Abstract
Spirotryprostatin alkaloids, a class of alkaloids with a unique spirocyclic indoledionepiperazine structure, were first extracted from the fermentation broth of Aspergillus fumigatus and have garnered significant attention in the fields of biology and pharmacology. The investigation into the pharmacological potential of this class of alkaloids has unveiled promising applications in drug discovery and development. Notably, certain spirotryprostatin alkaloids have demonstrated remarkable anti-cancer activity, positioning them as potential candidates for anti-tumor drug development. In recent years, organic synthetic chemists have dedicated efforts to devise efficient and viable strategies for the total synthesis of spirotryprostatin alkaloids, aiming to meet the demands within the pharmaceutical domain. The construction of the spiro-C atom within the spirotryprostatin scaffold and the chirality control at the spiro atomic center emerge as pivotal aspects in the synthesis of these compounds. This review categorically delineates the synthesis of spirotryprostatin alkaloids based on the formation mechanism of the spiro-C atom.
1. Introduction
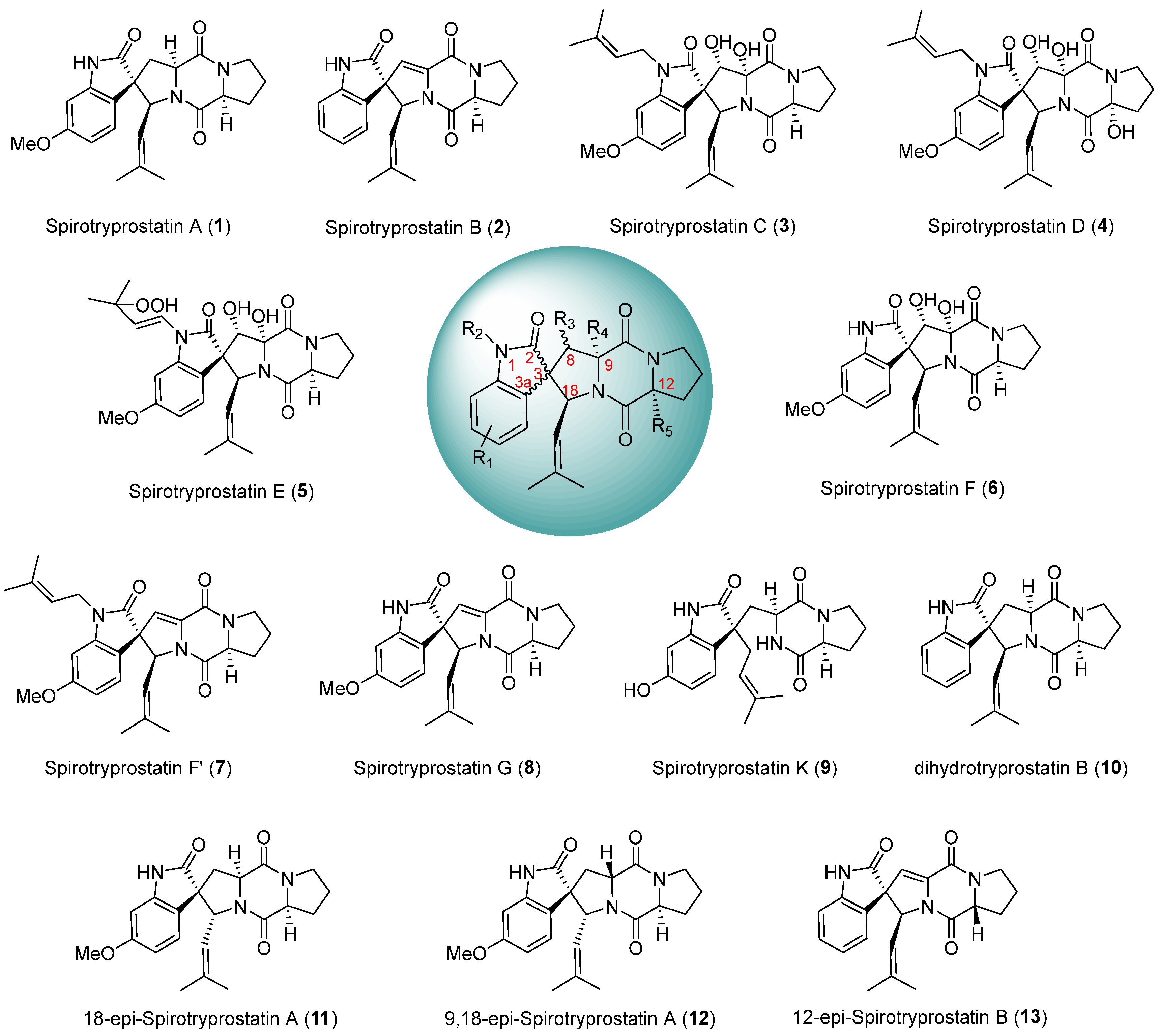
The spirotryprostatin class of alkaloids represents a significant category of spiroindolinone-piperazine alkaloids, characterized by a fundamental pentacyclic structure composed of a spiroindolinone moiety and a dioxopiperazine moiety, featuring multiple chiral centers. Initially isolated by Osada et al. [1,2] from the fermentation broth of Aspergillus fumigatus, two distinct spirotryprostatin alkaloids were first identified and designated spirotryprostatin A (1) and spirotryprostatin B (2). In the course of activity assays, it was observed that spirotryprostatins A and B exhibit inhibitory effects on murine breast cancer cells. Furthermore, their cytotoxic activity against human chronic myeloid leukemia cells and human acute promyelocytic leukemia cells was confirmed, suggesting the potential of spirotryprostatin-class alkaloids as lead compounds for novel drug screening endeavors [3,4,5,6]. This underscores the prospect of spirotryprostatins being promising candidates in the identification of novel therapeutic agents. The spiroindolinone and dioxopiperazine motifs within the structural framework of spirotryprostatin-class alkaloids represent pivotal elements found in numerous synthetic and natural compounds [7,8,9,10]. Compounds featuring the spiroindolinone moiety have demonstrated diverse biological activities [11,12,13], including anti-tumor, antimicrobial, anti-HIV, and anti-malarial properties. Similarly, those containing the dioxopiperazine moiety exhibit bioactivities [14,15,16] such as cytotoxicity, antimicrobial effects, antifungal properties, and thrombosis prevention. Notably, spirotryprostatin-class alkaloids uniquely combine both of these critical motifs, thereby manifesting significant and versatile biological activities. As of the present moment, ten principal structures of spirotryprostatin alkaloids [17,18,19,20,21] have been isolated (Scheme 1), and their pharmacological activities [22,23,24], including anti-tumor, anti-HIV, insecticidal [24], and analgesic effects have been substantiated. Consequently, these compounds have garnered extensive attention from the chemical community. However, it is noteworthy that spiroindolinone compounds, despite their promising pharmacological profiles, remain at the cellular experimentation stage in clinical research [25,26], with no current reports documenting their progression into marketable products.
