

Hirudin in the Treatment of Chronic Kidney Disease


Abstract
:1. Introduction
2. Pathophysiology of CKD
3. Hirudin
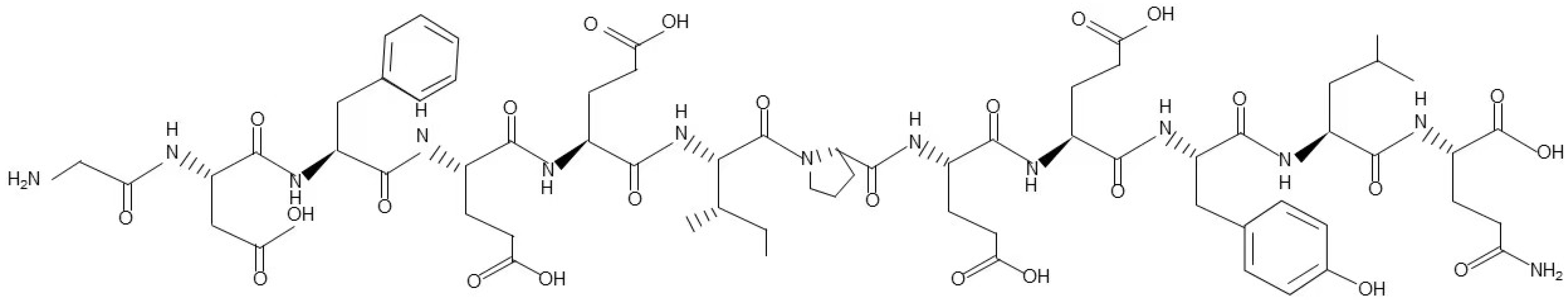
3.1. Structure and Derivatives of Hirudin
3.2. Isolation Procedures of Hirudin
3.3. Pharmacokinetics of Hirudin
3.4. Toxicology of Hirudin
3.5. Clinical Use of Hirudin Derivatives
4. Hirudin in CKD
4.1. Hirudin in Diabetic Nephrology
4.2. Hirudin in Nephrotic Syndrome
4.3. Hirudin in Renal Interstitial Fibrosis
4.4. Hirudin in Other Renal Disorders
4.4.1. Acute Kidney Injury
4.4.2. Metastatic Kidney Cancer
4.4.3. Arteriovenous Fistula Stenosis
4.4.4. IgA Nephropathy
5. Adverse Effects of Hirudin
5.1. Bleeding and Anticoagulant Monitoring
5.2. Immunization and Anaphylaxis
5.3. Drug Interactions
6. Prospects and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Osborn, J.W.; Tyshynsky, R.; Vulchanova, L. Function of Renal Nerves in Kidney Physiology and Pathophysiology. Annu. Rev. Physiol. 2021, 83, 429–450. [Google Scholar] [CrossRef]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Glassock, R.J.; Warnock, D.G.; Delanaye, P. The Global Burden of Chronic Kidney Disease: Estimates, Variability and Pitfalls. Nat. Rev. Nephrol. 2017, 13, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.T.; Xu, Y.; Zhang, W.; Bundy, J.D.; Chen, C.-S.; Kelly, T.N.; Chen, J.; He, J. A Systematic Analysis of Worldwide Population-Based Data on the Global Burden of Chronic Kidney Disease in 2010. Kidney Int. 2015, 88, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Brück, K.; Stel, V.S.; Gambaro, G.; Hallan, S.; Völzke, H.; Ärnlöv, J.; Kastarinen, M.; Guessous, I.; Vinhas, J.; Stengel, B. CKD Prevalence Varies across the European General Population. J. Am. Soc. Nephrol. 2016, 27, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.-W.; et al. Forecasting Life Expectancy, Years of Life Lost, and All-Cause and Cause-Specific Mortality for 250 Causes of Death: Reference and Alternative Scenarios for 2016–40 for 195 Countries and Territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Coresh, J.; Sang, Y.; Chalmers, J.; Fox, C.; Guallar, E.; Jafar, T.; Jassal, S.K.; Landman, G.W.; Muntner, P. Estimated Glomerular Filtration Rate and Albuminuria for Prediction of Cardiovascular Outcomes: A Collaborative Meta-Analysis of Individual Participant Data. Lancet Diabetes Endocrinol. 2015, 3, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Gansevoort, R.T.; Matsushita, K.; Van Der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.S.; De Jong, P.E.; Coresh, J. Lower Estimated GFR and Higher Albuminuria Are Associated with Adverse Kidney Outcomes. A Collaborative Meta-Analysis of General and High-Risk Population Cohorts. Kidney Int. 2011, 80, 93–104. [Google Scholar] [CrossRef]
- Vanholder, R.; Annemans, L.; Bello, A.K.; Bikbov, B.; Gallego, D.; Gansevoort, R.T.; Lameire, N.; Luyckx, V.A.; Noruisiene, E.; Oostrom, T. Fighting the Unbearable Lightness of Neglecting Kidney Health: The Decade of the Kidney. Clin. Kidney J. 2021, 14, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G.; Remuzzi, G.; Mendis, S.; Tonelli, M. The Contribution of Chronic Kidney Disease to the Global Burden of Major Noncommunicable Diseases. Kidney Int. 2011, 80, 1258–1270. [Google Scholar] [CrossRef]
- Faria, M.; de Pinho, M.N. Challenges of Reducing Protein-Bound Uremic Toxin Levels in Chronic Kidney Disease and End Stage Renal Disease. Transl. Res. 2021, 229, 115–134. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the Progression of Chronic Kidney Disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; de Zeeuw, D. Novel Drugs and Intervention Strategies for the Treatment of Chronic Kidney Disease. Br. J. Clin. Pharmacol. 2013, 76, 536–550. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural Products: A Continuing Source of Novel Drug Leads. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Chen, D.-Q.; Hu, H.-H.; Wang, Y.-N.; Feng, Y.-L.; Cao, G.; Zhao, Y.-Y. Natural Products for the Prevention and Treatment of Kidney Disease. Phytomedicine 2018, 50, 50–60. [Google Scholar] [CrossRef]
