Abstract
Proton exchange membrane water electrolysis is hindered by the sluggish kinetics of the anodic oxygen evolution reaction. RuO2 is regarded as a promising alternative to IrO2 for the anode catalyst of proton exchange membrane water electrolyzers due to its superior activity and relatively lower cost compared to IrO2. However, the dissolution of Ru induced by its overoxidation under acidic oxygen evolution reaction (OER) conditions greatly hinders its durability. Herein, we developed a strategy for stabilizing RuO2 in acidic OER by the incorporation of high-valence metals with suitable ionic electronegativity. A molten salt method was employed to synthesize a series of high-valence metal-substituted RuO2 with large specific surface areas. The experimental results revealed that a high content of surface Ru4+ species promoted the OER intrinsic activity of high-valence doped RuO2. It was found that there was a linear relationship between the ratio of surface Ru4+/Ru3+ species and the ionic electronegativity of the dopant metals. By regulating the ratio of surface Ru4+/Ru3+ species, incorporating Re, with the highest ionic electronegativity, endowed Re0.1Ru0.9O2 with exceptional OER activity, exhibiting a low overpotential of 199 mV to reach 10 mA cm−2. More importantly, Re0.1Ru0.9O2 demonstrated outstanding stability at both 10 mA cm−2 (over 300 h) and 100 mA cm−2 (over 25 h). The characterization of post-stability Re0.1Ru0.9O2 revealed that Re promoted electron transfer to Ru, serving as an electron reservoir to mitigate excessive oxidation of Ru sites during the OER process and thus enhancing OER stability. We conclude that Re, with the highest ionic electronegativity, attracted a mass of electrons from Ru in the pre-catalyst and replenished electrons to Ru under the operating potential. This work spotlights an effective strategy for stabilizing cost-effective Ru-based catalysts for acidic OER.
1. Introduction
Hydrogen is one of the most promising energy carriers in the world and is considered as the ultimate energy in the 21st century with the advantages of high-mass energy density and environment-friendly energy [1]. Nowadays, hydrogen production mainly depends on fossil fuels, leading to global warming and severe air pollution [2]. In light of this, electricity-driven water splitting has great potential for hydrogen production because the electricity can be collected from intermittent renewable energy such as sunlight and wind [3]. There are three types of water electrolysis technologies: alkaline water electrolysis (AWE), proton exchange membrane water electrolysis (PEMWE), and solid oxide electrolysis cell (SOEC) [4]. Among these various water electrolysis technologies, proton exchange membrane water electrolysis attracts more attention due to its high purity (>99.9999 vol%) of hydrogen production, immediate response, and low ohmic losses [5].
Proton exchange membrane water electrolysis is an efficient approach for renewable hydrogen production to facilitate a carbon neutral future [6,7,8]. However, PEMWE stacks are usually carried at current densities exceeding 500 mA cm−2 [9]. To meet the need of faster proton transfer, the acidity close to the anode is stronger than 1 M H2SO4 when only using distilled water as the electrolyte, and this effect is aggravated when using acidic electrolytes or high current densities [10]. As a consequence, rare materials demonstrate desirable activity and stability at the anode of PEMWE due to the highly acidic and oxidative working conditions [11,12], where a sluggish oxygen evolution reaction (OER) involving four proton transfer steps takes place [5,13]. To date, IrO2 is currently the most widely used OER catalyst in acidic conditions, but its high cost (Ir: 140 USD g−1) [14] and scarcity have challenged its popularized application in PEMWE [15,16]. Due to the high overpotential of Ir in catalyzing the oxygen evolution reaction, Ir-based catalysts fail to meet the application requirements. RuO2, with relatively high earth abundance, lower price (Ru: 16 USD g−1) [14], and superior OER activity, is considered as a promising alternative to IrO2 [17,18]. Nevertheless, Ru-based catalysts suffer unavoidable overoxidation of Ru sites to form soluble RuO4 species at high overpotential, resulting in the loss of Ru active sites and degradation of OER performance [19,20,21]. Therefore, the stabilization of Ru species is pivotal for the development of highly active and cost-effective OER catalysts for PEMWE.
Formation of lattice oxygen vacancies (VO) and overoxidation of active Ru during acidic OER together accelerate RuO2 crystal structure collapse and surface Ru loss [21,22]. Given this, introducing electron-donating elements to stabilize low-charge Ru at fixed potential or improving the intrinsic activity of catalysts to decrease the operation potential at the controlled water-splitting current are two main methods [23]. Element doping has great potential to combine the advantages of the above two methods. Meanwhile, tuning the electron structure of Ru sites by foreign metal dopants has been considered as an effective strategy to hinder the dissolution of Ru [24,25,26]. Recently, many efforts have been aimed at improving the stability of Ru-based catalysts via high-valence metal substitution [24,27,28]. For example, Sun’s group reported that the incorporation of high-valence Nb in RuO2 weakened the covalency of the Ru–O bond with increased electron density around Ru sites, resulting in high OER stability (360 h at 200 mA cm−2) in acidic conditions [14]. Liu’s group developed a tungsten oxide matrix-confined Ru catalyst to stabilize low-valence Ru by facilitating electron transfer from oxidized W through the O bridge during an OER stability test (45 h at 10 mA cm−2) [29]. These studies demonstrated the impressive effect of stabilizing active Ru during OER by the modification of high-valence metal [30,31]. However, Zhang et al. revealed that the incorporation of different types of high-valence metals in RuO2 led to distinct OER activity and stability [27]. Therefore, the enhanced stability of these catalysts could be governed by underlying factors rather than solely relying on modification with high-valence metals (>+4). In a previous study, the ionic electronegativity of foreign metal dopants was correlated with the active site’s charge, which reflected the local metal–O bonding structure and the bonding strength [23]. On the other hand, Nørskov et al. reported that ‘stable’ RuO2 exhibited unsatisfactory catalytic activity due to the lack of unstable high-valence Run>4+ species [32,33]. In this consideration, the ionic electronegativity of the incorporated high-valence metals may be the deeper factor that affects the activity and stability of Ru. In addition, the role of ionic electronegativity in stabilizing the Ru site needs to be elucidated as well.
