

Synthesis of a Non-Symmetrical Disorazole C1-Analogue and Its Biological Activity



Abstract
: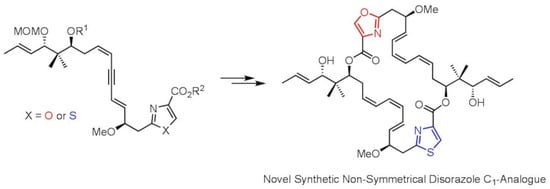
1. Introduction
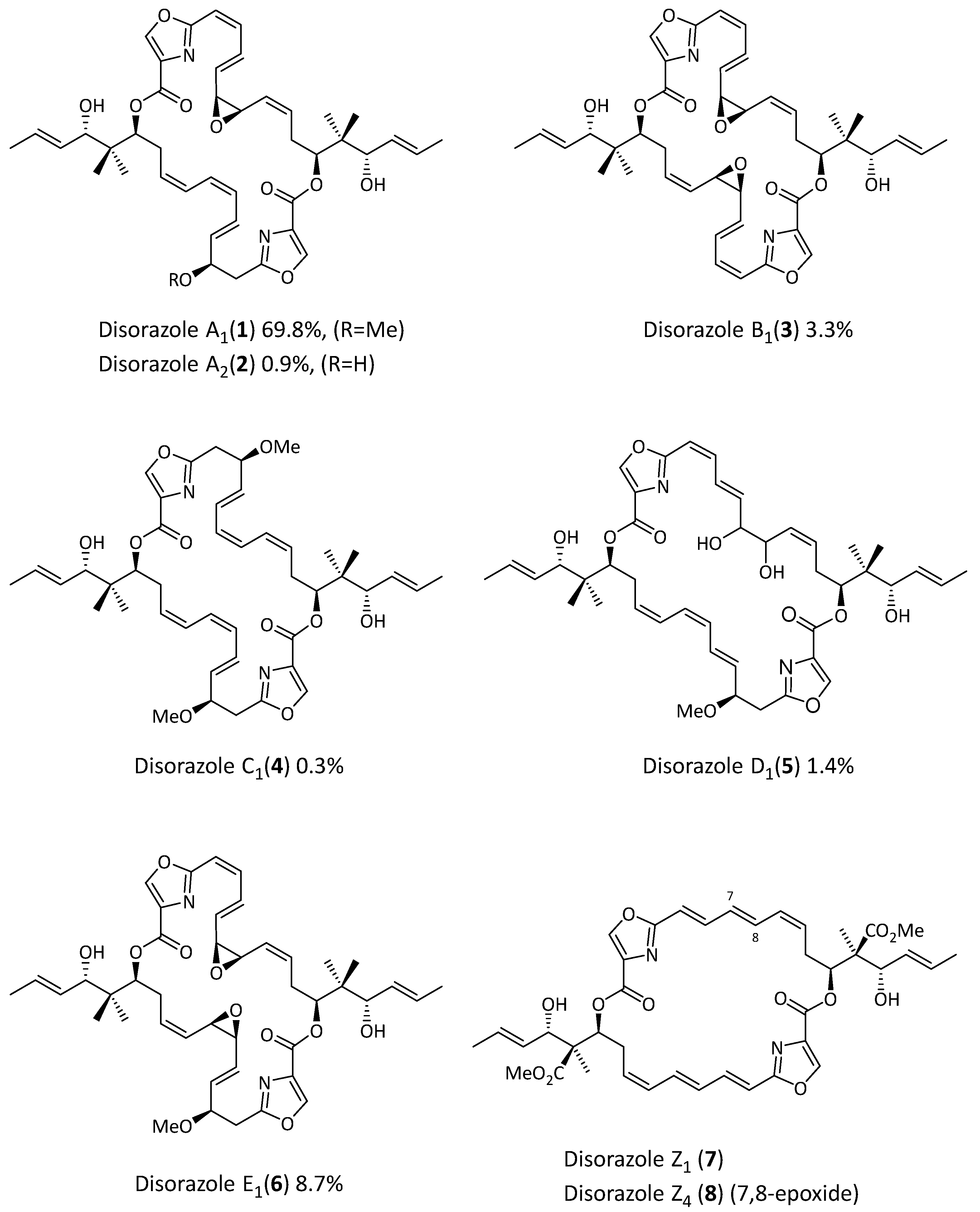