- Dong, H.; Ren, J.-X.; Wang, J.-J.; Ding, L.-S.; Zhao, J.-J.; Liu, S.-Y.; Gao, H.-M. Chinese Medicinal Leech: Ethnopharmacology, Phytochemistry, and Pharmacological Activities. Evid. Based Complement. Altern. Med. 2016, 2016, 7895935. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liao, C.; Guo, Q.; Wu, L.; Zhang, L.; Wang, X. Combined Systems Pharmacology and Fecal Metabonomics to Study the Biomarkers and Therapeutic Mechanism of Type 2 Diabetic Nephropathy Treated with Astragalus and Leech. RSC Adv. 2018, 8, 27448–27463. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.R.; Hofsteenge, J. Kinetics of the Inhibition of Thrombin by Hirudin. Biochemistry 1986, 25, 4622–4628. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Haase, M.; Lemke, S.; Hildebrandt, J.-P. Hirudins and Hirudin-like Factors in Hirudinidae: Implications for Function and Phylogenetic Relationships. Parasitol. Res. 2017, 116, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Zoldhelyi, P.; Webster, M.W.; Fuster, V.; Grill, D.E.; Gaspar, D.; Edwards, S.J.; Cabot, C.F.; Chesebro, J.H. Recombinant Hirudin in Patients with Chronic, Stable Coronary Artery Disease. Safety, Half-Life, and Effect on Coagulation Parameters. Circulation 1993, 88, 2015–2022. [Google Scholar] [CrossRef]
- Levin, L.-Å.; Bergqvist, D. Cost Effectiveness of Desirudin Compared with a Low Molecular Weight Heparin in the Prevention of Deep Vein Thrombosis after Total Hip Replacement Surgery. Pharmacoeconomics 2001, 19, 589–597. [Google Scholar] [CrossRef]
- Peng, L.; Pan, X.; Yin, G. Natural Hirudin Increases Rat Flap Viability by Anti-Inflammation via PARs/P38/NF-κB Pathway. BioMed Res. Int. 2015, 2015, 597264. [Google Scholar] [CrossRef]
- Yingxin, G.; Guoqian, Y.; Jiaquan, L.; Han, X. Effects of Natural and Recombinant Hirudin on VEGF Expression and Random Skin Flap Survival in a Venous Congested Rat Model. Int. Surg. 2013, 98, 82–87. [Google Scholar] [CrossRef]
- Zhao, B.; Wu, M.; Hu, Z.; Wang, T.; Yu, J.; Ma, Y.; Wang, Q.; Zhang, Y.; Chen, D.; Li, T. A Novel Oncotherapy Strategy: Direct Thrombin Inhibitors Suppress Progression, Dissemination and Spontaneous Metastasis in Non-small Cell Lung Cancer. Br. J. Pharmacol. 2022, 179, 5056–5073. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ye, H.; Zhang, D.; Yang, M.; Ji, Y.; Tang, L.; Zhu, X.; Yuan, L. The Role of Exosomal CDC6 in the Hirudin-Mediated Suppression of the Malignant Phenotype of Bladder Cancer Cells. Gene 2022, 821, 146269. [Google Scholar] [CrossRef]
- Li, W.-Q.; Qin, Z.-S.; Chen, S.; Cheng, D.; Yang, S.-C.; Choi, Y.M.M.; Chu, B.; Zhou, W.-H.; Zhang, Z.-J. Hirudin Alleviates Acute Ischemic Stroke by Inhibiting NLRP3 Inflammasome-Mediated Neuroinflammation: In Vivo and in Vitro Approaches. Int. Immunopharmacol. 2022, 110, 108967. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Lei, S.; Huang, L.; Li, S.; Yi, S.; Breitzig, M.; Huang, M.; Mo, X.; Sun, H.; Zheng, Q. Therapeutic Effects of the rhSOD2-Hirudin Fusion Protein on Bleomycin-Induced Pulmonary Fibrosis in Mice. Eur. J. Pharmacol. 2019, 852, 77–89. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and Molecular Mechanisms of Renal Fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Tan, R.J.; Liu, Y. Myofibroblast in Kidney Fibrosis: Origin, Activation, and Regulation. In Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 253–283. [Google Scholar] [CrossRef]
- Sol, M.; Kamps, J.A.; van den Born, J.; van den Heuvel, M.C.; van der Vlag, J.; Krenning, G.; Hillebrands, J.-L. Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 2020, 11, 573557. [Google Scholar] [CrossRef] [PubMed]
