBC correlation was observed from H-3 to C-9′, supporting the substitution of oleanolic acid at the C-3 position. In addition, the large coupling constant between H-2ax and H-3 (3JH-2ax–H-3 = 11.5 Hz) confirmed that H-3 occupied an α-axial position, thereby indicating that the O-caffeoyl moiety was β-oriented. Thus, compound R1 was elucidated as 3-O-caffeoyl oleanolic acid (Figure 3).
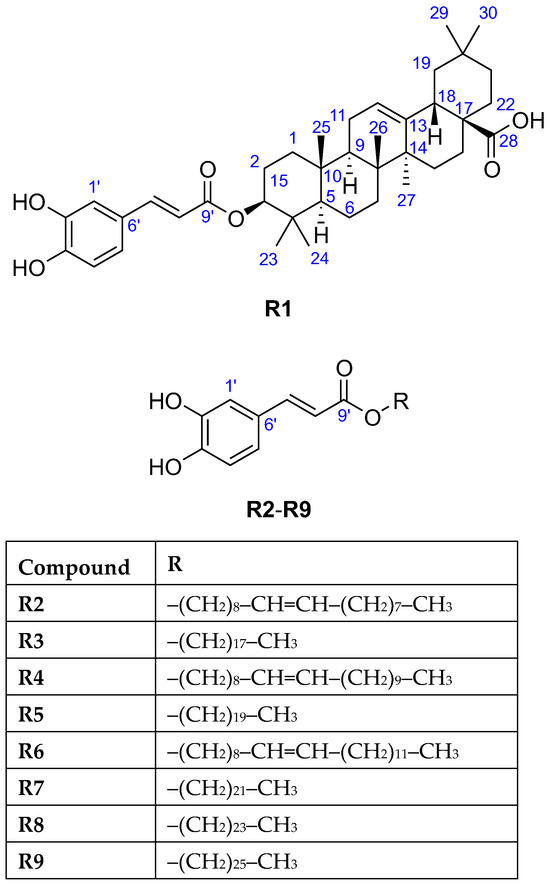
Figure 3.
The chemical structures of the isolated compounds R1–R9.
The 1H NMR spectra of the four isolated compounds R3, R5, R7, and R8 were remarkably similar, which was consistent with literature [32,33,34,35,36], displaying another set of signals at δH 4.17 (t, J ≈ 6.5 Hz, 2H, H-1), 1.70 (p, J ≈ 7 Hz, 2H, H-2), 1.41 (m, 2H, H-3), 1.29 (br s, varied integrals), 0.90 (t, J ≈ 7 Hz, 3H, terminal CH3) ppm suggestive for the presence of a saturated fatty acid moiety in their structures. It was verified by the observed 13C signals at δC 65.6 (CH2, C-1), 33.2–33.1 (CH2, C-16(R3)/C-18(R5)/C-20(R7)), 30.9–30.0 (CH2, C-4–C-15(R3)/C-17(R5)/C-19(R7)), 29.9–29.8 (CH2, C-2), 27.1 (CH2, C-3), 23.7 (CH2, C-17(R3)/C-19(R5)/C-21(R7)), 14.4 (CH3, C-18(R3)/C-20(R5)/C-22(R7)) ppm [28,37]. In the HESI-HRMS spectra, deprotonated molecules were observed at m/z 431.3162 [M−H]− (R3), m/z 459.3475 [M−H]− (R5), m/z 487.3788 [M−H]− (R7), and m/z 515.4101 [M−H]− (R8), corresponding to the molecular formulae C27H44O4 (R3), C29H48O4 (R5), C31H52O4 (R7), and C33H56O4 (R8) (Table 1) that suspected a long-chain series with a two methylene group difference between adjacent members. The chemical formula of the aliphatic chains could be determined based on the fact that R3, R5, R7, and R8 were caffeate esters: C18H37, C20H41, C22H45, and C24H49, respectively. Thus, the isolates were identified as octadecyl caffeate (R3), eicosanyl caffeate (R5), docosyl caffeate (R7), and tetracosyl caffeate (R8), respectively (Figure 3).
Due to the low purity and isolated amount of compound R9, only limited structural information could be inferred from its 1H NMR spectrum revealing the characteristic signals of O-caffeoyl moiety at δH 7.53 (d, J = 15.7 Hz, 1H, H-7′), 7.04 (d, J = 1.8 Hz, 1H, H-1′), 6.94 (dd, J = 7.8 and 1.9 Hz, 1H, H-5′), 6.78 (d, J = 8.2 Hz, 1H, H-4′), and 6.25 (d, J = 15.8 Hz, 1H, H-8′) ppm. The HESI-HRMS spectrum displayed a deprotonated molecule peak at m/z 543.4419 [M−H]− indicating the molecular formula as C35H60O4 (Table 1). Being a caffeate ester, the chemical formula C26H53 was deduced for the fatty alcohol moiety, thus compound R9 was assigned as hexacosanyl caffeate (Figure 3) [38].
2.5. GC–MS for Assignment of the Double Bond Position
Based on the HESI-HRMS analyses (Table 1), deprotonated molecules were detected at m/z 429.3005 [M−H]− (R2), 457.3318 [M−H]− (R4), and m/z 485.3631 [M−H]− (R6), corresponding to the molecular formulae C27H42O4 (R2), C29H46O4 (R4), and C31H50O4 (R6), indicative of one degree of unsaturation in the fatty alcohol moiety compared to R3, R5, R7, and R8. The 1H NMR spectra of compounds R2, R4, and R6 were similar to each other and that of R3, R5, R7, and R8, with the additional resonances at δH 5.34 (m, 2H) and 2.03 (m, 4H) consistent with one carbon-carbon double bond. The presence of a monounsaturated long chain was supported by additional 13C signals at δC 130.7–130.5 (CH) and 27.9–27.7 (CH2) ppm, indicating the chemical formulae C18H35 (R2), C20H39 (R4), and C22H43 (R6) for aliphatic chains. However, the position of the double bond in the side chain could not be elucidated by NMR spectroscopy; therefore, GC–MS analyses were conducted. Compounds R2 and R4 were identified as oleyl and gadoleyl caffeate, respectively, as their aliphatic chains (oleyl and gadoleyl alcohol) were recognized by the NIST mass spectral library search, showing an excellent agreement between the experimental and theoretical EI-MS spectra (Figures S42 and S43). However, the long fatty alcohol chain of compound R6 does not have a mass spectrum in the databases (Figure S44); therefore, we used its chromatographic retention property for its identification. The measured retention times for R2, R4, and R6 were 7.5, 8.4, and 9.3 min, respectively (Figure S45). The identical position and configuration of the double bond (9Z) in the fatty alcohol chain of R6 were confirmed by the linear relationship between the number of carbon atoms and logarithmic retention times (R2 = 0.998) (Figure S46), indicating that R2, R4, and R6 belong to the same homologous series. Thus, compound R6 was determined as (Z)-9-docosenyl caffeate.