In this work, we introduced high-valence metals as electron reservoirs with different ionic electronegativities in rutile RuO2 to screen out a catalyst with remarkable activity and stability. We prepared the high-valence metal-substituted RuO2 using a modified molten salt method. The obtained catalysts exhibited a nanosheet morphology consisting of small nanoparticles with high surface areas, resulting in outstanding apparent activity of all samples. We correlated the ratio of surface Ru4+/Ru3+ species and the intrinsic activity and stability of the as-prepared catalysts with the ionic electronegativity of the incorporated cations. It was found that the substitution of high-valence metals with different ionic electronegativities could regulate the ratio of surface Ru4+/Ru3+ species where the oxygen evolution reaction takes place. RuO2 doped with the metal with the highest ionic electronegativity, Re, exhibited remarkable OER performance, characterized by a low overpotential of 199 mV, long-term stability exceeding 300 h at a current density of 10 mA cm−2, and even survived a 25 h operation at 100 mA cm−2 in a 0.1 M HClO4 electrolyte. We conclude that Re, with the highest ionic electronegativity, is equipped with a larger electron reservoir, which can serve as an electron accepter from Ru and induce high OER activity in pre-catalysts, and turn into an electron donor at high potential to inhibit the overoxidation of Ru during the OER process, according to the XPS results. Tuning the surface Ru4+/Ru3+ species content of rutile Ru-based oxides via the different ionic electronegativities of high-valence metal dopants is an effective strategy for designing high-performance Ru-based oxide OER catalysts in acidic electrolyte.
2. Results and Discussion
2.1. Characterization of the M0.1Ru0.9O2 Catalysts
We are inspired by the fact that the ionic electronegativity of metal dopants can affect the Ru-O bonding nature in RuO2 [23]. In this work, we introduce high-valence metal dopants with different ionic electronegativities to serve as electron reservoirs in M-RuO2 to transfer electrons under different potentials (Figure 1a). As a proof of concept, Re7+, Mo6+, and Ta5+ with ionic electronegativities of 2.505, 2.101, and 1.925 were selected to partially substitute Ru in RuO2 [34]. To retain the rutile structure of the catalysts, the substituted amount was controlled at a small proportion of 0.1, which was the mole ratio of the foreign cation to the total metal in M-RuO2 (Figure 1b). High-valence metal (Re, Mo, Ta)-substituted RuO2 nanosheets (denoted as M0.1Ru0.9O2) were synthesized by a modified molten salt method. We used Brunauer–Emmett–Teller (BET) derived from N2 adsorption–desorption experiments to evaluate the specific surface area of the pre-catalysts. All homemade catalysts displayed high specific surface areas in the range from 154 m2 g−1 to 201 m2 g−1 (Figure S1), which was beneficial to the exposure of sufficient active sites to ensure high apparent activity. Typically, Re0.1Ru0.9O2 exhibited the highest BET surface area of 201 m2 g−1.
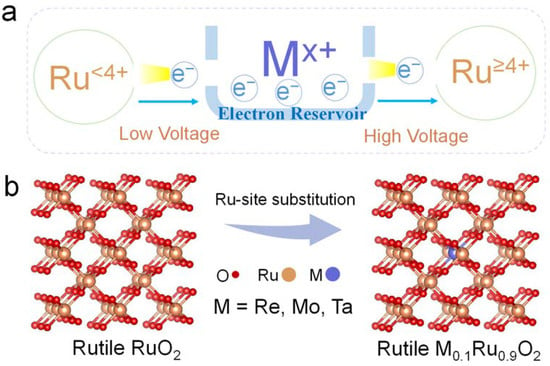
Figure 1.
(a) Schematic diagram of the electron transfer process under different potentials. (b) Schematic image of partial substitution of Ru with Re, Mo, and Ta.
The X-ray diffraction (XRD) patterns (Figure 2a) showed that the M0.1Ru0.9O2 catalysts maintained the same crystal structure and phase as rutile RuO2 without any impurities. Three characteristic peaks at 28.1°, 35.3°, and 54.1° were attributed to the (110), (101), and (211) planes for rutile RuO2, respectively [35]. The (110) peak in the XRD spectra of M0.1Ru0.9O2 exhibited a negative shift compared to that of RuO2, which could be attributed to the larger radius of the incorporated M cation, resulting in lattice expansion (Figure S2). According to the transmission electron microscopy (TEM) analysis, Re0.1Ru0.9O2 presented a unique morphology of porous nanosheets assembled with ~5-nanometer particles (Figure 2b and Figure S3a). Such ultrasmall nanoparticle morphology favored the exposure of highly active sites to improve the OER activity. In addition, the rutile structure was also confirmed by high-resolution TEM, wherein the well-defined lattice fringes with interplanar distances of 0.33 nm were assigned to the (110) plane of the structure (Figure 2c) and displayed a broader lattice distance in contrast with homemade RuO2 (HM-RuO2). The expanded lattice distance in Re0.1Ru0.9O2 was consistent with the negative shift of the XRD peak (Figure S3d). Similar morphologies and crystal structures could be observed in HM-RuO2, Ta0.1Ru0.9O2, and Mo0.1Ru0.9O2 (Figures S3b–d, S4a–c, and S5a–c). These results indicated that high-valence metal substitution barely changed the morphology along with high-valence metals equipped into the rutile RuO2 lattice matrix. Moreover, TEM elemental mapping images presented uniform distributions of Ru, O, and high-valence foreign metals in the particles (Figure 2d–g, Figure S4d–g and S5d–g). The above results indicated the successful substitution of high-valence foreign metals in RuO2.