- Cove-Smith, A.; Hendry, B.M. The Regulation of Mesangial Cell Proliferation. Nephron Exp. Nephrol. 2008, 108, e74–e79. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M. Podocyte Injury and Its Consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-C.; Livingston, M.J.; Liang, X.-L.; Dong, Z. Cell Apoptosis and Autophagy in Renal Fibrosis. In Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 557–584. [Google Scholar] [CrossRef]
- Zhao, J.-H. Mesangial Cells and Renal Fibrosis. In Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 165–194. [Google Scholar] [CrossRef]
- Lu, C.-C.; Wang, G.-H.; Lu, J.; Chen, P.-P.; Zhang, Y.; Hu, Z.-B.; Ma, K.-L. Role of Podocyte Injury in Glomerulosclerosis. In Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 195–232. [Google Scholar] [CrossRef]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal Tubule Injury: A Driving Force toward Chronic Kidney Disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Schelling, J.R. Tubular Atrophy in the Pathogenesis of Chronic Kidney Disease Progression. Pediatr. Nephrol. 2016, 31, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L. How Tubular Epithelial Cell Injury Contributes to Renal Fibrosis. In Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; pp. 233–252. [Google Scholar] [CrossRef]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse Origins of the Myofibroblast—Implications for Kidney Fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Fields, W.S. The History of Leeching and Hirudin. Pathophysiol. Haemost. Thromb. 1991, 21, 3–10. [Google Scholar] [CrossRef]
- De Filippis, V.; Acquasaliente, L.; Pontarollo, G.; Peterle, D. Noncoded Amino Acids in Protein Engineering: Structure–Activity Relationship Studies of Hirudin–Thrombin Interaction. Biotechnol. Appl. Biochem. 2018, 65, 69–80. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, N. Hirudin Variants Production by Genetic Engineered Microbial Factory. Biotechnol. Genet. Eng. Rev. 2018, 34, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Wüstenhagen, D.A.; Lukas, P.; Müller, C.; Aubele, S.A.; Hildebrandt, J.-P.; Kubick, S. Cell-Free Synthesis of the Hirudin Variant 1 of the Blood-Sucking Leech Hirudo Medicinalis. Sci. Rep. 2020, 10, 19818. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Eriksson, B.I.; Jaffer, A.K.; Smith, J. Impact of Stage 3B Chronic Kidney Disease on Thrombosis and Bleeding Outcomes after Orthopedic Surgery in Patients Treated with Desirudin or Enoxaparin: Insights from a Randomized Trial. J. Thromb. Haemost. 2012, 10, 1515–1520. [Google Scholar] [CrossRef]
- Peng, X.; Li, Z.; Li, D.; Li, Z.; Lu, Z.; Luo, C.; Ji, Z. Bivalirudin Presents a Favorable Safety Profile Regarding Adverse Drug Reactions, Thrombocytopenia, and Bleeding in Chinese Patients With High Bleeding Risk Undergoing Percutaneous Coronary Intervention: A Prospective, Multi-Center, Intensive Monitoring Study. Front. Cardiovasc. Med. 2022, 9, 1314. [Google Scholar] [CrossRef]
- Mongirdienė, A.; Liuizė, A.; Kašauskas, A. Novel Knowledge about Molecular Mechanisms of Heparin-Induced Thrombocytopenia Type II and Treatment Targets. Int. J. Mol. Sci. 2023, 24, 8217. [Google Scholar] [CrossRef]
- Montinari, M.R.; Minelli, S. From Ancient Leech to Direct Thrombin Inhibitors and beyond: New from Old. Biomed. Pharmacother. 2022, 149, 112878. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, F. Die Isolierung und Chemische Charakterisierung des Hirudins; Walter de Gruyter: Berlin, Germany, 1957. [Google Scholar]
- Walsmann, P. Isolation and Characterization of Hirudin from Hirudo medicinalis. Semin. Thromb. Hemost. 1991, 17, 83–87. [Google Scholar] [CrossRef]
- Nowak, G. Pharmacology of Recombinant Hirudin. Semin. Thromb. Hemost. 2002, 28, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Nowak, G. Pharmacokinetics of Hirudin. Semin. Thromb. Hemost. 1991, 17, 145–149. [Google Scholar] [CrossRef]