2.6. Equivalency Calculation of the Antioxidant Activity of the Isolates by DPPH• Microplate Assay
All isolated compounds exhibited antioxidant effects using the DPPH• microplate assay (Table 3), which confirmed the initial screening results obtained from the RP-HPTLC–DPPH• assay and the assignment by RP-HPTLC–HESI-HRMS. Antioxidant activity of R1–R8 was compared to that of caffeic acid and found to be 0.10–0.35 mg caffeic acid equivalents per mg isolate, corresponding to 0.29–1.20 mol caffeic acid equivalents per mol isolate. The R1 displayed the strongest antioxidant activity, surpassing caffeic acid at its molar level (1.20 mol caffeic acid equivalent/mol R1), while the R8 demonstrated the weakest activity (0.29 mol caffeic acid equivalent/mol R8). The antioxidant effect of R9 was detected but not quantified due to its low purity.
Table 3.
Antioxidant activity of the isolated fatty alcohol caffeates expressed as caffeic acid equivalents (mean of triplicates with standard deviation SD).
2.7. Progress Achieved in Comparison to Literature
3-O-Caffeoyl oleanolic acid (R1) has been isolated from different plant organs such as the seeds of Oenothera biennis [39], the whole plant of Leptopus lolonum [40], the leaves of Elaeagnus oldhamii [41], the barks of Betula platyphylla var. japonica [42], the skins of apples and pears [43], and the stem bark of R. pseudoacacia [44]. This compound demonstrated cytotoxic [41,45], antineoplastic [40,42], antibacterial against Mycobacterium tuberculosis [46], anti-inflammatory [43,47], anticoronavirus [45], and antioxidant [39] effects.
Phenolic esters with long-chain saturated fatty alcohols (R3, R5, R7, R8, and R9) were described in various plant species but not from the Robinia genus. Among others, the root bark of Paeonia suffruticosa [48], leaves of Artemisia argyi [49], bark of Acacia species [50], and roots of Ipomoea asarifolia [51] were reported as a source of octadecyl caffeate (R3) that displayed α-glucosidase and α-amylase inhibition [48], antioxidant [36,49], cytotoxic [51], antiproliferative [37], anti-HIV (Human immunodeficiency virus) [52], and anti-inflammatory [37] activities. Eicosanyl caffeate (R5) and docosyl caffeate (R7) were found in stems of Wikstroemia scytophylla [53], roots of Glycyrrhiza glabra [33], and Sophora species [54,55]. Both exhibited chymotrypsin-like elastase inhibition [33], antiproliferative [37], anti-inflammatory [37], and antioxidant [33,49,56] effects. The isolation of docosyl caffeate (R7) from Thymelaea hirsute [57], the bark of Acacia species [50], its antineoplastic effect [57], and moderate activity against acetyl- and butyrylcholinesterase enzymes [58] have also been reported. Tetracosyl caffeate (R8) was described as a constituent of wigs of Hypericum oblongifolium [59], the whole plant of Caragana conferta [60], roots of Caesalpinia mimosoides [61], and bark of Acacia species [50], and as a urease inhibitor [62], anti-inflammatory [61], antineoplastic [34], antimicrobial [63], and cytotoxic [61] agent. The stem bark of Pongamia glabra [38], bark of Acacia species [50], stem bark and leaves of Inga edulis [64], and stems of Hibiscus taiwanensis [65] were sources of hexacosanyl caffeate (R9) that showed antioxidant activity [66]. Synthetic oleyl caffeate (R2) exerted inhibitory activity against HIV-1 [67]. To the best of our knowledge, oleyl caffeate (R2), gadoleyl caffeate (R4), and (Z)-9-docosenyl caffeate (R6) has not been reported previously as natural product constituents.
Phenolic compounds with hydrogen- or electron-donating properties are potential free radical scavengers that protect biomolecules from oxidative stress. Their antioxidant capacity is structure-related, mainly depending on the number and position of hydroxyl groups attached to the aromatic ring and the presence of sugar or other substituents [68]. Caffeic acid, with its dihydroxylated aromatic ring in ortho position, is one of the strongest phenolic antioxidants. Its half-maximal effective concentration (EC50) value in the DPPH• assay was similar to that of flavonoid aglycones (quercetin, kaempferol, and epicatechin) and lower than that of the well-known potent antioxidant ascorbic acid or other phenolic acids (e.g., 3-O-chlorogenic acid, ferulic acid, p-coumaric acid, and p-hydroxybenzoic acid) [69,70]. In this study, the antioxidant activity of the isolated compounds was compared to that of caffeic acid, and it was found that 3-O-caffeoyl oleanolic acid (R1) was stronger, while other isolates were similar or slightly weaker than caffeic acid. These results are in agreement with literature data, as 3-O-caffeoyl oleanolic acid (R1) exerted lower free radical scavenging activity than ascorbic acid [39], octadecyl caffeate (R3) showed an antioxidant effect comparable to caffeic acid and higher than ferulic acid and sinapic acid [36], and hexacosanyl caffeate (R9) exhibited a slightly lower activity than caffeic acid [66]. However, in the DPPH• assay, eicosanyl caffeate (R5) and docosyl caffeate (R7) displayed weaker antioxidant activity (10–15 times higher EC50) than gallic acid [33,49], which was found to be a stronger free radical scavenger (two times lower EC50) than caffeic acid in the same assay [71].