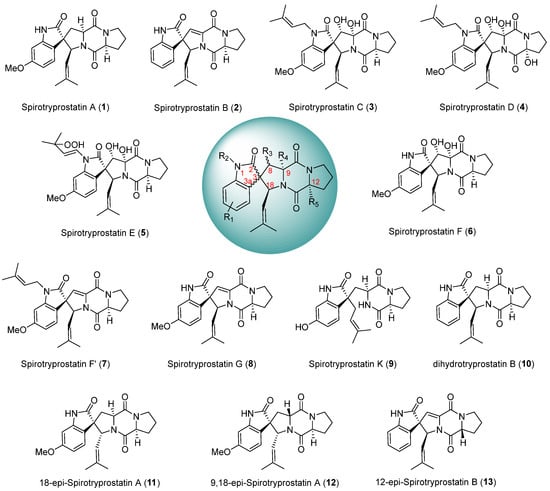
Scheme 1.
Main structures and diastereomers of spirotryprostatin alkaloids.
Since the discovery of spirotryprostatin-class alkaloids, numerous research groups have undertaken diverse synthetic approaches to their total synthesis (Table 1). The scope of these synthetic endeavors extends beyond the original spirotryprostatin alkaloids isolated and encompasses the total synthesis of modified derivatives [15,27,28,29,30,31,32,33,34,35]. Through structural modifications, the objective is not only to recreate the originally isolated spirotryprostatin alkaloids but also to generate a novel cohort of spirotryprostatin-class alkaloids with enhanced pharmacological efficacy. This synthetic modification approach aims to yield new pharmaceutical agents within the spirotryprostatin class with augmented therapeutic potential. The construction of the spiro-C atom within the spirotryprostatin scaffold and the chiral control at the spiro atomic center are pivotal in the synthesis of such compounds. The spiro-C atom located at the indole nucleus’s C3 position serves as the chiral center for the entire molecular framework, formed by the shared carbon atom between the oxidized indole and the pyrrolidine. Owing to the stereochemical selectivity inherent in chemical reactions, the total synthesis of certain analogous compounds often yields diastereoisomers of the target product (Scheme 1). In summary, the synthesis of compounds of this nature encounters challenges primarily in the construction of quaternary carbon atoms and the chiral control at the spiro-C center. Current synthetic methodologies for such compounds predominantly involve the rational design of substrates functionalized at the C3 position of the indole moiety, followed by ring-closure processes to form the spiro-indole-pyrrolidine framework. This review categorically reports the synthesis of spirotryprostatin-class alkaloids based on the manner in which the spiro-C atom is formed.

Table 1.
Summary of total syntheses of spirotryprostatin alkaloids.
2. The Synthetic Approach Employing Tryptophan Derivatives as the Starting Material
2.1. The Oxidative Rearrangement Methodology
2.1.1. Danishefsky’s Total Synthesis
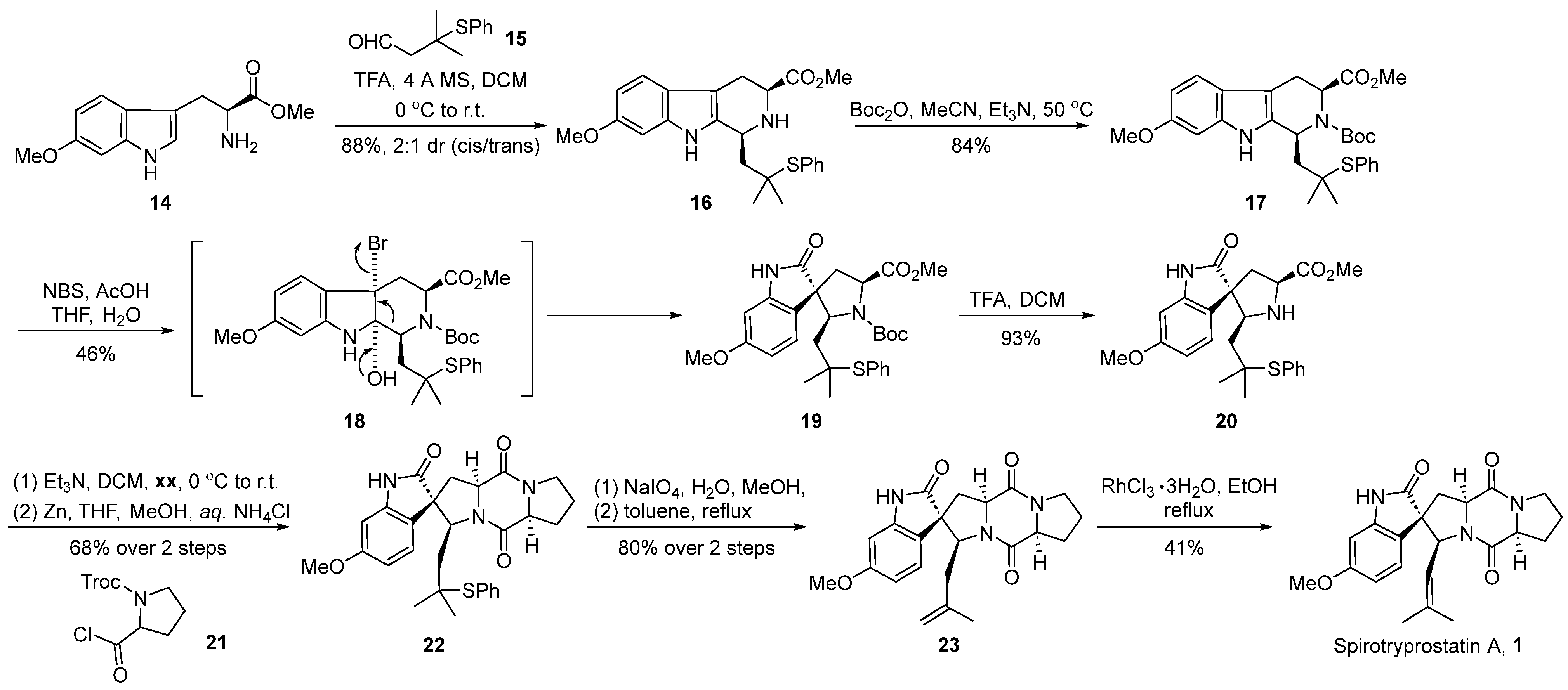
In 1998, Danishefsky’s group [36] first reported the total synthesis pathway of spirotryprostatin A. Subsequently, they modified the reaction pathway, successfully achieving the total synthesis of spirotryprostatin A (1) and its derivative dihydrotryprostatin B (10) (Scheme 2). A pivotal step in the reaction involved the oxidative rearrangement of a β-carboline derivative facilitated by NBS, leading to the formation of the spirocyclic moiety. The reaction commenced with 6-methoxytryptophan methyl ester 14 and tert-butylthioacetaldehyde 15 as starting materials. Initially, subjected to a Pictet–Spengler cyclization reaction, the process yielded the indole and hexacyclic intermediate 16 with a diastereomeric ratio of 2:1. The subsequent protection of the amino group with a Boc moiety led to the formation of intermediate 17. Furthermore, under the influence of NBS and acetic acid, an oxidative rearrangement ensued, culminating in the formation of the crucial pyrrolidine-indole ketone spirocyclic intermediate 19. Subsequently, intermediate 19 underwent deprotection facilitated by trifluoroacetic acid, followed by a two-step condensation with proline derivative 21 to form the dioxopiperazine structural intermediate 22. Finally, intermediate 22 underwent oxidation with sodium periodate, followed by the elimination of the phenylthioether to generate terminal alkene 23. Under the catalysis of rhodium trichloride hydrate, the double bond underwent isomerization, resulting in the target product spirotryprostatin A (1). The overall synthesis spanned eight steps with a total yield of 6.5%.