Figure 2.
Morphological characterization of M0.1Ru0.9O2 (M = Re, Mo, Ta) and HM-RuO2. (a) XRD patterns, (b) TEM image, (c) magnified TEM image, and (d–g) TEM and corresponding elemental mapping images of Re0.1Ru0.9O2.
The surface compositions and the corresponding electronic structures were further investigated by X-ray photoelectron spectroscopy (XPS), as the electrochemical reaction process takes place at the surface of the catalyst. As shown in Figure 3a and Table S1, the Ru 3p3/2 signals could be deconvoluted into two doublets. The two typical peaks of Ru 3p3/2 obtained for all samples were centered at 462.3 and 465.5, indexed to Ru4+ and Ru3+ species, respectively [14,36]. This result revealed that the oxidation state of Ru in the pre-catalysts was an Ru4+/Ru3+ mixture, which offered the potential for electron transfer. A linear relationship between the ratio of Ru4+/Ru3+ and ionic electronegativity was found, as shown in Figure 3b. The areal ratio of Ru4+/Ru3+ increased as the ionic electronegativity of the dopant metal increased from 1.925 (Ta5+) to 2.505 (Re7+), indicating the intensified valence state of Ru because the high-valence foreign metal with higher ionic electronegativity induced a reduced electron density in Ru. This trend of the variation in Ru valence was consistent with a previous report [23]. It was reported that Ru-based catalysts with a higher oxidation state of Ru exhibited higher OER activity due to optimized binding energy with oxygen intermediates [37]. In addition, the existence of high-valence foreign metals was also verified by their corresponding XPS spectra (Figure S6). According to previous reports, the oxidation states of Re, Mo, and Ta in the corresponding oxides were determined to be +7, +6, and +5, respectively, which were similar to our XPS results [38,39,40,41].
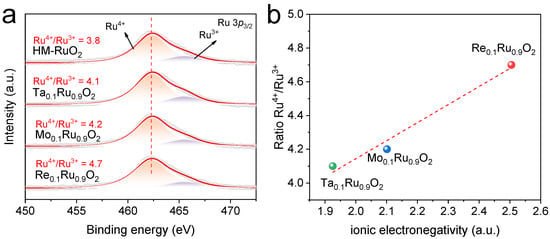
Figure 3.
Electronic structures of M0.1Ru0.9O2 (M = Re, Mo, Ta) and HM-RuO2. (a) Ru 3p XPS spectra (The orange part indicates the content of Ru4+ and the purple part indicates the content of Ru3+). (b) Scaling between the ionic electronegativity [34] of foreign high-valence metals and the areal ratio of Ru4+/Ru3+ species.
2.2. Acidic OER Performance
The electrocatalytic performance of all catalysts for OER in 0.1 M HClO4 electrolyte was evaluated on a rotating disk electrode (RDE) in a typical three-electrode electrolytic cell. We used commercial RuO2 (denoted as C-RuO2) and commercial IrO2 (denoted as C-IrO2) as the benchmarks to investigate the effect of ionic electronegativity on acidic OER performance (Figure 4). It can be seen that the OER activities of all of the obtained catalysts far outperformed those of C-RuO2 and C-IrO2 (Figure 4a). Re-doped RuO2 with the best OER activity achieved a current density of 10 mA cm−2 with the lowest overpotential of 199 mV, while Mo- and Ta-substituted RuO2 catalysts delivered slightly larger overpotentials of 218 mV and 223 mV, respectively. Additionally, Re0.1Ru0.9O2 possessed the lowest Tafel slope of 51.3 mV dec−1 (Figure 4b,c), indicating that Re replacement accelerated the kinetics of the OER process. Electrochemical impedance spectroscopy (EIS) was also used to evaluate the OER kinetics. The obtained Nyquist plots were fitted using Autolab software “Nova 2.1.4 Version” (Figure S7), employing a consistent Randles equivalent circuit, including series and charge transfer resistance (Rs and Rct) as well as capacitance and phase elements [42,43]. It was found that Re-doped RuO2 exhibited the lowest charge transfer resistance with a small value of 16.0 Ω at an overpotential of 230 mV compared to Mo0.1Ru0.9O2 (19.6 Ω) and Ta0.1Ru0.9O2 (19.8 Ω), again implying its enhanced OER kinetics, which followed the same trend as the Tafel slope. The specific activities of the catalysts were determined by measuring their BET surface areas derived from nitrogen adsorption–desorption analysis. The ECSA-normalized OER activities were calculated to compare the intrinsic activities of M0.1Ru0.9O2. As can be seen in Figure 4d, the specific activities of those catalysts followed the order of Re0.1Ru0.9O2 > Mo0.1Ru0.9O2 > Ta0.1Ru0.9O2.
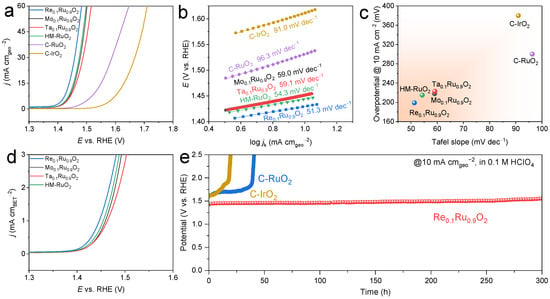
Figure 4.
OER performance of the prepared electrocatalysts in 0.1 M HClO4. (a) LSV curves. (b) Tafel plots. (c) Comparison of overpotential and Tafel slope among M0.1Ru0.9O2 (M = Re, Mo, Ta), HM-RuO2, commercial RuO2, and commercial IrO2. (d) Specific activities of the prepared catalysts (OER currents normalized by BET surface area). (e) Chronopotentiometry tests of Re0.1Ru0.9O2, commercial RuO2, and commercial IrO2 at 10 mA cm−2.