3. Materials and Methods
3.1. Materials
HPTLC plates, silica gel 60 RP18, and methanol (MS grade) were purchased from Merck (Darmstadt, Germany). Solvents for extraction and HPTLC (analytical grade) were obtained from Th. Geyer (Renningen, Germany) or Reanal (Budapest, Hungary). The 2,2-diphenyl-1-picrylhydrazyl radical (DPPH•) and caffeic acid (98%) were acquired from Sigma-Aldrich (Steinheim, Germany), and Natural Product reagent A (diphenylboryloxyethylamine or diphenylboric acid β-ethylamino ester, 98%) was purchased from Carl Roth (Karlsruhe, Germany). Methanol-d4 (CD3OD, 99.8 atom% D) for NMR measurements was purchased from VWR (Budapest, Hungary), and gradient-grade methanol and acetonitrile for isolation were supplied by Fisher Scientific (Pittsburg, PA, USA).
3.2. Sample Origin and Preparation
The stem bark of R. pseudoacacia L. was collected in October 2016 in Harta (46°41′45″ N 19°02′26″ E, altitude: 93 m) in the Great Plain of Hungary and dried at room temperature. A voucher sample (PPI-MA-RB1) has been deposited at the herbarium of the Plant Protection Institute, Centre for Agricultural Research, Budapest, Hungary. The dried material was powdered by a coffee grinder (Bosch MKM6000, Stuttgart, Germany) and was extracted with methanol (150 mg/mL) using an ultrasound-assisted extraction for 10 min (Sonorex Super RK 106, Bandelin, Berlin, Germany) and centrifuged for 1 min at 5000× g (Dlab D1008, Beijing, China).
3.3. High-Performance Thin-Layer Chromatography, Derivatization, and DPPH• Assay
The crude extract (3 µL) was applied onto the RP18 HPTLC plate by the Automatic TLC Sampler 4 (ATS4, CAMAG, Muttenz, Switzerland) as a 7 mm band with an 8 mm distance from the lower edge. HPTLC separation was carried out with a mobile phase of acetonitrile—ethanol 3:2 V/V in a Twin Trough Chamber (10 cm × 10 cm, CAMAG) up to 80 mm from the lower edge of the plate. The dried chromatogram was detected at 254 nm and 365 nm with the TLC Visualizer (CAMAG), and the UV spectra of selected zones were recorded by a TLC Scanner 4 (CAMAG). To detect phenolics (e.g., flavonoids, anthocyanidines, hydroxyl- and methoxycinnamic acids [72]), the plate was dipped into a 0.5% methanolic solution of Natural Product reagent A, dried, and documented at 365 nm. Free radical scavenging activity was visualized by the HPTLC–DPPH• assay. The chromatogram was immersed into a 0.02% methanolic solution of DPPH•, and the bright zones of antioxidants against a lilac background were documented under white light illumination in the transmittance mode (TLC Visualizer).
3.4. HPTLC–HESI-HRMS
For HPTLC–HRMS analysis [73,74], a TLC–MS Interface (CAMAG) or a PlateExpress interface (Advion, Ithaca, NY, USA) equipped with an oval elution head (4 mm × 2 mm) was integrated online between a quaternary pump (Ultimate LPG-3400 XRS, Dionex Softron, Germering, Germany) and a hybrid quadrupole-orbitrap mass spectrometer operated with a heated electrospray ionization probe (HESI-II, Q Exactive Plus, Thermo Fisher Scientific, Bremen, Germany). MS-grade methanol at a flow rate of 0.1 mL/min was used to elute selected zones. The following conditions were applied: spray voltage 3.5 kV, capillary temperature 270 °C, and nitrogen sheath and auxiliary gas with 20 and 10 arbitrary units, respectively, produced by an SF2 compressor (Atlas Copco Kompressoren und Drucklufttechnik, Essen, Germany). HRMS full scan spectra were recorded in both negative and positive ionization modes in the range of m/z 80–1200 with a resolution of 280,000; the automatic gain control target (AGCT) was set to 3 × 106, and the maximum injection time (IT) was 100 ms. Xcalibur 3.0.63 software (Thermo Fisher Scientific) provided the instrument control and data analysis.
3.5. Fractionation by Solid-Phase Extraction
The bark powder (20 g) was extracted three times with 100 mL of methanol by ultrasound-assisted extraction. The combined extracts were filtered (Whatman No. 2 filter paper, Sigma), concentrated by a rotary evaporator to 20 mL (Büchi Rotavapor R-134, Flawil, Switzerland), and diluted with water to 40 mL. This methanol-water 1:1 crude extract was purified by solid-phase extraction using Strata-XL cartridges (10 portions, 200 mg 100 µm polymeric RP, Phenomenex, Torrance, CA, USA). The cartridge was rinsed with 4 mL of methanol, conditioned with 4 mL of 50% aqueous methanol, loaded with 4 mL of sample, washed with 4 mL of acetonitrile, and then eluted with 4 mL of ethanol. The whole 10 eluates (from 10 cartridges) were pooled, concentrated by a rotary evaporator to 2 mL, and transferred to HPLC analysis.
3.6. Compound Isolation by HPLC–DAD–ESI-MS
The antioxidant compounds were isolated by HPLC using an LCMS-2020 system (Shimadzu, Kyoto, Japan) consisting of a binary gradient solvent pump, a vacuum degasser, a thermostated autosampler, a column oven, a photodiode detector, and a mass analyzer using an electrospray ionization (ESI) interface. The instrument control, data acquisition, and data processing were carried out by LabSolutions 5.42v software (Shimadzu). Separation was achieved on a Gemini C18 column (250 mm length, 4.6 mm ID, 5 µm particle size, Phenomenex, Torrance, CA, USA) at 35 °C with a linear gradient of 5% aqueous acetonitrile with 0.05% formic acid (A) and methanol with 0.05% formic acid (B). The gradient program was as follows: 0–16 min, 92% B; 16–25 min, 92–100% B; 25–35 min, 100% B; and 35.1–40 min, 92% B. The flow rate of the mobile phase was adjusted to 1.2 mL/min. The injection volume was set to 1 µL for method development and 100 µL for isolation. The appropriate peaks were collected based on the UV chromatogram at 323 nm, and the fractionation protocol was repeated 15 times. The combined 15 fractions were dried with a rotary evaporator at 40 °C and transferred to NMR spectroscopy. The MS conditions were as follows: nebulizer gas (N2) flow rate 1.5 L/min, drying gas (N2) flow rate 15 L/min, interface temperature 350 °C, heat block temperature 400 °C, desolvation line temperature 250 °C, and detector voltage 4.5 kV. Full mass scan spectra were recorded in the negative ionization mode in the range of m/z 150–1000 with a scan speed of 883 u/s.