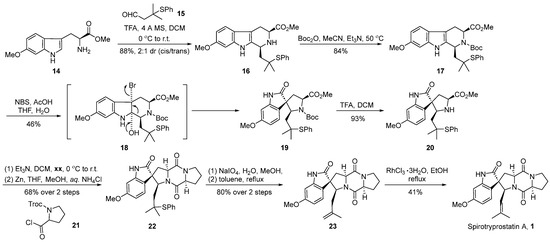
Scheme 2.
Danishefsky’s total synthesis of spirotryprostatin A via oxidative rearrangement.
2.1.2. Granesan’s Total Synthesis
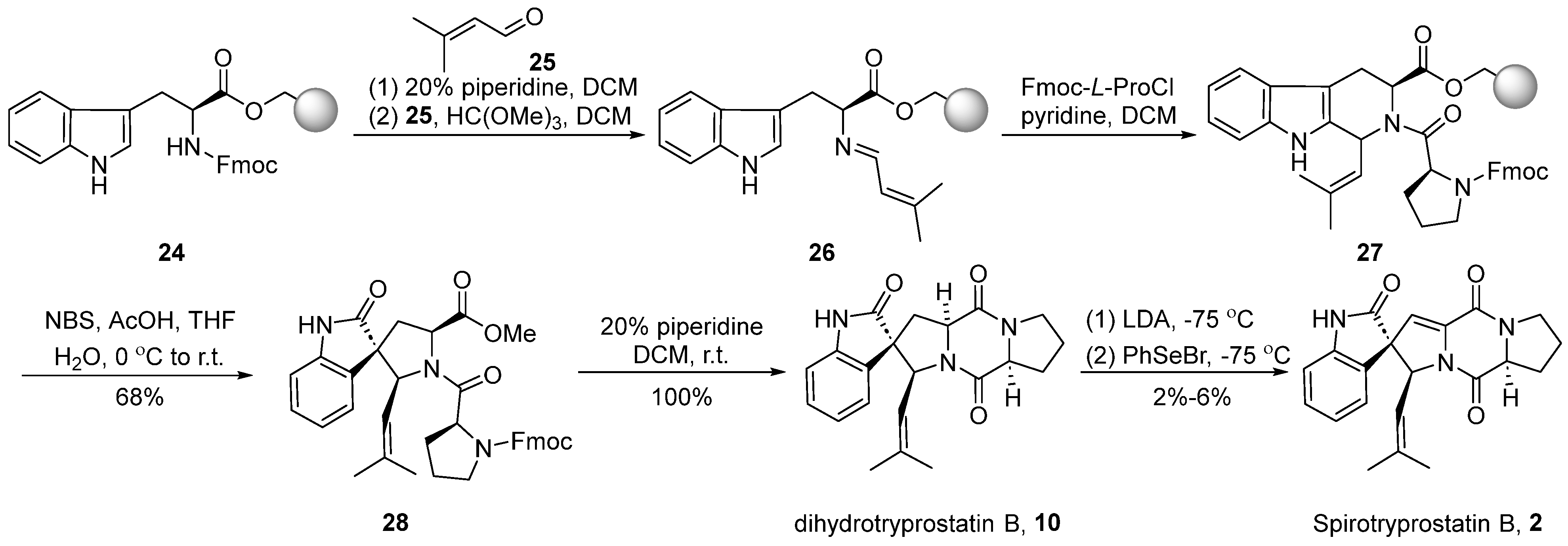
Around the year 2000, Granesan’s group [37,38,39] applied the PhSeBr reagent elimination reaction to the dioxopiperazine system for the construction of double bonds. Simultaneously, oxidative rearrangement was employed as a crucial step in spirocyclic formation. This strategic approach led to the successful total synthesis of dihydrospirotryprostatin B (10) and spirotryprostatin B (2). In the initial stages of Granesan’s synthesis, the traditional starting point still involved L-tryptophan methyl ester. However, in the optimized approach, the starting material was modified to utilize L-tryptophan 24 protected with N-Fmoc and anchored to polystyrene resin [38] (Scheme 3). In the presence of piperidine, the Fmoc protecting group was removed from the starting material, liberating the amino group. This amino group then reacted with isovaleraldehyde 25 and methyl formate to form an imine intermediate 26. Subsequently, imine 26 underwent a Pictet-Spengler reaction with N-Fmoc-L-proline acyl chloride, leading to the formation of dipeptide intermediate 27 with a cis/trans ratio of 1:1 at the C18 position. Intermediate 27, under the influence of NBS and acetic acid, underwent an oxidative rearrangement to yield the spirocyclic intermediate 28. The subsequent removal of the Fmoc protecting group from the proline moiety, followed by a ring-closure process, led to the formation of dihydrospirotryprostatin B (10). The latter underwent a PhSeBr-mediated elimination reaction to yield the final product spirotryprostatin B (2). The entire reaction sequence encompassed five steps with an overall yield ranging from 2–6%, accompanied by the generation of several byproducts.
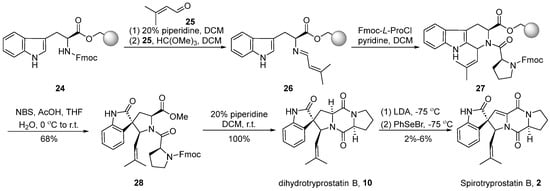
Scheme 3.
Granesan’s total synthesis of spirotryprostatin B via oxidative rearrangement.