Electrocatalytic stability is a more critical indicator for Ru-based catalysts. Chronopotentiometry (CP) tests were performed to investigate the OER durability of Re0.1Ru0.9O2, C-RuO2, and C-IrO2 in 0.1 M HClO4 at constant current densities of 10 mA cm−2 and 100 mA cm−2 (Figure 4e and Figure 5a). It is worth noting that Re0.1Ru0.9O2 exhibited highly desirable stability with a small degradation rate of 0.35 mV h−1 during 300 h continuous operation at 10 mA cm−2 and survived a 25 h CP test at 100 mA cm−2. Such stable performance was competitive with previously reported advanced Ru-based catalysts under similar conditions (Table S2). In contrast, commercial RuO2 suffered a sharp decline during the 50 h test at the current density of only 10 mA cm−2. Commercial IrO2 also failed to survive the long-term stability test at 10 mA cm−2. After the 300 h test, only 9.17 wt% Ru in Re0.1Ru0.9O2 was dissolved into the electrolyte, with a releasing rate of 0.015 μgRu h−1 based on ICP-MS analysis. The corresponding characterizations of post-stability Re0.1Ru0.9O2 were performed to observe the possible change during the OER test (Figure S8). The post-stability sample maintained the same morphology as that in Figure 2b. Further, the rutile crystal structure was preserved according to the HR-TEM image, which presented numerous lattice fringes indexed to the RuO2 (110) plane (Figure S8b). Moreover, the element compositions were still uniformly distributed, as seen in Figure S8c–f. The above results indicated that the Re0.1Ru0.9O2 catalyst maintained the bulk crystal structure well, suggesting the high acid corrosion resistance of Re0.1Ru0.9O2.

Figure 5.
(a) Chronopotentiometry tests of M0.1Ru0.9O2 (M = Re, Mo, Ta, Ru) at 100 mA cm−2. (b,c) Comparison of specific activity (current density normalized by BET at 1.50 V vs. RHE) and potential variation at 100 mA cm−2 in a 2 h stability test as the ionic electronegativity increases. (d) XPS spectra of Re0.1Ru0.9O2 before and after the OER stability test. (e) XPS spectra of HM-RuO2 before and after the OER stability test. (The orange part indicates the content of Ru4+ and the purple part indicates the content of Ru3+). (f) Schematic diagram of electron transfer in Re0.1Ru0.9O2 and HM-RuO2 during the OER process.
2.3. Investigation of Relationship between OER Stability and Electron Structure
Obtaining insights into the noticeable stability is significant for practical applications. It has been reported that high-valence metals with multiple oxidation states enable variations in Ru valence under different potentials. That is, the role of the high-valence metals in M0.1Ru0.9O2 can transform from electron receptors at on-site potential to electron donors at high potential [31,44]. According to the XPS results, dopants with high ionic electronegativity attracted a mass of electrons from Ru in the pre-catalysts and then generated active high-valence Ru species. The prepared M0.1Ru0.9O2 catalysts possessed high intrinsic activity compared to commercial RuO2, which helped to reduce the working potential at the controlled current density. The stability at a high current density of 100 mA cm−2 for the M0.1Ru0.9O2 catalysts was assessed to compare their acid tolerance at a relatively high potential (Figure 5a). The acid resistance of Re-doped RuO2 was superior to that of the homemade remainder. We increased the operation potential to 100 mA cm−2 in 2 h to quantify the acid tolerance at relatively high potential. Both the specific activity and the acid tolerance of high-valence metal-doped RuO2 improved as the ionic electronegativity increased (Figure 5b,c). XPS analysis of the post-catalysts after a 2 h stability test at 100 mA cm−2 was carried out to explore the variations in the ratio of surface Ru species (Figure 5d,e and Table S3). The ratio of Ru4+/Ru3+ decreased from 4.7 to 3.4 for Re0.1Ru0.9O2, while the ratio of Ru4+/Ru3+ increased from 3.8 to 4.3 for HM-RuO2, implying that electrons were transferred from Re with high ionic electronegativity to Ru during the OER process (Figure 5f). Consequently, introducing Re with high ionic electronegativity as an electron reservoir into RuO2 made a great contribution to the enhancement of OER stability.
3. Materials and Methods
3.1. Chemicals
Sodium nitrate (NaNO3), ruthenium (III) chloride hydrate (RuCl3·3H2O), molybdenum pentachloride (MoCl5), and tantalum (V) chloride (TaCl5) were purchased from Macklin Ltd. Commercial RuO2, commercial IrO2, and sodium perrhenate (NaReO4) were purchased from Energy Chemical Ltd. The 5 wt% Nafion® ionomer was purchased from DuPont Co. All of the reagents and chemicals were used as received without further purification.
3.2. Preparation of M0.1Ru0.9O2 Catalysts
A family of M0.1Ru0.9O2 (M = Re, Ta, Mo, Ru) catalysts was synthesized by a modified molten salt method [45]. Generally, 0.45 mmol RuCl3, 0.05 mmol NaReO4, and 10 g NaNO3 powder were ground evenly in a mortar, and then the mixed powder was transferred into a 50 mL porcelain crucible. After the precursor salts were melted in a muffle furnace at 350 °C for 2 h, the catalysts were obtained in the flowing black molten salt. The mixture was removed for quick cooling to room temperature. The obtained product was rinsed with Milli-Q ultra-pure water thrice and dried in an oven at 60 °C. Taking HM-RuO2 samples as reference, 0.45 mmol RuCl3 and 10 g NaNO3 powder were ground evenly in a mortar. Other samples were prepared by the same procedure, except the foreign M salt was replaced with the corresponding precursor salts (TaCl5 and MoCl5).