3.7. NMR Spectroscopy
The isolated compounds R1–R9 were dissolved in methanol-d4, and the samples were transferred to a standard 5 mm NMR tube for measurements. NMR spectra were collected on a Varian DDR 600 (1H: 599.9 MHz, 13C: 150.9 MHz; 14.1 T) spectrometer equipped with a dual 5 mm inverse-detection pulsed-field gradient (IDPFG) probehead at 298 K. The instrument was operated and controlled by VnmrJ 3.2C software. All applied pulse sequences were obtained from the Chempack 5.1 standard pulse program library of the instrument. 1H and 13C chemical shifts (δ) are provided on the δ-scale, reported in ppm and referenced to the NMR solvent used (CHD2OD residual peak at δH = 3.31 ppm and CD3OD at δC = 49.0 ppm), whereas spin-spin coupling constants (J) are given in Hz. The signal multiplicities are denoted as s—singlet, br s—broad singlet, d—doublet, t—triplet, p—pentet; m—multiplet; dd—doublet of doublets; td—triplet of doublets. The full 1H and 13C NMR resonance assignments were performed by means of comprehensive one- (1H and 13C) and two-dimensional homonuclear (1H–1H COSY and 1H–1H TOCSY) and heteronuclear (1H–13C edHSQC (1JC–H = 140 Hz) and 1H–13C HMBC (nJC–H = 8 Hz), both of them gradient-enhanced with adiabatic pulses) NMR experiments. In the case of compound 1, band-selective HSQC (bsHSQC) and HMBC (bsHMBC) spectra were also recorded to enhance the spectral resolution in the F1 dimension.
3.8. ATR-FTIR Spectroscopy
The ATR-FTIR spectra were recorded by a Perkin Elmer Spectrum 400 FT-IR/FT-NIR spectrometer (Waltham, MA, USA) equipped with a diamond/ZnSe ATR crystal and a MIR TGS detector. Spectra were collected in the range of 4000–650 cm−1 with a spectral resolution of 4 cm–1. A few drops of the isolates (1 mg/mL in ethanol) were placed onto the ATR crystal, then the solvent was completely evaporated and the spectra were obtained by averaging 8–32 scans after background subtraction. Data processing and analysis were performed by Perkin Elmer Spectrum Software version 6.3.1, which included baseline correction and Savitzky-Golay smoothing.
3.9. GC–MS
The isolated compounds R2, R4, and R6 were dissolved in ethanol (1 mg/mL). For the GC–MS analysis, a Shimadzu GCMS-TQ8040 NX instrument was applied using a Rtx-5 (30 m × 250 µm i.d.; film thickness: 0.32 µm, Restek, Bellefonte, PA, USA) capillary column. Helium was used as a carrier gas with a linear velocity of 50 cm/s. The solution of each compound (1 µL) was injected in split mode (split ratio 1:20) at 300 °C. The column oven temperature was programmed to increase from 80 °C to 320 °C at 20 °C/min, and the final temperature was held for 10.5 min. The ionization in the electron impact ion source was performed with an electron beam of 70 eV. The triple quadrupole analyzer operated in full scan mode (m/z range 29–600, scan speed 3333 amu/s). The interface and the ion source temperatures were maintained at 280 °C, and the accelerating and detector voltages were set to 4.0 kV and 0.9 kV, respectively. The data were acquired and evaluated with GCMSsolutions 4.52 software (Shimadzu). The identification of the compounds was aided by the NIST 17 mass spectral library.
3.10. DPPH• Microplate Assay of Isolated Compounds
The antioxidant activity of the isolated compounds (1 mg/mL in ethanol) was evaluated using 96-well microplates and expressed as caffeic acid equivalents (mg caffeic acid/mg isolates and mol caffeic acid equivalent/mol isolates). Caffeic acid (10, 9, 8, 7, 6, 5, 4, 3, 2, 1 µL, 1 mg/mL in ethanol) and isolated compounds (10 µL) were pipetted to the wells in triplicate (on two separate occasions). After evaporation of the ethanol, 100 µL of DPPH• solution (0.3 M in methanol) was added to each well. After incubating the microplate at 25 °C for 10 min in the dark, the deep-violet stable free radical DPPH• was reduced to the pale-yellow 2,2-diphenyl-1-picrylhydrazine in the presence of antioxidants, resulting in a decrease in absorbance measured at 517 nm (Clariostar® Plus microplate reader, BMG LABTECH, Ortenberg, Germany).
4. Conclusions
This study identified nine antioxidant caffeate esters from the stem bark of R. pseudoacacia using RP-HPTLC–DPPH• assay, RP-HPTLC–UV/VIS/FLD–HESI-HRMS, HPLC–DAD–ESI-MS, GC–MS, ATR–FTIR, and NMR spectroscopy. It led to the identification of 3-O-caffeoyl oleanolic acid (R1), oleyl caffeate (R2), octadecyl caffeate (R3), gadoleyl caffeate (R4), eicosanyl caffeate (R5), (Z)-9-docosenyl caffeate (R6), docosyl caffeate (R7), tetracosyl caffeate (R8), and hexacosanyl caffeate (R9). This is the first report for natural compounds R2, R4, and R6, while R3, R5, R7, R8, and R9 were obtained from this genus for the first time. The antioxidant effects of the isolated compounds were confirmed using the DPPH• microplate assay. The stem bark of black locust holds significant potential as a candidate for pharmaceutical applications, as the known isolates display a range of other bioactivities such as antimicrobial, cytotoxic, antiproliferative, and anti-inflammatory properties.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29235673/s1, Figure S1: UV-VIS spectra (190–600 nm) of the compounds R1–R9 recorded by RP-HPTLC-densitometry; Figure S2: HPTLC chromatogram of black locust bark isolates (R1–R9) and extract (E) on RP18 plates with acetonitrile-ethanol 3:2 V/V after derivatization with natural product reagent at UV 365 nm; Figures S3–S35: 1D and 2D NMR spectra of compounds R1–R9; Figures S36–S41: ATR FTIR spectra of compounds R1–R7; Figure S42: The experimental EI-MS spectrum of compound R2 and the theoretical EI-MS spectrum of oleyl alcohol; Figure S43: The experimental EI-MS spectrum of compound R4 and the theoretical EI-MS spectrum of gadoleyl alcohol; Figure S44: The experimental EI-MS spectrum of compound R6; Figure S45: GC–MS TIC chromatograms of compounds R2, R4, and R6; Figure S46: Plot of the logarithmic retention time (tR) versus the number of carbon atoms for compounds R2, R4, and R6.