2.1.3. Zhang’s Total Synthesis
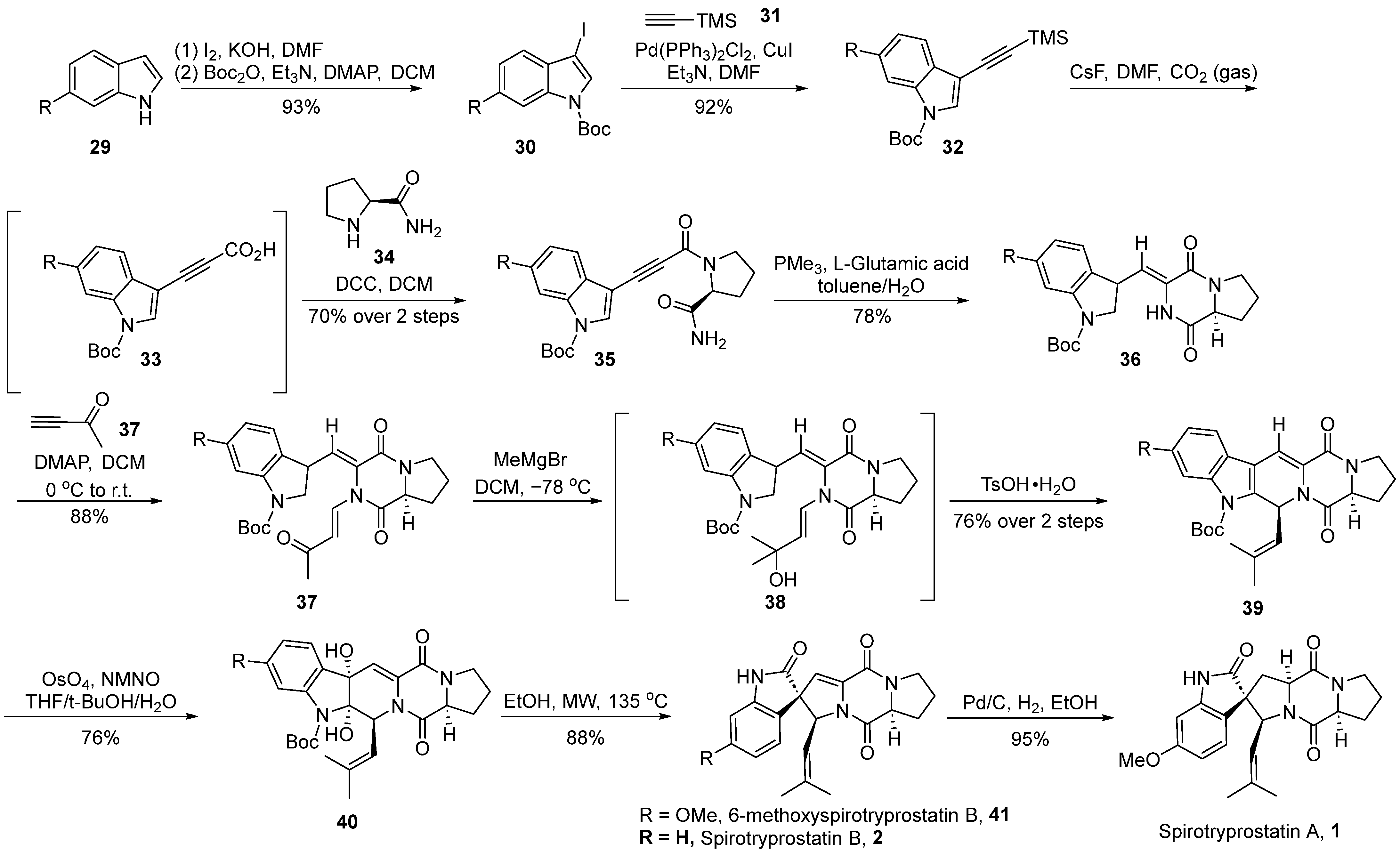
In 2019, Zhang et al. [40] reported the total synthesis of spirotryprostatin A (1), achieved through intramolecular cyclization and osmium tetroxide-mediated oxidative rearrangement processes (Scheme 4). The authors initiated the synthesis using 6-substituted indole 29 as the starting material, which underwent iodination followed by Boc protection, resulting in N-Boc-iodinated indole compound 30. Compound 30 was then subjected to Sonogashira coupling with trimethylsilylacetylene 31, yielding the alkyne-silane intermediate 32. Alkyne-silane 32 underwent carboxylation with CO2, forming unstable intermediate 33. Subsequently, direct condensation with (S)-prolyl amide 34 led to the formation of alkyne amide 35. Alkyne amide 35 underwent intramolecular cyclization to generate dioxopiperazine intermediate 36 with E/Z = 7:1. Subsequent nucleophilic addition with 3-buten-2-one 37, followed by further addition with a Grignard reagent, resulted in tertiary alcohol intermediate 38. The dehydration of intermediate 38, catalyzed by TsOH, formed a double bond, which, upon selective oxidation with osmium tetroxide, led to the formation of diol compound 40. Subsequently, compound 40 underwent rearrangement under high-temperature microwave conditions. When the R-group was hydrogen, spirotryprostatin B (2) was directly obtained, whereas the use of methoxy-substituted indole as the starting material yielded 6-methoxyspirotryprostatin B 41. The latter, upon the Pd/C-catalyzed reduction of the double bond, afforded the natural product spirotryprostatin A (1). The entire reaction sequence comprised 11 steps, resulting in a 20% overall yield.
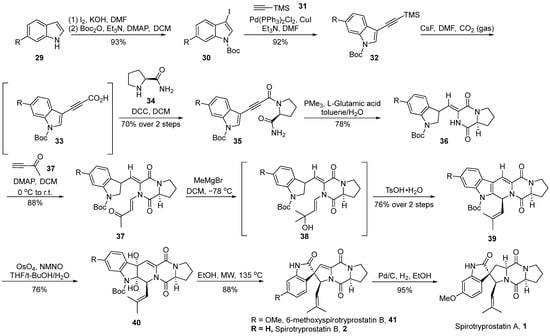
Scheme 4.
Zhang’s total synthesis of spirotryprostatin alkaloids via OsO4-mediated oxidative rearrangement.