- Bichler, J.; Gemmerli, R.; Fritz, H. Studies for Revealing a Possible Sensitization to Hirudin after Repeated Intravenous Injections in Baboons. Thromb. Res. 1991, 61, 39–51. [Google Scholar] [CrossRef]
- Eichler, P.; Friesen, H.-J.; Lubenow, N.; Jaeger, B.; Greinacher, A. Antihirudin Antibodies in Patients with Heparin-Induced Thrombocytopenia Treated with Lepirudin: Incidence, Effects on aPTT, and Clinical Relevance. Blood J. Am. Soc. Hematol. 2000, 96, 2373–2378. [Google Scholar]
- Eriksson, B.I.; Wille-Jørgensen, P.; Kälebo, P.; Mouret, P.; Rosencher, N.; Bösch, P.; Baur, M.; Ekman, S.; Bach, D.; Lindbratt, S. A Comparison of Recombinant Hirudin with a Low-Molecular-Weight Heparin to Prevent Thromboembolic Complications after Total Hip Replacement. N. Engl. J. Med. 1997, 337, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Coppens, M.; Eikelboom, J.W.; Gustafsson, D.; Weitz, J.I.; Hirsh, J. Translational Success Stories: Development of Direct Thrombin Inhibitors. Circ. Res. 2012, 111, 920–929. [Google Scholar] [CrossRef]
- Linkins, L.-A.; Dans, A.L.; Moores, L.K.; Bona, R.; Davidson, B.L.; Schulman, S.; Crowther, M. Treatment and Prevention of Heparin-Induced Thrombocytopenia: Antithrombotic Therapy and Prevention of Thrombosis: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141, e495S–e530S. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.I.; Kälebo, P.; Zachrisson, B.; Ekman, S.; Bach, D.; Close, P. Prevention of Deep-Vein Thrombosis after Total Hip Replacement: Direct Thrombin Inhibition with Recombinant Hirudin, CGP 39393. Lancet 1996, 347, 635–639. [Google Scholar] [CrossRef]
- Zaleski, K.L.; DiNardo, J.A.; Nasr, V.G. Bivalirudin for Pediatric Procedural Anticoagulation: A Narrative Review. Anesth. Analg. 2019, 128, 43–55. [Google Scholar] [CrossRef]
- Stone, G.W.; McLaurin, B.T.; Cox, D.A.; Bertrand, M.E.; Lincoff, A.M.; Moses, J.W.; White, H.D.; Pocock, S.J.; Ware, J.H.; Feit, F. Bivalirudin for Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2006, 355, 2203–2216. [Google Scholar] [CrossRef]
- Stone, G.W.; Witzenbichler, B.; Guagliumi, G.; Peruga, J.Z.; Brodie, B.R.; Dudek, D.; Kornowski, R.; Hartmann, F.; Gersh, B.J.; Pocock, S.J. Heparin plus a Glycoprotein IIb/IIIa Inhibitor versus Bivalirudin Monotherapy and Paclitaxel-Eluting Stents versus Bare-Metal Stents in Acute Myocardial Infarction (HORIZONS-AMI): Final 3-Year Results from a Multicentre, Randomised Controlled Trial. Lancet 2011, 377, 2193–2204. [Google Scholar] [CrossRef] [PubMed]
- Augoustides, J.G. Update in Hematology: Heparin-Induced Thrombocytopenia and Bivalirudin. J. Cardiothorac. Vasc. Anesth. 2011, 25, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, G.; Zhao, W.; Peng, W. Anticoagulation Strategies in Patients with Extracorporeal Membrane Oxygenation: A Network Meta-analysis and Systematic Review. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2023, 43, 1084–1093. [Google Scholar] [CrossRef]
- Navaei, A.; Kostousov, V.; Teruya, J. Is It Time to Switch to Bivalirudin for ECMO Anticoagulation? Front. Med. 2023, 10, 1237601. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhou, L. The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxid. Med. Cell. Longev. 2019, 2019, 7495629. [Google Scholar] [CrossRef]
- Letelier, C.E.M.; Ojeda, C.A.S.M.; Provoste, J.J.R.; Zaror, C.J.F. Pathophysiology of Diabetic Nephropathy: A Literature Review. Medwave 2017, 17, e6839. [Google Scholar] [CrossRef]
- Pichler, R.; Afkarian, M.; Dieter, B.P.; Tuttle, K.R. Immunity and Inflammation in Diabetic Kidney Disease: Translating Mechanisms to Biomarkers and Treatment Targets. Am. J. Physiol. Ren. Physiol. 2017, 312, F716–F731. [Google Scholar] [CrossRef]
- Xu, B.-H.; Sheng, J.; You, Y.-K.; Huang, X.-R.; Ma, R.C.; Wang, Q.; Lan, H.-Y. Deletion of Smad3 Prevents Renal Fibrosis and Inflammation in Type 2 Diabetic Nephropathy. Metabolism 2020, 103, 154013. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed]