Author Contributions
Conceptualization, resources, and writing—review and editing, Á.M.M. and G.E.M.; methodology, and investigation, Á.M.M.; data curation, and writing—original draft preparation, M.B. and Á.M.M.; A.D. performed the spectral analyses; J.B. performed the GC–MS analyses. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially funded by the National Research, Development, and Innovation Office of Hungary (SNN139496, Á.M.M.). Instrumentation at JLU Giessen was partially funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—INST 162/471-1 FUGG; INST 162/536-1 FUGG.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are available upon reasonable request.
Acknowledgments
The authors express their gratitude to Judit Nyiri (Pharmaceutical Chemistry and Technology Department, National Institute of Pharmacy and Nutrition, Budapest, Hungary) for her assistance in recording the ATR-FTIR spectra.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Puchałka, R.; Dyderski, M.K.; Vítková, M.; Sádlo, J.; Klisz, M.; Netsvetov, M.; Prokopuk, Y.; Matisons, R.; Mionskowski, M.; Wojda, T.; et al. Black Locust (Robinia pseudoacacia L.) Range Contraction and Expansion in Europe under Changing Climate. Glob. Chang. Biol. 2021, 27, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Csiszár, Á.; Winkler, D.; Bartha, D.; Zagyvai, G. Diverse Interactions: Root-Nodule Formation and Herb-Layer Composition in Black Locust (Robinia pseudoacacia) Stands. Plants 2023, 12, 3253. [Google Scholar] [CrossRef]
- Botta-Dukat, Z.; Balogh, L.; Feher, A. The Most Important Invasive Plants in Hungary; Institute of Ecology and Botany; Hungarian Academy of Sciences: Budapest, Hungary, 2008. [Google Scholar]
- Vítková, M.; Müllerová, J.; Sádlo, J.; Pergl, J.; Pyšek, P. Black Locust (Robinia pseudoacacia) Beloved and Despised: A Story of an Invasive Tree in Central Europe. For. Ecol. Manage. 2017, 384, 287–302. [Google Scholar] [CrossRef]
- Motti, R.; Paura, B.; Cozzolino, A.; de Falco, B. Edible Flowers Used in Some Countries of the Mediterranean Basin: An Ethnobotanical Overview. Plants 2022, 11, 3272. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.; Zhao, X.; Yang, Y.; Li, B.; Zhu, F.; Zhu, R. Immune Enhancement of Taishan Robinia pseudoacacia Polysaccharide on Recombinant Proteus Mirabilis OmpA in Chickens. Int. Immunopharmacol. 2014, 22, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.; Kim, E.; Oh, K.; Kim, H.-J.; Dhakal, R.; Kim, Y.; Baek, K.-H. Bactericidal Effect of Extracts and Metabolites of Robinia pseudoacacia L. on Streptococcus mutans and Porphyromonas gingivalis Causing Dental Plaque and Periodontal Inflammatory Diseases. Molecules 2015, 20, 6128–6139. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, E. Quantitative and Qualitative Identification of Bioactive Compounds in Edible Flowers of Black and Bristly Locust and Their Antioxidant Activity. Biomolecules 2020, 10, 1603. [Google Scholar] [CrossRef]
- Uzelac, M.; Sladonja, B.; Šola, I.; Dudaš, S.; Bilić, J.; Famuyide, I.M.; McGaw, L.J.; Eloff, J.N.; Mikulic-Petkovsek, M.; Poljuha, D. Invasive Alien Species as a Potential Source of Phytopharmaceuticals: Phenolic Composition and Antimicrobial and Cytotoxic Activity of Robinia pseudoacacia L. Leaf and Flower Extracts. Plants 2023, 12, 2715. [Google Scholar] [CrossRef]
- Obistioiu, D.; Cocan, I.; Tîrziu, E.; Herman, V.; Negrea, M.; Cucerzan, A.; Neacsu, A.-G.; Cozma, A.L.; Nichita, I.; Hulea, A.; et al. Phytochemical Profile and Microbiological Activity of Some Plants Belonging to the Fabaceae Family. Antibiotics 2021, 10, 662. [Google Scholar] [CrossRef]
- Truchado, P.; Ferreres, F.; Bortolotti, L.; Sabatini, A.G.; Tomás-Barberán, F.A. Nectar Flavonol Rhamnosides Are Floral Markers of Acacia (Robinia pseudacacia) Honey. J. Agric. Food Chem. 2008, 56, 8815–8824. [Google Scholar] [CrossRef]
- Tian, F.; Chang, C.-J.; Grutzner, J.B.; Nichols, D.E.; McLaughlin, J.L. Robinlin: A Novel Bioactive Homo-Monoterpene from Robinia pseudoacacia L. (Fabaceae). Bioorg. Med. Chem. Lett. 2001, 11, 2603–2606. [Google Scholar] [CrossRef] [PubMed]
- Amorim, C.; Silvério, S.C.; Prather, K.L.J.; Rodrigues, L.R. From Lignocellulosic Residues to Market: Production and Commercial Potential of Xylooligosaccharides. Biotechnol. Adv. 2019, 37, 107397. [Google Scholar] [CrossRef] [PubMed]
- Kamperidou, V.; Terzopoulou, P.; Barboutis, I. Marginal Lands Providing Tree-crop Biomass as Feedstock for Solid Biofuels. Biofuels Bioprod. Biorefining 2021, 15, 1395–1405. [Google Scholar] [CrossRef]
- Pérez-Pérez, A.; Gullón, B.; Lobato-Rodríguez, Á.; Garrote, G.; del Río, P.G. Microwave-Assisted Extraction of Hemicellulosic Oligosaccharides and Phenolics from Robinia pseudoacacia Wood. Carbohydr. Polym. 2023, 301, 120364. [Google Scholar] [CrossRef]