3.3. Physical Characterization
Nitrogen adsorption–desorption analysis was carried out on a Tristar II 3020 gas adsorption analyzer (Micromeritics, Atlanta, GA, USA) to obtain the specific surface area of samples. X-ray diffraction (XRD) patterns were obtained from a Miniflex-600 powder diffractometer (Rigaku, Shibuya-ku, Tokyo, Japan) with Cu Kα radiation to determine the crystal structure of samples. The data were collected with a step size of 0.01 s and a dwell time of 0.1 s. Transmission electron microscopy (TEM) and the corresponding energy-dispersive X-ray analysis (EDX) were conducted using a Talos F200x field emission microscope (Thermo Fisher, Waltham, MA, USA) to detect the morphology and the element distribution of the samples. X-ray photoelectron spectroscopy (XPS) results were collected from a K-Alpha+ photoelectron spectrometer (Thermo Fisher Scientific) to analyze the surface composition and the valence of elements in the samples. All binding energies of the elements were referred to the C 1 s peak at 284.8 eV. Inductively coupled plasma (ICP) measurements were completed on an IRIS Intrepid instrument (Thermo Fisher, Waltham, MA, USA) to detect the content of the leaching element in the electrolyte after a long-term stability test. The moles of dissolved Ru (nRu) in Re0.1Ru0.9O2 were determined by: nRu = mRu/MRu, where mRu is the mass of dissolved Ru after a 300 h OER stability test, and MRu is the molar mass of Ru (101.07 g mol−1).
3.4. Electrochemical Measurements
The electrochemical performance of the as-prepared catalysts was evaluated on a WaveDriver electrochemical instrument using a standard three-electrode system in 0.1 M HClO4, with the working electrode rotating at a speed of 2500 rpm and a graphite rod (diameter = 6 mm) serving as the counter electrode. Ag/AgCl (saturated KCl) was used as the reference electrode and catalyst-supported glassy carbon (GC, diameter = 5 mm) was regarded as the working electrode. All of the samples were measured based on independent tests. Typically, 5 mg catalyst powder, 5 mg carbon powder (XC-72), and 50 μL 5% Nafion were dispersed in 950 μL isopropanol and then sonicated for 1 h to form a homogeneous ink. Carbon powder served to increase the conductivity for the oxide catalysts. A 10 μL sample of the catalyst dispersion was dropped onto the glassy carbon electrode (0.25 mg cm−1). All potentials, except the potential of the stability tests, were calibrated on glassy carbon versus the reversible hydrogen electrode (RHE), using the following equation:
where 0.26 V is the potential difference between the Ag/AgCl reference electrode and RHE in 0.1 M HClO4 and calibrated in H2-saturated 0.1 M HClO4 prior to electrocatalytic measurement.
E (vs.RHE) = E (vs. Ag/AgCl) + 0.26 V
The linear sweep voltammetry (LSV) curves of all catalysts were collected in 0.1 M HClO4 within the potential range of 1.0~1.8 V vs. Ag/AgCl at a scanning rate of 10 mV s−1. Electrochemical impedance spectroscopy (EIS) analysis was conducted at 1.2 V vs. Ag/AgCl over the frequency range of 0.1~100 kHz. The obtained electrochemical data with iR compensation were denoted as in the following equation: Ecorrection = E − iRsolution, where Rsolution is the solution resistance from the EIS measurements. The stability tests were carried out on 0.5 cm × 0.5 cm carbon paper with a mass loading of 1 mgoxide cm−2 under 0.1 M HClO4 at 10 mA cm−2 or 100 mA cm−2. The potentials of the stability tests were referenced to the RHE by using pure hydrogen calibration and corrected with iR loss on carbon paper.
4. Conclusions
In summary, high-valence metal dopants with high ionic electronegativity were employed as electron reservoirs to stabilize the Ru sites in Ru-based oxide lattice matrices for an efficient acidic OER process. A molten-salt method was used to synthesize the rutile RuO2 catalysts doped by M (Re, Ta, Mo) cations with different ionic electronegativities. We correlated ionic electronegativity with the ratio of surface Ru4+/Ru3+ species, the intrinsic activity, and the durability in acid of the catalysts. The high ionic electronegativities of the dopants induced high ratios of surface Ru4+/Ru3+ species, along with enhancements in both the intrinsic activity and the durability of the catalysts. Among all of the studied catalysts, Re0.1Ru0.9O2 exhibited excellent OER performance with a low overpotential of 199 mV and a negligible decay over a 300 h stability test at 10 mA cm−2. It was found that Re, with the highest ionic electronegativity, served as an electron donor in Re0.1Ru0.9O2 to inhibit the overoxidation of Ru at high potential and thus stabilize active Ru species. This work provides insight into the role of substitution with a high-valence metal with high ionic electronegativity as an electron reservoir to improve the stability of Ru-based materials.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29040785/s1. References [46,47,48,49,50,51,52,53] are cite in the Supplementary Materials.