- Junren, C.; Xiaofang, X.; Huiqiong, Z.; Gangmin, L.; Yanpeng, Y.; Xiaoyu, C.; Yuqing, G.; Yanan, L.; Yue, Z.; Fu, P. Pharmacological Activities and Mechanisms of Hirudin and Its Derivatives-a Review. Front. Pharmacol. 2021, 12, 660757. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Zhang, Y.; Shi, X.; Peng, Z.; Xing, Y.; Jiarui, H. Hirudin Reduces the Expression of Markers of the Extracellular Matrix in Renal Tubular Epithelial Cells in a Rat Model of Diabetic Kidney Disease Through the Hypoxia-Inducible Factor-1α (HIF-1α)/Vascular Endothelial Growth Factor (VEGF) Signaling Pathway. Med. Sci. Monit. 2020, 26, e921894. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pang, X.; Zhang, Y.; Peng, Z.; Shi, X.; Xing, Y. Hirudin Protects Against Kidney Damage in Streptozotocin-Induced Diabetic Nephropathy Rats by Inhibiting Inflammation via P38 MAPK/NF-κB Pathway. Drug Des. Devel. Ther. 2020, 14, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Zhang, Y.; Peng, Z.; Shi, X.; Han, J.; Xing, Y. Hirudin Reduces Nephropathy Microangiopathy in STZ-Induced Diabetes Rats by Inhibiting Endothelial Cell Migration and Angiogenesis. Life Sci. 2020, 255, 117779. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zuo, Z.; Shi, X.; Zhang, Y.; Peng, Z.; Xing, Y.; Pang, X. Hirudin Ameliorates Diabetic Nephropathy by Inhibiting Gsdmd-Mediated Pyroptosis. Cell Biol. Toxicol. 2021, 39, 573–589. [Google Scholar] [CrossRef] [PubMed]
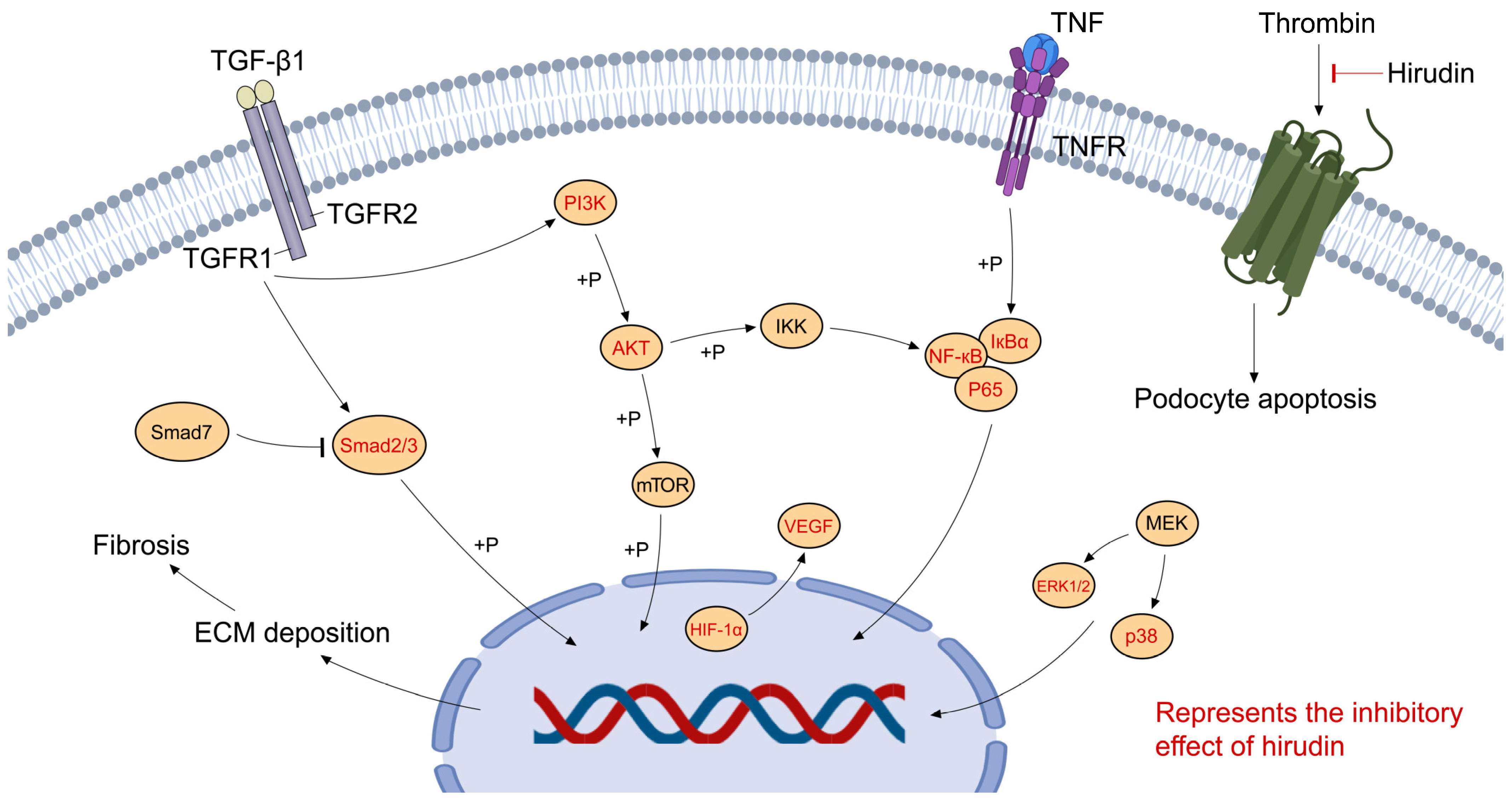
- Yang, K.; Fan, B.; Zhao, Q.; Ji, Y.; Liu, P.; Gao, S.; Ren, T.; Dou, Y.; Pei, M.; Yang, H. Hirudin Ameliorates Renal Interstitial Fibrosis via Regulating TGF- β 1/Smad and NF- κ B Signaling in UUO Rat Model. Evid. Based Complement. Altern. Med. 2020, 2020, 7291075. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Xiong, W.; Gao, X.; Xiong, Y.; Sun, W. A Molecular Mechanism Study to Reveal Hirudin’s Downregulation to PI3K/AKT Signaling Pathway through Decreasing PDGFRβ in Renal Fibrosis Treatment. BioMed Res. Int. 2022, 2022, 5481552. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-X.; Lin, W.; Yang, K.; Wei, L.-J.; Chen, J.-L.; Liu, X.-Y.; Zhong, K.; Chen, X.; Pei, M.; Yang, H.-T. Transcriptome-Based Network Analysis Reveals Hirudin Potentiates Anti-Renal Fibrosis Efficacy in UUO Rats. Front. Pharmacol. 2021, 12, 741801. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Waller, A.P.; Agrawal, S.; Wolfgang, K.J.; Luu, H.; Shahzad, K.; Isermann, B.; Smoyer, W.E.; Nieman, M.T.; Kerlin, B.A. Thrombin-Induced Podocyte Injury Is Protease-Activated Receptor Dependent. J. Am. Soc. Nephrol. 2017, 28, 2618–2630. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Lin, Q.; Mo, J.; Xiao, Y.; Xie, Y. Hirudin Attenuates Puromycin Aminonucleoside-induced Glomerular Podocyte Injury by Inhibiting MAPK-mediated Endoplasmic Reticulum Stress. Drug Dev. Res. 2022, 83, 1047–1056. [Google Scholar] [CrossRef]
- Sevastos, J.; Kennedy, S.E.; Davis, D.R.; Sam, M.; Peake, P.W.; Charlesworth, J.A.; Mackman, N.; Erlich, J.H. Tissue Factor Deficiency and PAR-1 Deficiency Are Protective against Renal Ischemia Reperfusion Injury. Blood 2007, 109, 577–583. [Google Scholar] [CrossRef]
- Knowles, L.M.; Gurski, L.A.; Maranchie, J.K.; Pilch, J. Fibronectin Matrix Formation Is a Prerequisite for Colonization of Kidney Tumor Cells in Fibrin. J. Cancer 2015, 6, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ding, X.; Yang, Y. Hirudin Regulates Vascular Function in Chronic Renal Failure through Modulating Macrophage Polarization. BioMed Res. Int. 2022, 2022, 6043698. [Google Scholar] [CrossRef]
- Chen, J.; Shi, W.; Xu, Y.; Zhang, H.; Chen, B. Hirudin Prevents Vascular Endothelial Cell Apoptosis and Permeability Enhancement Induced by the Serum from Rat with Chronic Renal Failure through Inhibiting RhoA/ROCK Signaling Pathway. Drug Dev. Res. 2021, 82, 553–561. [Google Scholar] [CrossRef]