- Sergent, T.; Kohnen, S.; Jourez, B.; Beauve, C.; Schneider, Y.-J.; Vincke, C. Characterization of Black Locust (Robinia pseudoacacia L.) Heartwood Extractives: Identification of Resveratrol and Piceatannol. Wood Sci. Technol. 2014, 48, 1005–1017. [Google Scholar] [CrossRef]
- Vek, V.; Poljanšek, I.; Oven, P. Efficiency of Three Conventional Methods for Extraction of Dihydrorobinetin and Robinetin from Wood of Black Locust. Eur. J. Wood Wood Prod. 2019, 77, 891–901. [Google Scholar] [CrossRef]
- Magel, E.; Jay-Allemand, C.; Ziegler, H. Formation of Heartwood Substances in the Stemwood of Robinia pseudoacacia L. II. Distribution of Nonstructural Carbohydrates and Wood Extractives across the Trunk. Trees 1994, 8, 165–171. [Google Scholar] [CrossRef]
- Putman, L.J.; Laks, P.E.; Pruner, M.S. Chemical Constituents of Black Locust Bark and Their Biocidal Activity. Holzforschung 1989, 43, 219–224. [Google Scholar] [CrossRef]
- Dai, J.; Mumper, R.J. Plant Phenolics: Extraction, Analysis and Their Antioxidant and Anticancer Properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Saeed-ur-Rahman; Bilal, M.; Huang, D. Role of Flavonoids in Plant Interactions with the Environment and against Human Pathogens—A Review. J. Integr. Agric. 2019, 18, 211–230. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, X.; Zhao, Y.; Wang, L.; Wang, Y. Adaptive Response of Flavonoids in Robinia pseudoacacia L. Affected by the Contamination of Cadmium and Elevated CO2 to Arbuscular Mycorrhizal Symbiosis. Ecotoxicol. Environ. Saf. 2023, 263, 115379. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A. Concept, Mechanism, and Applications of Phenolic Antioxidants in Foods. J. Food Biochem. 2020, 44, e13394. [Google Scholar] [CrossRef] [PubMed]
- Krüger, S.; Urmann, O.; Morlock, G.E. Development of a Planar Chromatographic Method for Quantitation of Anthocyanes in Pomace, Feed, Juice and Wine. J. Chromatogr. A 2013, 1289, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Krüger, S.; Hüsken, L.; Fornasari, R.; Scainelli, I.; Morlock, G.E. Effect-Directed Fingerprints of 77 Botanical Extracts via a Generic High-Performance Thin-Layer Chromatography Method Combined with Assays and Mass Spectrometry. J. Chromatogr. A 2017, 1529, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Inarejos-Garcia, A.M.; Heil, J.; Martorell, P.; Álvarez, B.; Llopis, S.; Helbig, I.; Liu, J.; Quebbeman, B.; Nemeth, T.; Holmgren, D.; et al. Effect-Directed, Chemical and Taxonomic Profiling of Peppermint Proprietary Varieties and Corresponding Leaf Extracts. Antioxidants 2023, 12, 476. [Google Scholar] [CrossRef]
- Móricz, Á.M.; Ott, P.G.; Häbe, T.T.; Darcsi, A.; Böszörményi, A.; Alberti, Á.; Krüzselyi, D.; Csontos, P.; Béni, S.; Morlock, G.E. Effect-Directed Discovery of Bioactive Compounds Followed by Highly Targeted Characterization, Isolation and Identification, Exemplarily Shown for Solidago virgaurea. Anal. Chem. 2016, 88, 8202–8209. [Google Scholar] [CrossRef] [PubMed]
- Uwai, K.; Osanai, Y.; Imaizumi, T.; Kanno, S.; Takeshita, M.; Ishikawa, M. Inhibitory Effect of the Alkyl Side Chain of Caffeic Acid Analogues on Lipopolysaccharide-Induced Nitric Oxide Production in RAW264.7 Macrophages. Bioorg. Med. Chem. 2008, 16, 7795–7803. [Google Scholar] [CrossRef]
- Zhang, W.-D.; Bang Tam, H.T.; Chen, W.-S.; Kong, D.-Y.; Li, H.-T.; Wang, Y.-H.; Fouraste, I. Two New Caffeoyl Conjugation from Erigeron breviscapus. J. Asian Nat. Prod. Res. 2000, 2, 283–288. [Google Scholar] [CrossRef]
- Marealle, A.I.; Innocent, E.; Andrae-Marobela, K.; Qwarse, M.; Machumi, F.; Nondo, R.S.O.; Heydenreich, M.; Moshi, M.J. Safety Evaluation and Bioassay-Guided Isolation of Antimycobacterial Compounds from Morella salicifolia Root Ethanolic Extract. J. Ethnopharmacol. 2022, 296, 115501. [Google Scholar] [CrossRef]
- Pan, H.; Lundgren, L.N.; Andersson, R. Triterpene Caffeates from Bark of Betula pubescens. Phytochemistry 1994, 37, 795–799. [Google Scholar] [CrossRef]
- Menezes, J.C.J.M.D.S.; Edraki, N.; Kamat, S.P.; Khoshneviszadeh, M.; Kayani, Z.; Mirzaei, H.H.; Miri, R.; Erfani, N.; Nejati, M.; Cavaleiro, J.A.S.; et al. Long Chain Alkyl Esters of Hydroxycinnamic Acids as Promising Anticancer Agents: Selective Induction of Apoptosis in Cancer Cells. J. Agric. Food Chem. 2017, 65, 7228–7239. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Deepak, M.; Setty, M.; D’Souza, P.; Agarwal, A.; Sangli, G.K. Bioactive Caffeic Acid Esters from Glycyrrhiza glabra. Nat. Prod. Res. 2009, 23, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Velazquez Cruz, M.; Salinas-Arellano, E.; Castro Dionicio, I.; Jeyaraj, J.G.; Mirtallo Ezzone, N.P.; Carcache de Blanco, E.J. Bioactive Compounds Isolated from the Bark of Aesculus glabra Willd. Phytochem. Lett. 2024, 61, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Tabakam, G.T.; Kodama, T.; Donfack, A.R.N.; Nguekeu, Y.M.M.; Nomin-Erdene, B.; Htoo, Z.P.; Do, K.M.; Ngouela, S.A.; Tene, M.; Morita, H.; et al. A New Caffeic Acid Ester and a New Ceramide from the Roots of Eriosema glomeratum. Phytochem. Lett. 2021, 45, 82–87. [Google Scholar] [CrossRef]