2.2. Mannich Reaction
Danishefsky’s Total Synthesis
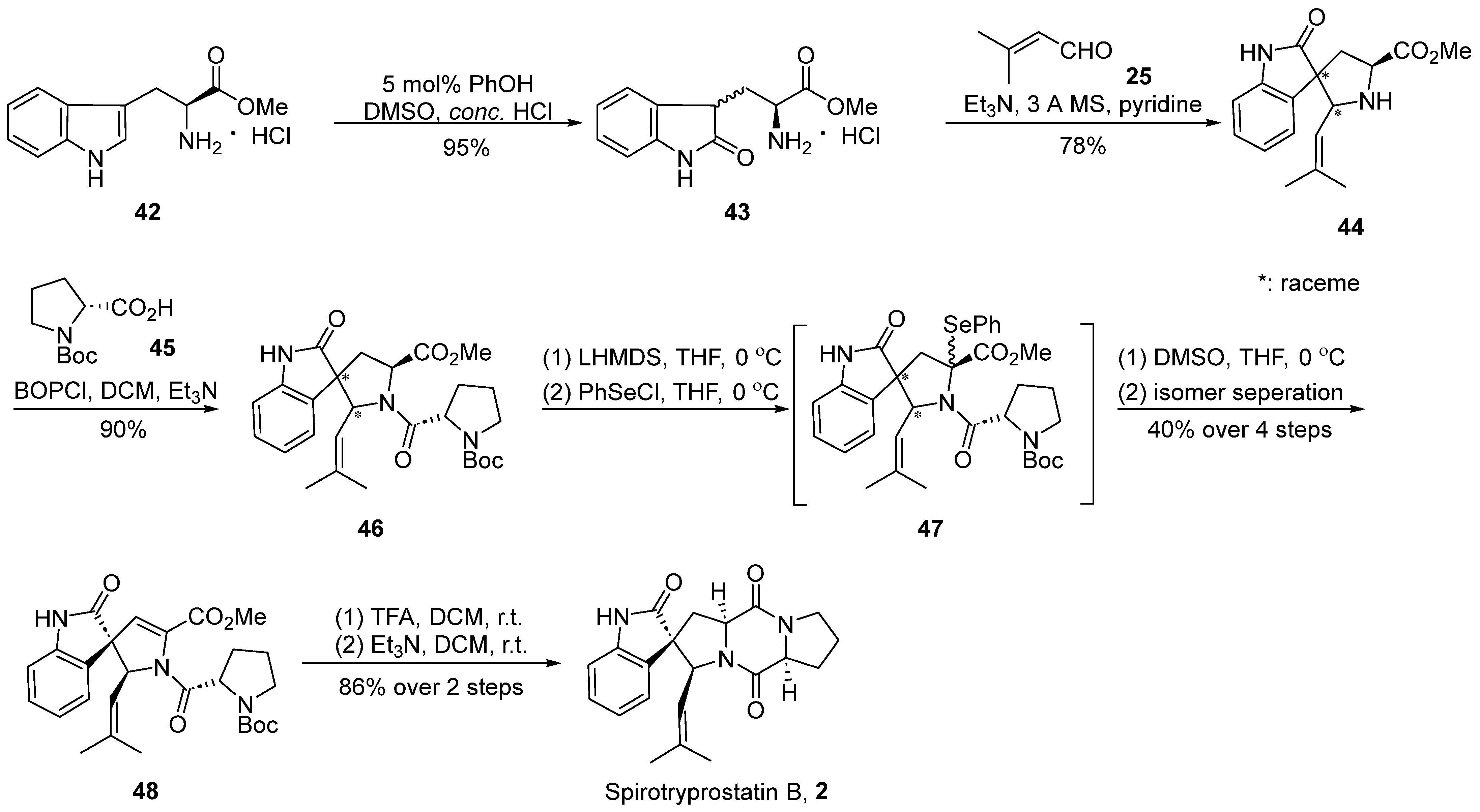
In the year 2000, Danishefsky’s group [41] accomplished the inaugural total synthesis of spirotryprostatin B (2), employing the Mannich reaction as a pivotal step in the formation of the spirocyclic moiety (Scheme 5). The reaction commenced with L-tryptophan methyl ester hydrochloride 42 and, under the influence of phenol and concentrated hydrochloric acid, underwent transformation into the oxidized indole structure compound 43. Subsequently, through a Mannich reaction with 3-buten-2-one 25, four distinct spirocyclic indole ketone intermediate-like compounds 44, each possessing an isobutene side chain, were obtained. Without the isolation of the four isomers, they were directly condensed with Boc-protected proline 45, yielding a mixture of compound 46. In the final step, a method involving the elimination of PhSeCl reagent under the influence of LHMDS was employed, leading to the introduction of a double bond at positions C8 and C9. The reaction yielded a mixture of isomers, from which a singular isomer 48 was isolated. Following the removal of protective groups using TFA and a subsequent cyclization step catalyzed by triethylamine, the target compound spirotryprostatin B (2) was obtained. The entire reaction sequence encompassed eight steps, resulting in an overall yield of 4.6%.
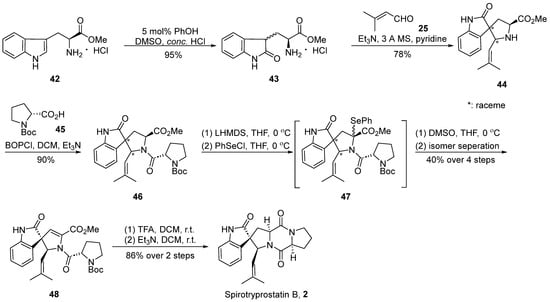
Scheme 5.
Danishefsky’s total synthesis of spirotryprostatin B via Mannich reaction.
The comprehensive synthesis strategy involved the Mannich reaction between an L-tryptophan derivative and an aldehyde to construct the envisaged spirocyclic indole scaffold. While the desired product was successfully obtained, the approach introduced significant stereochemical challenges, inevitably resulting in a mixture of diastereomers. Despite subsequent attempts to assist the separation of diastereomeric mixtures using L-Boc-protected proline in later reactions, the overall yield of spirotryprostatin B was notably diminished.
2.3. Intramolecular N-Acyliminium Ion Spirocyclic Cyclization
Horne’s Total Synthesis
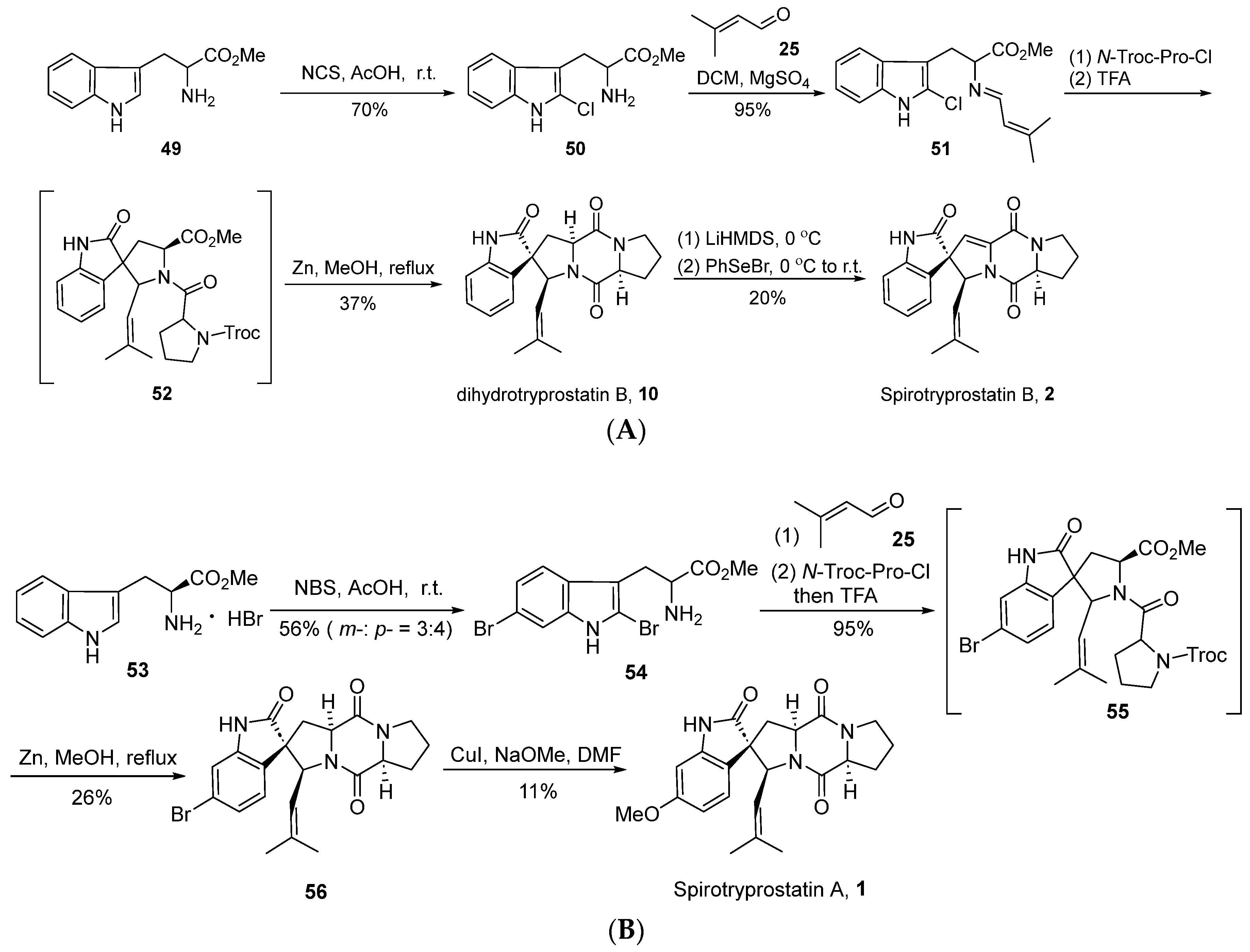