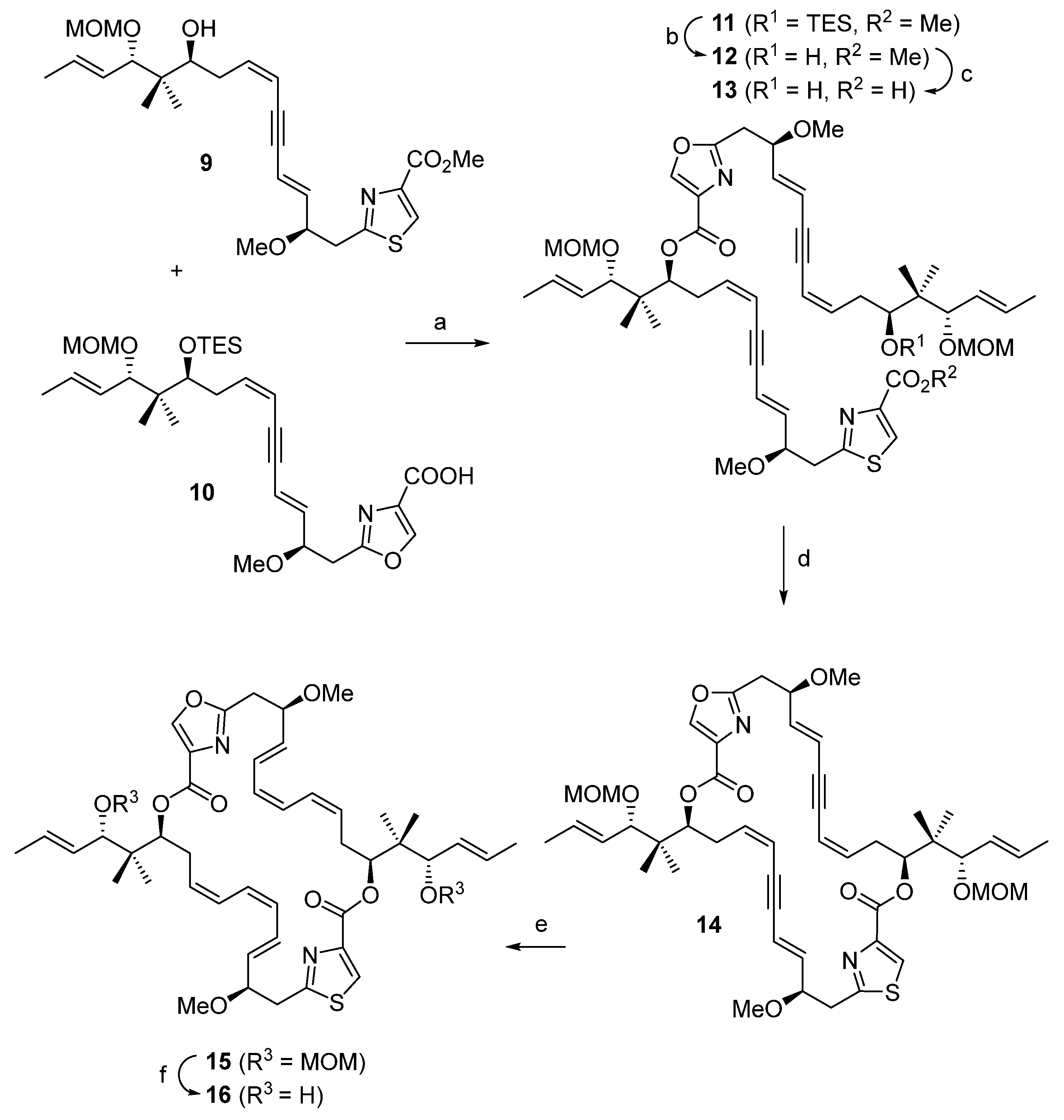
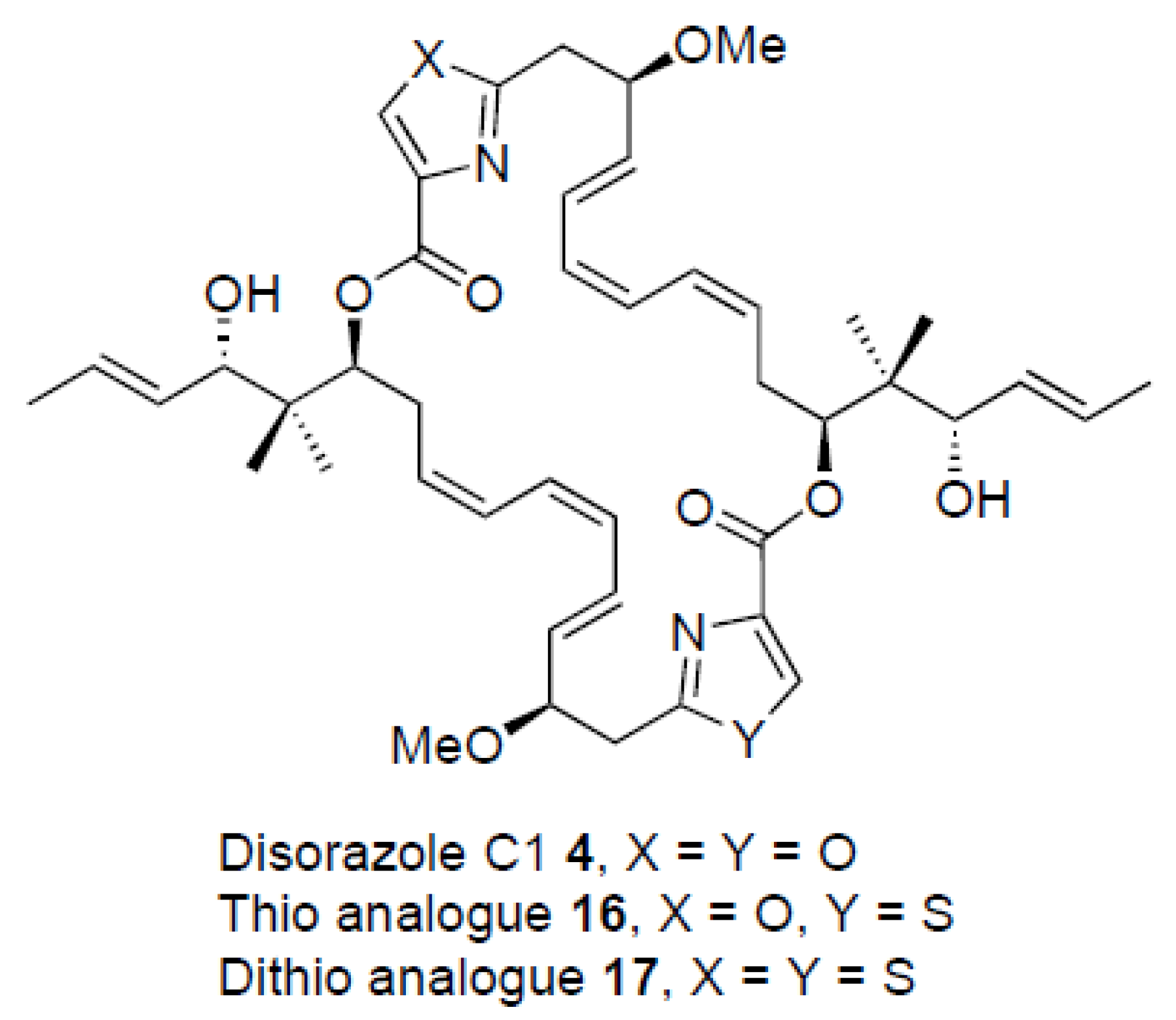
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jahresbericht GBF 2001. Available online: https://www.helmholtz-hzi.de/fileadmin/user_upload/Infothek/Ueber_das_HZI/Jahresberichte/Ergebnisberichte/Annual_Report_2001.pdf (accessed on 7 February 2024).