Author Contributions
Conceptualization, J.W., Z.Q., J.Z. and L.D.; methodology, J.W. and J.Z.; investigation, J.W., Z.Q., J.Z. and L.D.; data curation, J.W.; writing—original draft preparation, J.W.; writing—review and editing, J.W., Z.Q. and J.Z.; supervision, H.S., Z.C., J.Z. and L.D.; project administration, L.D.; funding acquisition, J.Z. and L.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 22302071 and 21975080), the Guangdong Basic and Applied Basic Research Foundation for Distinguished Young Scholars (No. 2021B1515020025), the Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515111097), the China Postdoctoral Science Foundation (2022M711195), and Fundamental Research Funds for the Central Universities, SCUT.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gasteiger, H.A.; Marković, N.M. Just a Dream—Or Future Reality? Science 2009, 324, 48–49. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A Comprehensive Review on PEM Water Electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Montoya, J.H.; Seitz, L.C.; Chakthranont, P.; Vojvodic, A.; Jaramillo, T.F.; Nørskov, J.K. Materials for Solar Fuels and Chemicals. Nat. Mater. 2016, 16, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liang, X.; Wang, L.; Sun, K.; Wang, Y.; Xie, Z.; Wu, Q.; Bai, X.; Hamdy, M.S.; Chen, H.; et al. Status and Perspectives of Key Materials for PEM Electrolyzer. Nano Res. Energy 2022, 1, 9120032. [Google Scholar] [CrossRef]
- Wang, Y.; Pang, Y.; Xu, H.; Martinez, A.; Chen, K.S. PEM Fuel Cell and Electrolysis Cell Technologies and Hydrogen Infrastructure Development—A Review. Energy Environ. Sci. 2022, 15, 2288–2328. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and Challenges for a Sustainable Energy Future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.A. Sustainable Hydrogen Production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, 146. [Google Scholar] [CrossRef] [PubMed]
- Millet, P.; Ngameni, R.; Grigoriev, S.A.; Mbemba, N.; Brisset, F.; Ranjbari, A.; Etiévant, C. PEM Water Electrolyzers: From Electrocatalysis To Stack Development. Int. J. Hydrogen Energy 2010, 35, 5043–5052. [Google Scholar] [CrossRef]
- Wang, Q.; Cheng, Y.; Tao, H.B.; Liu, Y.; Ma, X.; Li, D.S.; Yang, H.B.; Liu, B. Long-Term Stability Challenges and Opportunities in Acidic Oxygen Evolution Electrocatalysis. Angew. Chem. Int. Ed. 2023, 62, 202216645. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Q.; Wang, A.; Duan, J.; Zhou, W.; Sang, Y.; Liu, H. Iron Oxide Embedded Titania Nanowires—An Active and Stable Electrocatalyst for Oxygen Evolution in Acidic Media. Nano Energy 2018, 45, 118–126. [Google Scholar] [CrossRef]
- Gao, J.; Tao, H.; Liu, B. Progress of Nonprecious-Metal-Based Electrocatalysts for Oxygen Evolution in Acidic Media. Adv. Mater. 2021, 33, 2003786. [Google Scholar] [CrossRef] [PubMed]
- Schuler, T.; Kimura, T.; Schmidt, T.J.; Büchi, F.N. Towards a Generic Understanding of Oxygen Evolution Reaction Kinetics in Polymer Electrolyte Water Electrolysis. Energy Environ. Sci. 2020, 13, 2153–2166. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Fang, J.; Li, M.; Sendeku, M.G.; Wang, X.; Wu, H.; Li, Y.; Ge, J.; Zhuang, Z.; et al. Eliminating Over-oxidation of Ruthenium Oxides by Niobium for Highly Stable Electrocatalytic Oxygen Evolution in Acidic Media. Joule 2023, 7, 558–573. [Google Scholar] [CrossRef]
- Li, N.; Cai, L.; Wang, C.; Lin, Y.; Huang, J.; Sheng, H.; Pan, H.; Zhang, W.; Ji, Q.; Duan, H.; et al. Identification of the Active-Layer Structures for Acidic Oxygen Evolution from 9R-BaIrO3 Electrocatalyst with Enhanced Iridium Mass Activity. J. Am. Chem. Soc. 2021, 143, 18001–18009. [Google Scholar] [CrossRef]
- Yin, J.; Jin, J.; Lu, M.; Huang, B.; Zhang, H.; Peng, Y.; Xi, P.; Yan, C.-H. Iridium Single Atoms Coupling with Oxygen Vacancies Boosts Oxygen Evolution Reaction in Acid Media. J. Am. Chem. Soc. 2020, 142, 18378–18386. [Google Scholar] [CrossRef]
- Su, J.W.; Ge, R.X.; Jiang, K.M.; Dong, Y.; Hao, F.; Tian, Z.Q.; Chen, G.X.; Chen, L. Assembling Ultrasmall Copper-Doped Ruthenium Oxide Nanocrystals into Hollow Porous Polyhedra: Highly Robust Electrocatalysts for Oxygen Evolution in Acidic Media. Adv. Mater. 2018, 30, 1801351. [Google Scholar] [CrossRef]
- Yu, J.; He, Q.; Yang, G.; Zhou, W.; Shao, Z.; Ni, M. Recent Advances and Prospective in Ruthenium-Based Materials for Electrochemical Water Splitting. ACS Catal. 2019, 9, 9973–10011. [Google Scholar] [CrossRef]