In 2004, Horne’s group [42] successfully achieved the total synthesis of spirotryprostatin B (2) through an innovative strategy based on intramolecular N-acyliminium ion spirocyclic cyclization, utilizing 2-halotryptophan esters as key intermediates (Scheme 6A). The reaction initiated with L-tryptophan methyl ester 49, which underwent a reaction with N-chlorosuccinimide (NCS) to yield 2-chlorotryptophan methyl ester 50. Subsequent condensation with isovaleraldehyde 25 resulted in the formation of imine intermediate 51. Under the influence of N-Troc-protected proline acyl chloride and trifluoroacetic acid, intermediate 51 underwent acylation, inducing spirocyclic cyclization and yielding indole-oxidized spirocyclic intermediate 52. In contrast to the conventional oxidative rearrangement reactions, a distinctive feature of this reaction lies in the hindrance of the indole’s 2-position by halogen, leading to electrophilic attack at the 3-position and subsequent spirocyclic formation. Subsequently, under the influence of zinc, the Troc-group underwent departure, yielding dihydrospirotryprostatin B and three distinct isomers. Finally, the authors employed a non-oxidative method to convert dihydrospirotryprostatin B (10) into spirotryprostatin B (2), involving a seven-step reaction sequence with an overall yield of 4.9%. This route innovatively utilized the strategy of N-acyliminium ion spirocyclic cyclization for constructing the oxidized indole spirocyclic moiety. However, the lack of control over enantioselectivity in this approach, coupled with the generation of relevant byproducts, limits the applicability of this route.
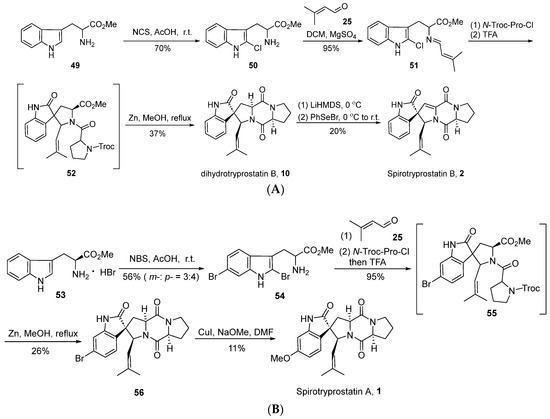
Scheme 6.
(A) Horne’s total synthesis of spirotryprostatin B via intramolecular N-acyliminium ion spirocyclic cyclization. (B) Horne’s total synthesis of spirotryprostatin A via intramolecular N-acyliminium ion spirocyclic cyclization.
In the same year, Horne et al. [43] successfully achieved the total synthesis of spirotryprostatin A (1) using a similar methodology (Scheme 6B). The route commenced with L-tryptophan methyl ester 53, wherein excess NBS was employed for bromination. However, this step exhibited limited regioselectivity, leading to bromination at both the C5 and C6 positions. Subsequently, employing a strategy analogous to the synthesis of spirotryprostatin B, intermediate compound 56 was obtained. Through a copper-catalyzed methoxylation strategy, the bromine moieties were converted into methoxy groups, culminating in the total synthesis of spirotryprostatin A (1). The overall reaction pathway requires only four steps, yielding a final overall efficiency of 6.2%. However, it is noted that the overall yield is relatively low, and configuration changes occur under alkaline conditions, resulting in the generation of byproducts.