- Irschik, H.; Jansen, R.; Gerth, K.; Höfle, G.; Reichenbach, H. Disorazole A1, an Efficient Inhibitor of Eukariotic Organisms Isolated from Myxobacteria. J. Antibiot. 1995, 48, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Weissman, K.J.; Mueller, R. Myxobacterial secondary metabolites: Bioactivities and modes-of-action. Nat. Prod. Rep. 2010, 27, 1276–1295. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Irschik, H.; Reichenbach, H.; Wray, V.; Höfle, G. Disorazoles, Highly Cytotoxyc Metabolites from the Sorangicin–Producing Bacterium Sorangium cellulosum. Liebigs Ann. Chem. 1994, 1994, 759–773. [Google Scholar] [CrossRef]
- Elnakady, Y.A.; Sasse, F.; Lünsdorf, H.; Reichenbach, H. Disorazol A1, a highly effective antimitotic agent acting on tubulin polymerization and inducing apoptosis in mammalian cells. Biochem. Pharmacol. 2004, 67, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Birkelbach, J.; Fu, C.; Herrmann, J.; Irschik, H.; Morgenstern, B.; Hirschfelder, K.; Li, R.; Zhang, Y.; Jansen, R.; et al. The Disorazole Z Family of Highly Potent Anticancer Natural Products from Sorangium cellulosum: Bioactivity, Biosynthesis, and Heterologous Expression. Microbiol. Spectr. 2023, 11, e00730-23. [Google Scholar] [CrossRef] [PubMed]
- Bold, C.P.; Altmann, K.-H. The Chemistry of Disorazoles and Structure-Activity Relationships: An Update. Tetrahedron 2024, in press. [Google Scholar] [CrossRef]
- Perez, H.L.; Cardaerelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Lizzadro, L.; Spieß, O.; Schinzer, D. Total Synthesis of (−)-Disorazole C1. Org. Lett. 2021, 23, 4543–4547. [Google Scholar] [CrossRef] [PubMed]
- Lizzadro, L.; Spieß, O.; Collisi, W.; Stadler, L.; Schinzer, D. Extending the Struture-Activity Relationship of Disorazole C1: Exchanging the Oxazole Ring by Thiazole and Influence of Chiral Centers within Disorazole Core on Cytotoxicity. ChemBioChem 2022, 23, e202200458. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Bellavance, G.; Buchman, M.; Pulikuri, K.K. Total Synthesis of Disorazoles A1 and B1 and full Structural Elucidation of Disorazole B1. J. Am. Chem. Soc. 2017, 139, 15636–15639. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Marin-Felix, Y.; Surup, F.; Stchigel, A.M.; Stadler, M. Seven New Cytotoxic and Antimicrobial Xanthoquinodins from Jugulospora vestita. J. Fungi 2020, 6, 188. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Wessel, A.-C.; Luangsa-ard, J.J.; Stadler, M. Viridistratins A-C, Antimicrobial and Cytotoxic Benzo[j]fluoranthenes from Stromata of Annulohypoxylon viridistratum (Hypoxylaceae, Ascomycota). Biomolecules 2020, 10, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Menchon, G.; Prota, A.E.; Lucena-Agell, D.; Bucher, P.; Jansen, R.; Irschik, H.; Müller, R.; Paterson, I.; Díaz, J.F.; Altmann, K.-H.; et al. A fluorescense anisotropy assay to discover and characterize ligands targeting the maytansine site of tubulin. Nat. Commun. 2018, 9, 2106. [Google Scholar] [CrossRef] [PubMed]
- Bold, C.P.; Lucena-Agnel, D.; Olive, M.A.; Diaz, J.F.; Altmann, K. Synthesis and Biological Evaluation of C(13)/C(13’)-Bis(desmethyl)disorazole Z. Angew. Chem. Int. Ed. 2023, 62, e202212190. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | ||||||
|---|---|---|---|---|---|---|
| L929 | KB3.1 | A431 | A549 | PC-3 | MCF-7 | |
| Compound | IC50 (ng/mL) | |||||
| Epothilone B | 0.24 | 0.017 | 0.026 | 0.034 | 0.048 | 0.015 |
| Disorazole C1 4 | 0.25 | 0.23 | 0.32 | 0.60 | 0,11 | 0.28 |
| Analogue 16 | 2.1 | 0.94 | 0.94 | 1.6 | 1.7 | 0.75 |
| Analogue 17 | 180 | 85 | 82 | 86 | 120 | 91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lizzadro, L.; Spieß, O.; Reinecke, S.; Stadler, M.; Schinzer, D. Synthesis of a Non-Symmetrical Disorazole C1-Analogue and Its Biological Activity. Molecules 2024, 29, 1123. https://doi.org/10.3390/molecules29051123
Lizzadro L, Spieß O, Reinecke S, Stadler M, Schinzer D. Synthesis of a Non-Symmetrical Disorazole C1-Analogue and Its Biological Activity. Molecules. 2024; 29(5):1123. https://doi.org/10.3390/molecules29051123
Chicago/Turabian StyleLizzadro, Luca, Oliver Spieß, Silke Reinecke, Marc Stadler, and Dieter Schinzer. 2024. "Synthesis of a Non-Symmetrical Disorazole C1-Analogue and Its Biological Activity" Molecules 29, no. 5: 1123. https://doi.org/10.3390/molecules29051123
APA StyleLizzadro, L., Spieß, O., Reinecke, S., Stadler, M., & Schinzer, D. (2024). Synthesis of a Non-Symmetrical Disorazole C1-Analogue and Its Biological Activity. Molecules, 29(5), 1123. https://doi.org/10.3390/molecules29051123