- Menezes, J.C.J.M.D.S.; Kamat, S.P.; Cavaleiro, J.A.S.; Gaspar, A.; Garrido, J.; Borges, F. Synthesis and Antioxidant Activity of Long Chain Alkyl Hydroxycinnamates. Eur. J. Med. Chem. 2011, 46, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakasam, B.; Vanisree, M.; Zhang, Y.; Dewitt, D.L.; Nair, M.G. Impact of Alkyl Esters of Caffeic and Ferulic Acids on Tumor Cell Proliferation, Cyclooxygenase Enzyme, and Lipid Peroxidation. J. Agric. Food Chem. 2006, 54, 5375–5381. [Google Scholar] [CrossRef]
- Saha, M.M.; Mallik, U.K.; Mallik, A.K. A Chromenoflavanone and Two Caffeic Esters from Pongamia glabra. Phytochemistry 1991, 30, 3834–3836. [Google Scholar] [CrossRef]
- Hamburger, M.; Riese, U.; Graf, H.; Melzig, M.F.; Ciesielski, S.; Baumann, D.; Dittmann, K.; Wegner, C. Constituents in Evening Primrose Oil with Radical Scavenging, Cyclooxygenase, and Neutrophil Elastase Inhibitory Activities. J. Agric. Food Chem. 2002, 50, 5533–5538. [Google Scholar] [CrossRef]
- Qi, S.-Z.; Liu, T.; Wang, M.; Zhang, X.-X.; Yang, Y.-R.; Jing, W.-H.; Long, L.-P.; Song, K.-R.; Jin, Y.; Gao, H.-Y. New Phenylpropanoid-Conjugated Pentacyclic Triterpenoids from the Whole Plants of Leptopus lolonum with Their Antiproliferative Activities on Cancer Cells. Bioorg. Chem. 2021, 107, 104628. [Google Scholar] [CrossRef]
- Liao, C.-R.; Kuo, Y.-H.; Ho, Y.-L.; Wang, C.-Y.; Yang, C.; Lin, C.-W.; Chang, Y.-S. Studies on Cytotoxic Constituents from the Leaves of Elaeagnus oldhamii Maxim. in Non-Small Cell Lung Cancer A549 Cells. Molecules 2014, 19, 9515–9534. [Google Scholar] [CrossRef]
- Eom, H.J.; Kang, H.R.; Choi, S.U.; Kim, K.H. Cytotoxic Triterpenoids from the Barks of Betula platyphylla var. japonica. Chem. Biodivers. 2017, 14, e1600400. [Google Scholar] [CrossRef] [PubMed]
- Andre, C.M.; Larsen, L.; Burgess, E.J.; Jensen, D.J.; Cooney, J.M.; Evers, D.; Zhang, J.; Perry, N.B.; Laing, W.A. Unusual Immuno-Modulatory Triterpene-Caffeates in the Skins of Russeted Varieties of Apples and Pears. J. Agric. Food Chem. 2013, 61, 2773–2779. [Google Scholar] [CrossRef] [PubMed]
- Zarev, Y. Isolation and Characterization of 3- O-Caffeoyloleanolic Acid from Robinia pseudoacacia Stem Bark. Pharmacia 2023, 70, 1209–1212. [Google Scholar] [CrossRef]
- Al Ibrahim, M.; Akissi, Z.L.E.; Desmarets, L.; Lefèvre, G.; Samaillie, J.; Raczkiewicz, I.; Sahpaz, S.; Dubuisson, J.; Belouzard, S.; Rivière, C.; et al. Discovery of Anti-Coronavirus Cinnamoyl Triterpenoids Isolated from Hippophae rhamnoides during a Screening of Halophytes from the North Sea and Channel Coasts in Northern France. Int. J. Mol. Sci. 2023, 24, 16617. [Google Scholar] [CrossRef] [PubMed]
- Tanachatchairatana, T.; Bremner, J.B.; Chokchaisiri, R.; Suksamrarn, A. Antimycobacterial Activity of Cinnamate-Based Esters of the Triterpenes Betulinic, Oleanolic and Ursolic Acids. Chem. Pharm. Bull. 2008, 56, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-G.; Li, H.-R.; Wang, L.-Y.; Li, Y.-H.; Lu, S.-G.; Wen, X.-F.; Wang, J.; Daikonya, A.; Kitanaka, S. Triterpenoids from Hippophae rhamnoides L. and Their Nitric Oxide Production-Inhibitory and DPPH Radical-Scavenging Activities. Chem. Pharm. Bull. 2007, 55, 15–18. [Google Scholar] [CrossRef]
- Chen, P.-C.; Dlamini, B.S.; Chen, C.-R.; Shih, W.-L.; Lee, C.-H.; Chang, C.-I. Inhibitory Potential of Chemical Constituents from Paeonia suffruticosa Against α-Glucosidase and α-Amylase. Pharm. Chem. J. 2022, 56, 821–826. [Google Scholar] [CrossRef]
- Lv, J.; Duan, J.; Shen, B.; Yin, Y. Caffeic Acid Esters from Artemisia argyi and Their Antioxidant Activities. Chem. Nat. Compd. 2013, 49, 8–11. [Google Scholar] [CrossRef]
- Freire, C.S.R.; Silvestre, A.J.D.; Neto, C.P. Demonstration of Long-chain n-Alkyl Caffeates and Δ7-steryl Glucosides in the Bark of Acacia Species by Gas Chromatography–Mass Spectrometry. Phytochem. Anal. 2007, 18, 151–156. [Google Scholar] [CrossRef]
- Saraux, N.; Imeri, D.; Quirós-Guerrero, L.; Karimou, S.; Christen, P.; Cuendet, M. Phytochemical Investigation of the Roots of Ipomoea asarifolia and Antiproliferative Activity of the Isolated Compounds against Multiple Myeloma Cells. J. Nat. Prod. 2022, 85, 56–62. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, X.; Li, X.; Jiang, H.; Ma, Q.; Wang, P.; Liu, Y.; Hu, J.; Zheng, Y.; Zhou, J.; et al. Phenols with Anti-HIV Activity from Daphne acutiloba. Planta Med. 2012, 78, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ma, Q.; Huang, S.; Liang, W.; Wang, P.; Hu, J.; Zhou, J.; Zhao, Y. A New Guaiane-type Sesquiterpene with 15 Known Compounds from Wikstroemia scytophylla Diels. Chin. J. Chem. 2012, 30, 1335–1338. [Google Scholar] [CrossRef]