- Deng, F.; Zhang, J.; Li, Y.; Wang, W.; Hong, D.; Li, G.; Feng, J. Hirudin Ameliorates Immunoglobulin A Nephropathy by Inhibition of Fibrosis and Inflammatory Response. Ren. Fail. 2019, 41, 104–112. [Google Scholar] [CrossRef]
- Xinxin, P.; Qing, Z.; Zining, P.; Yage, Z.; Xiujie, S.; Jiarui, H. Exploring the Mechanism of Hirudin in the Treatment of Diabetic Kidney Disease Using Network Pharmacology Combined with Molecular Docking Verification. J. Tradit. Chin. Med. 2022, 42, 586. [Google Scholar]
- Cha, D.R.; Kang, Y.S.; Han, S.Y.; Jee, Y.H.; Han, K.H.; Han, J.Y.; Kim, Y.S.; Kim, N.H. Vascular Endothelial Growth Factor Is Increased during Early Stage of Diabetic Nephropathy in Type II Diabetic Rats. J. Endocrinol. 2004, 183, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Mansoor, S.; Sharma, A.; Sapkal, A.; Sheth, J.; Falatoonzadeh, P.; Kuppermann, B.D.; Kenney, M.C. Diabetic Retinopathy and VEGF. Open Ophthalmol. J. 2013, 7, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Dhawan, V.; Holm, R.; Nagarsenker, M.S.; Perrie, Y. Liposomes: Advancements and Innovation in the Manufacturing Process. Adv. Drug Deliv. Rev. 2020, 154, 102–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.-J. Pathological Roles of MAPK Signaling Pathways in Human Diseases. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Pantano, S.; Natoli, G. P38-Dependent Marking of Inflammatory Genes for Increased NF-κB Recruitment. Nat. Immunol. 2002, 3, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Wan, J.; Liu, D.; Pan, S.; Zhou, S.; Liu, Z. NLRP3-Mediated Pyroptosis in Diabetic Nephropathy. Front. Pharmacol. 2022, 13, 4167. [Google Scholar] [CrossRef]
- Noone, D.G.; Iijima, K.; Parekh, R. Idiopathic Nephrotic Syndrome in Children. Lancet 2018, 392, 61–74. [Google Scholar] [CrossRef]
- System, U.R.D. USRDS 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Natl. Inst. Health Natl. Inst. Diabetes Dig. Dig. Kidney Dis. 2013, 2014, A7. [Google Scholar] [CrossRef]
- Murphy, S.L.; Xu, J.; Kochanek, K.D. Deaths: Final Data for 2010. National Vital Statistics Reports. Natl. Cent. Health Stat. 2013, 61, 1–117. [Google Scholar]
- Grahammer, F.; Schell, C.; Huber, T.B. The Podocyte Slit Diaphragm—From a Thin Grey Line to a Complex Signalling Hub. Nat. Rev. Nephrol. 2013, 9, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. Proteinuria with and without Renal Glomerular Podocyte Effacement. J. Am. Soc. Nephrol. 2006, 17, 2383–2389. [Google Scholar] [CrossRef] [PubMed]
- Kerlin, B.A.; Ayoob, R.; Smoyer, W.E. Epidemiology and Pathophysiology of Nephrotic Syndrome–Associated Thromboembolic Disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Baroni, E.A.; Costa, R.S.; da Silva, C.G.; Coimbra, T.M. Heparin Treatment Reduces Glomerular Injury in Rats with Adriamycin-Induced Nephropathy but Does Not Modify Tubulointerstitial Damage or the Renal Production of Transforming Growth Factor-Beta. Nephron 2000, 84, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Nakajima, K.; Ohno, Y.; Kaneshiro, Y.; Matsuo, T.; Tanaka, H.; Kaneko, K. Protective Effects of Antithrombin on Puromycin Aminonucleoside Nephrosis in Rats. Eur. J. Pharmacol. 2008, 589, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Madhusudhan, T.; He, T.; Hummel, B.; Schmidt, S.; Vinnikov, I.A.; Shahzad, K.; Kashif, M.; Muller-Krebs, S.; Schwenger, V. Low but Sustained Coagulation Activation Ameliorates Glucose-Induced Podocyte Apoptosis: Protective Effect of Factor V Leiden in Diabetic Nephropathy. Blood J. Am. Soc. Hematol. 2011, 117, 5231–5242. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The Unfolded Protein Response: Controlling Cell Fate Decisions under ER Stress and Beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Djudjaj, S.; Boor, P. Cellular and Molecular Mechanisms of Kidney Fibrosis. Mol. Asp. Med. 2019, 65, 16–36. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for Epithelial-Mesenchymal Transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Ren, Y.; Du, C.; Shi, Y.; Wei, J.; Wu, H.; Cui, H. The Sirt1 Activator, SRT1720, Attenuates Renal Fibrosis by Inhibiting CTGF and Oxidative Stress. Int. J. Mol. Med. 2017, 39, 1317–1324. [Google Scholar] [CrossRef]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.; Lee, H.B. Role of Reactive Oxygen Species in TGF-Β1-Induced Mitogen-Activated Protein Kinase Activation and Epithelial-Mesenchymal Transition in Renal Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2005, 16, 667–675. [Google Scholar] [CrossRef]
- Shirato, K. Thrombin Stimulates Production of Fibronectin by Human Proximal Tubular Epithelial Cells via a Transforming Growth Factor—Dependent Mechanism. Nephrol. Dial. Transplant. 2003, 18, 2248–2254. [Google Scholar] [CrossRef]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The Master Regulator of Fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Xie, Y.; Lan, F.; Zhao, J.; Shi, W. Hirudin Improves Renal Interstitial Fibrosis by Reducing Renal Tubule Injury and Inflammation in Unilateral Ureteral Obstruction (UUO) Mice. Int. Immunopharmacol. 2020, 81, 106249. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Pathways in TGF-β Signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Hassan, B.; Akcakanat, A.; Holder, A.M.; Meric-Bernstam, F. Targeting the PI3-Kinase/Akt/mTOR Signaling Pathway. Surg. Oncol. Clin. N. Am. 2013, 22, 641–664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jin, D.; Kang, X.; Zhou, R.; Sun, Y.; Lian, F.; Tong, X. Signaling Pathways Involved in Diabetic Renal Fibrosis. Front. Cell Dev. Biol. 2021, 9, 696542. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K.; Harnsirikarn, T.; Chokesuwattanaskul, S. Ferroptosis: A Potential Therapeutic Target in Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 6583. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, M.; Rosso, G.; Bertoni, E. Update on Ischemia-Reperfusion Injury in Kidney Transplantation: Pathogenesis and Treatment. World J. Transplant. 2015, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.; Costes, S.V.; Cho, E.H.; Lockett, S. Inhibition of Metastatic Outgrowth from Single Dormant Tumor Cells by Targeting the Cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef]
- Ikizler, T.A.; Schulman, G. Hemodialysis: Techniques and Prescription. Am. J. Kidney Dis. 2005, 46, 976–981. [Google Scholar] [CrossRef]
- Masengu, A.; Hanko, J. Patient Factors and Haemodialysis Arteriovenous Fistula Outcomes. J. Vasc. Access 2017, 18, S19–S23. [Google Scholar] [CrossRef]
- Gorecka, J.; Fereydooni, A.; Gonzalez, L.; Lee, S.R.; Liu, S.; Ono, S.; Xu, J.; Liu, J.; Taniguchi, R.; Matsubara, Y. Molecular Targets for Improving Arteriovenous Fistula Maturation and Patency. Vasc. Investig. Ther. 2019, 2, 33. [Google Scholar] [CrossRef]
- Guo, X.; Fereydooni, A.; Isaji, T.; Gorecka, J.; Liu, S.; Hu, H.; Ono, S.; Alozie, M.; Lee, S.R.; Taniguchi, R. Inhibition of the Akt1-mTORC1 Axis Alters Venous Remodeling to Improve Arteriovenous Fistula Patency. Sci. Rep. 2019, 9, 11046. [Google Scholar] [CrossRef]
- Guiteras, R.; Flaquer, M.; Cruzado, J.M. Macrophage in Chronic Kidney Disease. NDT Plus 2016, 9, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Gómez, G.I.; Velarde, V.; Sáez, J.C. Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage. Int. J. Mol. Sci. 2019, 20, 4408. [Google Scholar] [CrossRef] [PubMed]