- Spöri, C.; Kwan, J.T.H.; Bonakdarpour, A.; Wilkinson, D.P.; Strasser, P. The Stability Challenges of Oxygen Evolving Catalysts: Towards a Common Fundamental Understanding and Mitigation of Catalyst Degradation. Angew. Chem. Int. Ed. 2017, 56, 5994–6021. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.R.; Kolb, M.J.; Giordano, L.; Pedersen, A.F.; Katayama, Y.; Hwang, J.; Mehta, A.; You, H.; Lunger, J.R.; Zhou, H.; et al. Operando Identification of Site-dependent Water Oxidation Activity on Ruthenium Dioxide Single-crystal Surfaces. Nat. Catal. 2020, 3, 516–525. [Google Scholar] [CrossRef]
- Roy, C.; Rao, R.R.; Stoerzinger, K.A.; Hwang, J.; Rossmeisl, J.; Chorkendorff, I.; Shao-Horn, Y.; Stephens, I.E.L. Trends in Activity and Dissolution on RuO2 under Oxygen Evolution Conditions: Particles versus Well-defined Extended Surfaces. ACS Energy Lett. 2018, 3, 2045–2051. [Google Scholar] [CrossRef]
- Deka, N.; Jones, T.E.; Falling, L.J.; Sandoval-Diaz, L.-E.; Lunkenbein, T.; Velasco-Velez, J.-J.; Chan, T.-S.; Chuang, C.-H.; Knop-Gericke, A.; Mom, R.V. On the Operando Structure of Ruthenium Oxides during the Oxygen Evolution Reaction in Acidic Media. ACS Catal. 2023, 13, 7488–7498. [Google Scholar] [CrossRef]
- Shi, Z.; Li, J.; Wang, Y.; Liu, S.; Zhu, J.; Yang, J.; Wang, X.; Ni, J.; Jiang, Z.; Zhang, L.; et al. Customized Reaction Route for Ruthenium Oxide towards Stabilized Water Oxidation in High-Performance PEM Electrolyzers. Nat. Commun. 2023, 14, 843. [Google Scholar] [CrossRef]
- Wang, H.; Zhai, T.; Wu, Y.; Zhou, T.; Zhou, B.; Shang, C.; Guo, Z. High-Valence Oxides for High Performance Oxygen Evolution Electrocatalysis. Adv. Sci. 2023, 10, 2301706. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Yang, B.; Li, Z.; Qin, X.; Zhang, Q.; Lei, L.; Qiu, M.; Wu, G.; Hou, Y. Highly Active Ruthenium Sites Stabilized by Modulating Electron-feeding for Sustainable Acidic Oxygen-evolution Electrocatalysis. Energy Environ. Sci. 2022, 15, 2356–2365. [Google Scholar] [CrossRef]
- Hou, L.; Li, Z.; Jang, H.; Wang, Y.; Cui, X.; Gu, X.; Kim, M.G.; Feng, L.; Liu, S.; Liu, X. Electronic and Lattice Engineering of Ruthenium Oxide towards Highly Active and Stable Water Splitting. Adv. Energy Mater. 2023, 13, 2300177. [Google Scholar] [CrossRef]
- Wen, Y.; Liu, C.; Huang, R.; Zhang, H.; Li, X.; García de Arquer, F.P.; Liu, Z.; Li, Y.; Zhang, B. Introducing Brønsted Acid Sites to Accelerate the Bridging-oxygen-assisted Deprotonation in Acidic Water Oxidation. Nat. Commun. 2022, 13, 4871. [Google Scholar] [CrossRef]
- He, J.; Li, W.; Xu, P.; Sun, J. Tuning Electron Correlations of RuO2 by Co-doping of Mo and Ce for Boosting Electrocatalytic Water Oxidation in Acidic Media. Appl. Catal. B-Environ. 2021, 298, 120528. [Google Scholar] [CrossRef]
- Wang, X.; Jang, H.; Liu, S.; Li, Z.; Zhao, X.; Chen, Y.; Kim, M.G.; Qin, Q.; Liu, X. Enhancing the Catalytic Kinetics and Stability of Ru Sites for Acidic Water Oxidation by Forming Brønsted Acid Sites in Tungsten Oxide Matrix. Adv. Energy Mater. 2023, 13, 2301673. [Google Scholar] [CrossRef]
- Lee, G.R.; Kim, J.; Hong, D.; Kim, Y.J.; Jang, H.; Han, H.J.; Hwang, C.-K.; Kim, D.; Kim, J.Y.; Jung, Y.S. Efficient and Sustainable Water Electrolysis Achieved by Excess Electron Reservoir Enabling Charge Replenishment to Catalysts. Nat. Commun. 2023, 14, 5402. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Hung, S.F.; Lin, Z.Y.; Zhang, Y.; Zhang, C.; Hao, Y.; Liu, S.; Kuo, C.H.; Chen, H.Y.; Peng, J.; et al. Valence Oscillation of Ru Active Sites for Efficient and Robust Acidic Water Oxidation. Adv. Mater. 2023, 35, 2305939. [Google Scholar] [CrossRef] [PubMed]
- Danilovic, N.; Subbaraman, R.; Chang, K.-C.; Chang, S.H.; Kang, Y.J.; Snyder, J.; Paulikas, A.P.; Strmcnik, D.; Kim, Y.-T.; Myers, D.; et al. Activity–Stability Trends for the Oxygen Evolution Reaction on Monometallic Oxides in Acidic Environments. J. Phys. Chem. Lett. 2014, 5, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Dickens, C.F.; Nørskov, J.K. A Theoretical Investigation into the Role of Surface Defects for Oxygen Evolution on RuO2. J. Phys. Chem. C 2017, 121, 18516–18524. [Google Scholar] [CrossRef]
- Li, K.; Xue, D. Estimation of Electronegativity Values of Elements in Different Valence States. J. Phys. Chem. A 2006, 110, 11332–11337. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Yu, T.; Deng, S.; Zhou, X.-Y.; Lin, D.; Zhang, Q.; Jin, Z.; Zhang, D.; He, Y.-B.; Qiu, H.-J.; et al. RuO2 Electronic Structure and Lattice Strain Dual Engineering for Enhanced Acidic Oxygen Evolution Reaction Performance. Nat. Commun. 2022, 13, 3784. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, X.; Zhang, B.; Bai, B.; Das, P.; Azam, T.; Xiao, J.; Wu, Z.-S. Breaking the Ru-O-Ru Symmetry of a RuO2 Catalyst for Sustainable Acidic Water Oxidation. Angew. Chem. Int. Ed. 2024, 136, 202316903. [Google Scholar] [CrossRef]