3. 1,3-Dipolar Cycloaddition
3.1. Williams’s Total Synthesis
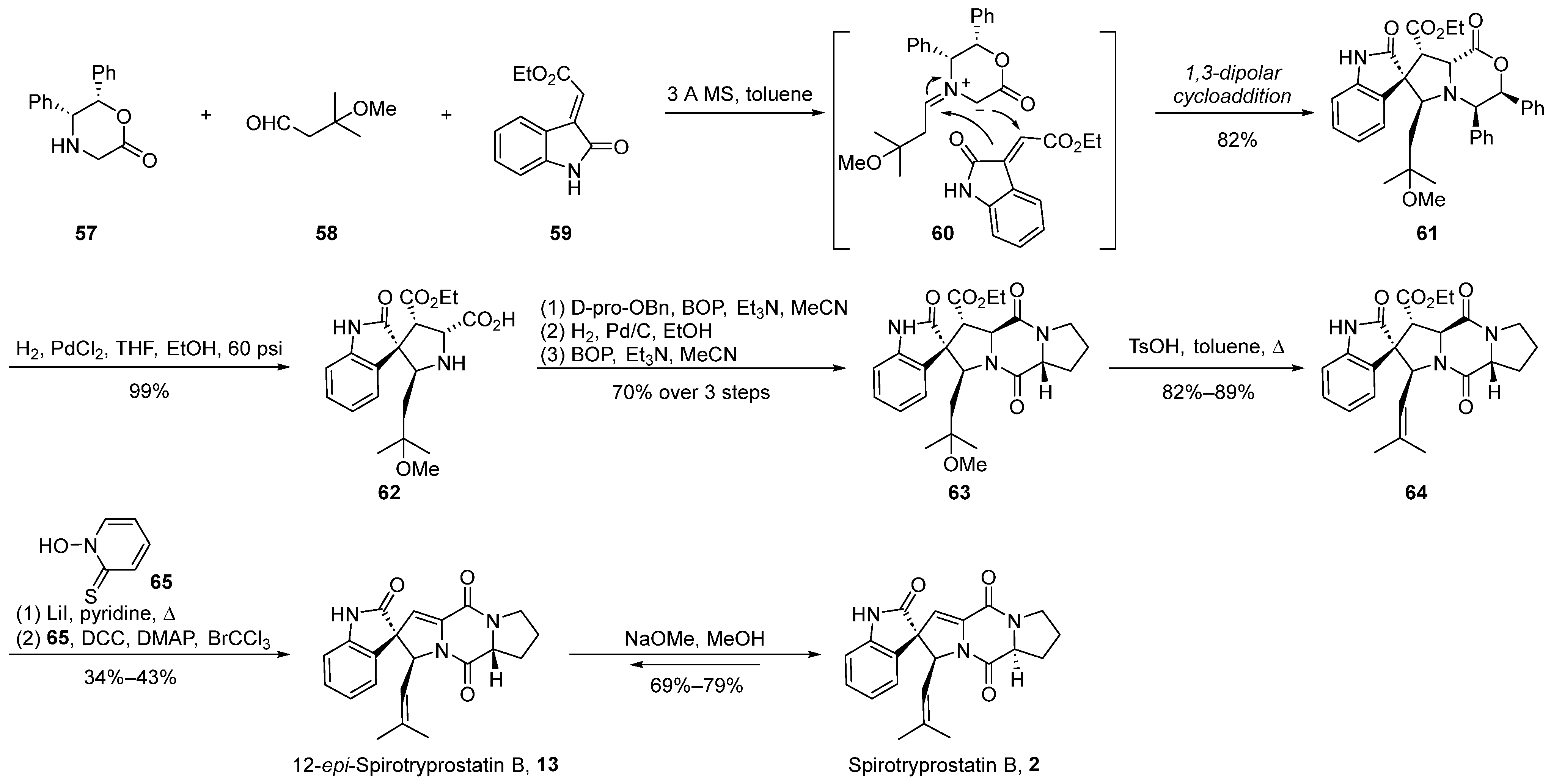
The 1,3-dipolar cycloaddition method has proven to be an exceedingly efficacious strategy for constructing helical stereocenters [44,45,46,47]. This approach has been widely applied in the total synthesis of numerous natural products and their analogs [48,49,50,51]. In the year 2000, Williams et al. [52] initially reported the application of the methylimine ylide-mediated 1,3-dipolar cycloaddition method for the total synthesis of spirotryprostatin B (2) and 12-epi-spirotryprostatin B (13). Subsequently, in 2002 [53], further refinements were introduced to enhance the efficacy of this methodology (Scheme 7). The reaction commenced with quinone 57, aldehyde 58, and oxindole-based ethyl oxindole-2-carboxylate 59 as substrates. Initially, under the influence of molecular sieves in toluene, quinone 57 and aldehyde 58 underwent condensation, forming imine ylide intermediate 60. Subsequent to this step, a 1,3-dipolar cycloaddition reaction ensued with ethyl oxindole-2-carboxylate 59, resulting in the formation of spirocyclic intermediate 61. The stereochemical assignment of compound 61 was elucidated through single-crystal X-ray crystallography, revealing the authors’ proposition that steric hindrance effected by the isoprene aldehyde precursor favors the adoption of the E-stereoisomer geometry. Consequently, achieving high stereochemical selectivity in spirotryprostatins bearing the isoprene moiety at carbon position C18 was feasible. The ensuing steps involved hydrogenation for benzyl group removal to yield 62, followed by ring opening of the lactone, amidation, elimination, and Hunsdiecker reaction, ultimately affording 12-epi-spirotryprostatin B (13). Upon treatment with methoxide in methanol, 12-epi-spirotryprostatin B (13) underwent isomerization, culminating in the target compound (-)-spirotryprostatin B (2). The overall synthetic route encompassed nine steps with a yield of 11%.
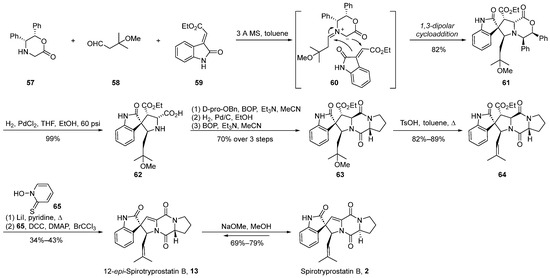