- Komatsu, M.; Yokoe, I.; Shirataki, Y. Studies on the Constituents of Sophora Species. XIII. Constituents of the Aerial Parts of Sophora tomentosa L. Chem. Pharm. Bull. 1978, 26, 3863–3870. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, M.; Tanaka, T.; Mizuno, M.; Lang, F.A. Two Flavanones from Roots of Sophora leachiana. Phytochemistry 1992, 31, 721–723. [Google Scholar] [CrossRef]
- Han, J.; Weng, X.; Bi, K. Antioxidants from a Chinese Medicinal Herb—Lithospermum erythrorhizon. Food Chem. 2008, 106, 2–10. [Google Scholar] [CrossRef]
- Badawy, A.; Hassanean, H.; Ibrahim, A.K.; Habib, E.S.; El-Magd, M.A.; Ahmed, S.A. Isolates from Thymelaea hirsuta Inhibit Progression of Hepatocellular Carcinoma In Vitro And In Vivo. Nat. Prod. Res. 2021, 35, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.; Tel-Çayan, G.; Öztürk, M.; Duru, M.E.; Nadeem, S.; Anis, I.; Ng, S.W.; Shah, M.R. Phytochemicals from Dodonaea viscosa and Their Antioxidant and Anticholinesterase Activities with Structure–Activity Relationships. Pharm. Biol. 2016, 54, 1649–1655. [Google Scholar] [CrossRef]
- Mumtaz, A.; Mohammad, A.; Khair, Z.; Habib, A.; Nazia, A.; Itrat, A.; Shah, M.R. Antiproliferative Activity and Chemical Constituents of Hypericum oblongifolium. J. Chem. Soc. Pak. 2011, 33, 772–777. [Google Scholar]
- Khan, R.; Malik, A.; Adhikari, A.; Qadir, M.I.; Choudhary, M.I. Conferols A and B, New Anti-Inflammatory 4-Hydroxyisoflavones from Caragana conferta. Chem. Pharm. Bull. 2009, 57, 415–417. [Google Scholar] [CrossRef][Green Version]
- Yodsaoue, O.; Karalai, C.; Ponglimanont, C.; Tewtrakul, S.; Chantrapromma, S. Potential Anti-Inflammatory Diterpenoids from the Roots of Caesalpinia mimosoides Lamk. Phytochemistry 2010, 71, 1756–1764. [Google Scholar] [CrossRef]
- Ibrar, A.; Khan, I.; Abbas, N. Structurally Diversified Heterocycles and Related Privileged Scaffolds as Potential Urease Inhibitors: A Brief Overview. Arch. Pharm. 2013, 346, 423–446. [Google Scholar] [CrossRef]
- Du, H.; Wang, Y.; Hao, X.; Li, C.; Peng, Y.; Wang, J.; Liu, H.; Zhou, L. Antimicrobial Phenolic Compounds from Anabasis aphylla L. Nat. Prod. Commun. 2009, 4, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Tchuenmogne, A.M.T.; Donfack, E.V.; Kongue, M.D.T.; Lenta, B.N.; Ngouela, S.; Tsamo, E.; Sidhu, N.; Dittrich, B.; Laatsch, H. Ingacamerounol, A New Flavonol and Other Chemical Constituents from Leaves and Stem Bark of Inga edulis Mart. Bull. Korean Chem. Soc. 2013, 34, 3859–3862. [Google Scholar] [CrossRef][Green Version]
- Wu, P.-L.; Wu, T.-S.; He, C.-X.; Su, C.-H.; Lee, K.-H. Constituents from the Stems of Hibiscus taiwanensis. Chem. Pharm. Bull. 2005, 53, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.G.H.; Martin, H.F. Antioxidants in Oats: Mono-esters of Caffeic and Ferulic Acids. J. Sci. Food Agric. 1967, 18, 589–595. [Google Scholar] [CrossRef]
- Sonar, V.P.; Corona, A.; Distinto, S.; Maccioni, E.; Meleddu, R.; Fois, B.; Floris, C.; Malpure, N.V.; Alcaro, S.; Tramontano, E.; et al. Natural Product-Inspired Esters and Amides of Ferulic and Caffeic Acid as Dual Inhibitors of HIV-1 Reverse Transcriptase. Eur. J. Med. Chem. 2017, 130, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Vuolo, M.M.; Lima, V.S.; Junior, M.R.M. Phenolic compounds: Structure, classification, and antioxidant power. In Bioactive Compounds; Elsevier: Amsterdam, The Netherlands, 2019; pp. 33–50. [Google Scholar]
- Gao, Q.; Li, Y.; Li, Y.; Zhang, Z.; Liang, Y. Antioxidant and Prooxidant Activities of Phenolic Acids Commonly Existed in Vegetables and Their Relationship with Structures. Food Sci. Technol. 2022, 42, e07622. [Google Scholar] [CrossRef]
- Gonçalves, A.C.; Falcão, A.; Alves, G.; Silva, L.R.; Flores-Félix, J.D. Antioxidant Activity of the Main Phenolics Found in Red Fruits: An in Vitro and in Silico Study. Food Chem. 2024, 452, 139459. [Google Scholar] [CrossRef]
- Nenadis, N.; Lazaridou, O.; Tsimidou, M.Z. Use of Reference Compounds in Antioxidant Activity Assessment. J. Agric. Food Chem. 2007, 55, 5452–5460. [Google Scholar] [CrossRef]
- Jork, H.; Funk, W.; Fischer, W.; Wimmer, H.; Burns, D.T. Thin-Layer Chromatography: Reagents and Detection Methods; VCH: Weinheim, Germany, 1990. [Google Scholar]
- Móricz, Á.M.; Ott, P.G.; Morlock, G.E. Discovered Acetylcholinesterase Inhibition and Antibacterial Activity of Polyacetylenes in Tansy Root Extract via Effect-Directed Chromatographic Fingerprints. J. Chromatogr. A 2018, 1543, 73–80. [Google Scholar] [CrossRef]
- Schreiner, T.; Sauter, D.; Friz, M.; Heil, J.; Morlock, G.E. Is Our Natural Food Our Homeostasis? Array of a Thousand Effect-Directed Profiles of 68 Herbs and Spices. Front. Pharmacol. 2021, 12, 755941. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).