- Luvizotto, M.J.; Menezes-Silva, L.; Woronik, V.; Monteiro, R.C.; Câmara, N.O.S. Gut-Kidney Axis in IgA Nephropathy: Role on Mesangial Cell Metabolism and Inflammation. Front. Cell Dev. Biol. 2022, 10, 993716. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R. The Gut–Kidney Axis in IgA Nephropathy: Role of Microbiota and Diet on Genetic Predisposition. Pediatr. Nephrol. 2018, 33, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Eichler, P.; Lubenow, N.; Kwasny, H.; Luz, M. Heparin-Induced Thrombocytopenia with Thromboembolic Complications: Meta-Analysis of 2 Prospective Trials to Assess the Value of Parenteral Treatment with Lepirudin and Its Therapeutic aPTT Range. Blood J. Am. Soc. Hematol. 2000, 96, 846–851. [Google Scholar]
- Warkentin, T.E. Bivalent Direct Thrombin Inhibitors: Hirudin and Bivalirudin. Best Pract. Res. Clin. Haematol. 2004, 17, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Eichler, P.; Albrecht, D.; Strobel, U.; Pötzsch, B.; Eriksson, B.I. Antihirudin Antibodies Following Low-Dose Subcutaneous Treatment with Desirudin for Thrombosis Prophylaxis after Hip-Replacement Surgery: Incidence and Clinical Relevance. Blood J. Am. Soc. Hematol. 2003, 101, 2617–2619. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Lubenow, N.; Eichler, P. Anaphylactic and Anaphylactoid Reactions Associated with Lepirudin in Patients with Heparin-Induced Thrombocytopenia. Circulation 2003, 108, 2062–2065. [Google Scholar] [CrossRef]
- Veach, S.A.; Franks, A.M.; Allan, M.C. Severe Anaphylactic Reaction after Repeated Intermittent Exposure to Lepirudin. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2007, 27, 760–765. [Google Scholar] [CrossRef]
- White, H.D.; Chew, D.P. Bivalirudin: An Anticoagulant for Acute Coronary Syndromes and Coronary Interventions. Expert Opin. Pharmacother. 2002, 3, 777–788. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Derivatives | Lepirudin | Desirudin | Bivalirudin |
|---|---|---|---|
| Indication | Treatment of thrombosis complicating HIT | Thrombosis prophylaxis after hip or knee arthroplasty | Patients with UA undergoing PTCA; PCI with temporary use of GPI; patients at risk of HIT undergoing PCI |
| Thrombin affinity (Ki) | 0.23 pM | 0.26 pM | 1.9 nM |
| Route of administration | Intravenous, subcutaneous | Intravenous, subcutaneous | Intravenous |
| Plasma half-life | 80 min | 60 min | 25 min |
| Clearance | Renal | Renal | Proteolysis (80%) |
| Kidney Diseases | Functions | References |
|---|---|---|
| Diabetic nephrology | Inhibiting the HIF-1α/VEGF signaling pathway; | [71,72] |
| Inhibiting the p38 MAPK/NF-κB signaling pathway; | [73,74] | |
| Inhibiting Gsdmd-mediated pyroptosis | [75] | |
| Renal interstitial fibrosis | Inhibiting the TGF-β1/Smad and NF-κB signaling pathways; | [76] |
| Inhibiting the PI3K/AKT signaling pathways | [77,78] | |
| Nephrotic syndrome | Inhibiting the interactions between thrombin and PAR; | [79] |
| Inhibiting p38 MAPK signaling-mediated ERS | [80] | |
| Acute kidney injury | Inhibiting the interactions between thrombin and PAR | [81] |
| Metastatic kidney cancer | inhibiting F-actin formation and reducing metastatic growth | [82] |
| Arteriovenous fistula stenosis | Promoting SMC hyperplasia; stimulating the differentiation of M1 macrophage towards M2 macrophage Regulates endothelial cell permeability and apoptosis, inhibiting RhoA/ROCK signaling | [83,84] |
| IgA nephrology | Reducing cell apoptosis; inflammatory response and keeping balance of T cells | [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.-J.; Cao, Y.-L.; Zhang, C. Hirudin in the Treatment of Chronic Kidney Disease. Molecules 2024, 29, 1029. https://doi.org/10.3390/molecules29051029
Liu S-J, Cao Y-L, Zhang C. Hirudin in the Treatment of Chronic Kidney Disease. Molecules. 2024; 29(5):1029. https://doi.org/10.3390/molecules29051029
Chicago/Turabian StyleLiu, Sai-Ji, Yi-Ling Cao, and Chun Zhang. 2024. "Hirudin in the Treatment of Chronic Kidney Disease" Molecules 29, no. 5: 1029. https://doi.org/10.3390/molecules29051029
APA StyleLiu, S.-J., Cao, Y.-L., & Zhang, C. (2024). Hirudin in the Treatment of Chronic Kidney Disease. Molecules, 29(5), 1029. https://doi.org/10.3390/molecules29051029