- Xue, Y.; Zhao, J.; Huang, L.; Lu, Y.-R.; Malek, A.; Gao, G.; Zhuang, Z.; Wang, D.; Yavuz, C.T.; Lu, X. Stabilizing Ruthenium Dioxide with Cation-anchored Sulfate for Durable Oxygen Evolution in Proton-exchange Membrane Water Electrolyzers. Nat. Commun. 2023, 14, 8093. [Google Scholar] [CrossRef]
- Hao, S.; Liu, M.; Pan, J.; Liu, X.; Tan, X.; Xu, N.; He, Y.; Lei, L.; Zhang, X. Dopants Fixation of Ruthenium for Boosting Acidic Oxygen Evolution Stability and Activity. Nat. Commun. 2020, 11, 5368. [Google Scholar] [CrossRef]
- Liu, J.; Banis, M.N.; Li, X.; Lushington, A.; Cai, M.; Li, R.; Sham, T.-K.; Sun, X. Atomic Layer Deposition of Lithium Tantalate Solid-State Electrolytes. J. Phys. Chem. C 2013, 117, 20260–20267. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, Y.; Peng, F.; Meng, F.; Zha, J.; Ma, L.; Du, Y.; Peng, N.; Ma, L.; Zhang, Q.; et al. Intercalation-Activated Layered MoO3 Nanobelts as Biodegradable Nanozymes for Tumor-Specific Photo-Enhanced Catalytic Therapy. Angew. Chem. Int. Ed. 2022, 61, 202115939. [Google Scholar] [CrossRef]
- Liu, X.; Xi, S.; Kim, H.; Kumar, A.; Lee, J.; Wang, J.; Tran, N.Q.; Yang, T.; Shao, X.; Liang, M.; et al. Restructuring Highly Electron-Deficient Metal-Metal Oxides for Boosting Stability in Acidic Oxygen Evolution Reaction. Nat. Commun. 2021, 12, 5676. [Google Scholar] [CrossRef]
- Oh, Y.; Theerthagiri, J.; Min, A.; Moon, C.J.; Yu, Y.; Choi, M.Y. Pulsed Laser Interference Patterning of Transition-Metal Carbides for Stable Alkaline Water Electrolysis Kinetics. Carbon Energy 2024, e448. [Google Scholar] [CrossRef]
- Oh, Y.; Theerthagiri, J.; Aruna Kumari, M.L.; Min, A.; Moon, C.J.; Choi, M.Y. Electrokinetic-mechanism of Water and Furfural Oxidation on Pulsed Laser-interlaced Cu2O and CoO on Nickel Foam. J. Energy Chem. 2024, 91, 145–154. [Google Scholar] [CrossRef]
- Jin, H.; Liu, X.; An, P.; Tang, C.; Yu, H.; Zhang, Q.; Peng, H.-J.; Gu, L.; Zheng, Y.; Song, T.; et al. Dynamic Rhenium Dopant Boosts Ruthenium Oxide for Durable Oxygen Evolution. Nat. Commun. 2023, 14, 354. [Google Scholar] [CrossRef]
- Zhao, Z.L.; Wang, Q.; Huang, X.; Feng, Q.; Gu, S.; Zhang, Z.; Xu, H.; Zeng, L.; Gu, M.; Li, H. Boosting the Oxygen Evolution Reaction Using Defect-rich Ultra-thin Ruthenium Oxide Nanosheets in Acidic Media. Energy Environ. Sci. 2020, 13, 5143–5151. [Google Scholar] [CrossRef]
- Wu, Y.; Yao, R.; Zhao, Q.; Li, J.; Liu, G. La-RuO2 Nanocrystals with Efficient Electrocatalytic Activity for Overall Water Splitting in Acidic Media: Synergistic Effect of La Doping and Oxygen Vacancy. Chem. Eng. J. 2022, 439, 135699. [Google Scholar] [CrossRef]
- Liu, C.X.; Jiang, Y.B.; Wang, T.; Li, Q.S.; Liu, Y.Z. Nano Si-doped Ruthenium Oxide Particles from Caged Precursors for High-performance Acidic Oxygen Evolution. Adv. Sci. 2023, 10, 2207429. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Jiang, H.; Chen, Z.Y.; Chen, Q.; Ma, M.Y.; Zhen, L.; Song, B.; Xu, C.Y. Bifunctional WC-supported RuO2 Nanoparticles for Robust Water Splitting in Acidic Media. Angew. Chem. Int. Ed. 2022, 61, 202202519. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-Y.; Chen, F.-Y.; Li, B.; Yu, S.-W.; Finfrock, Y.Z.; Meira, D.M.; Yan, Q.-Q.; Zhu, P.; Chen, M.-X.; Song, T.-W.; et al. Non-iridium-based Electrocatalyst for Durable Acidic Oxygen Evolution Reaction in Proton Exchange Membrane Water Electrolysis. Nat. Mater. 2022, 22, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Lin, C.L.; Yu, G.Q.; Du, P.; Xie, X.Y.; He, X.; Zheng, Z.C.; Sun, N.; Tang, H.L.; Li, X.B.; et al. Ru/Se-RuO2 Composites via Controlled Selenization Strategy for Enhanced Acidic Oxygen Evolution. Adv. Funct. Mater. 2023, 33, 2211102. [Google Scholar] [CrossRef]
- Feng, T.L.; Yu, G.T.; Tao, S.Y.; Zhu, S.J.; Ku, R.Q.; Zhang, R.; Zeng, Q.S.; Yang, M.X.; Chen, Y.X.; Chen, W.H.; et al. A Highly Efficient Overall Water Splitting Ruthenium-cobalt Alloy Electrocatalyst across a Wide pH Range Electronic Coupling with Carbon Dots. J. Mater. Chem. A 2020, 8, 9638–9645. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, R.; Ding, Y.; Zhang, B.; Li, H.; Bai, B.; Li, M.; Cui, Y.; Xiao, J.; Wu, Z.-S. Unraveling Oxygen Vacancy Site Mechanism of Rh-doped RuO2 Catalyst for Long-lasting Acidic Water Oxidation. Nat. Commun. 2023, 14, 1412. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wan, X.; Qin, X.; Chen, C.; Qian, X.; Guo, Y.; Xu, Q.; Cai, W.-B.; Yang, H.; Jiang, K. Electronic Structure Modulation of RuO2 by TiO2 Enriched with Oxygen Vacancies to Boost Acidic O2 Evolution. ACS Catal. 2022, 12, 9437–9445. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).