Scheme 7.
Williams’s total synthesis of spirotryprostatin B via 1,3-dipolar cycloaddition.
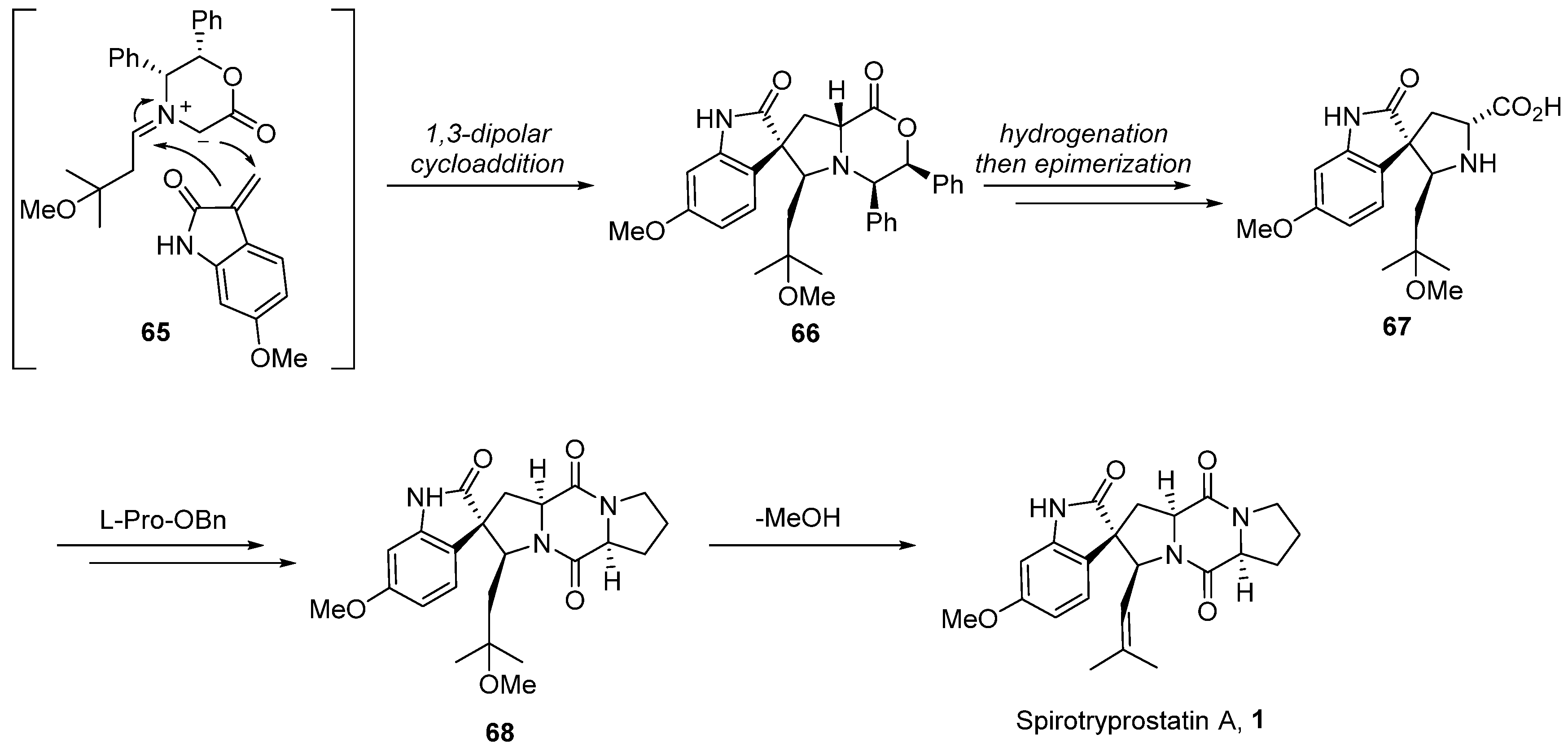
In 2004, Williams [54,55] extended the application of this methodology to the total synthesis route of spirotryprostatin A (1) (Scheme 8). Given the structural disparities between spirotryprostatins A and B, primarily attributed to the methoxy-substituted indole moiety on the benzene ring in spirotryprostatin A and the double bond in the pyrrolidine of spirotryprostatin B, the authors initiated synthesis with indole-2-ketone, possessing the methoxy substituent. A 1,3-dipolar cycloaddition reaction was employed to furnish spiro indoline ketopyrrolidine intermediate 66, featuring four adjacent chiral centers with methoxy substituents. Subsequent analogous treatment procedures led to the final product, spirotryprostatin A (1), with a yield of 2%. The primary challenge in achieving a higher yield was attributed to suboptimal chiral control during the 1,3-dipolar cycloaddition step, resulting in the formation of multiple stereoisomers.
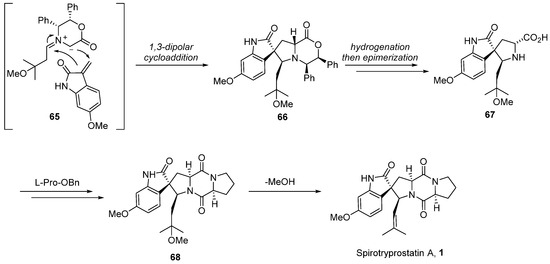
Scheme 8.
Williams’s total synthesis of spirotryprostatin A via 1,3-dipolar cycloaddition.
3.2. Waldmann’s Total Synthesis
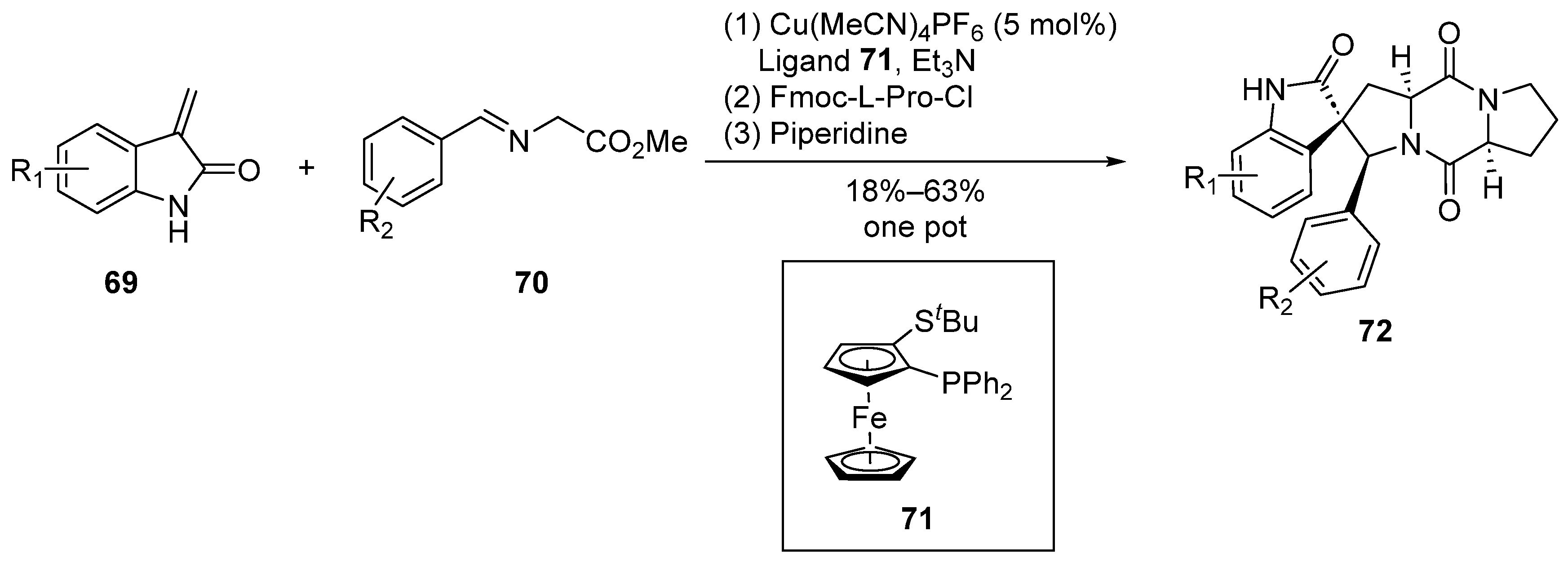
In 2011, Waldmann et al. [56] reported, for the first time, a Cu/N,P-bis(2-methylphenyl)ferrocenyl Lewis acid-catalyzed 1,3-dipolar cycloaddition reaction (Scheme 9). Employing a concise one-pot, three-step methodology, the researchers achieved high enantiomeric selectivity in the synthesis of spirocyclic pyrrolidine–indolone compounds. Subsequently, leveraging this structural motif, a series of derivatives based on the spirotryprostatin A scaffold were successfully synthesized. Notably, this synthetic route demonstrated remarkable enantiomeric selectivity, reaching up to 97%.
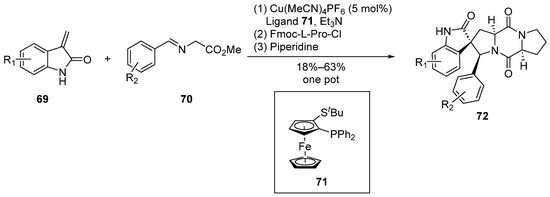
Scheme 9.
Waldmann’s total synthesis of spirotryprostatin A scaffold via 1,3-dipolar cycloaddition.
3.3. Gong’s Total Synthesis
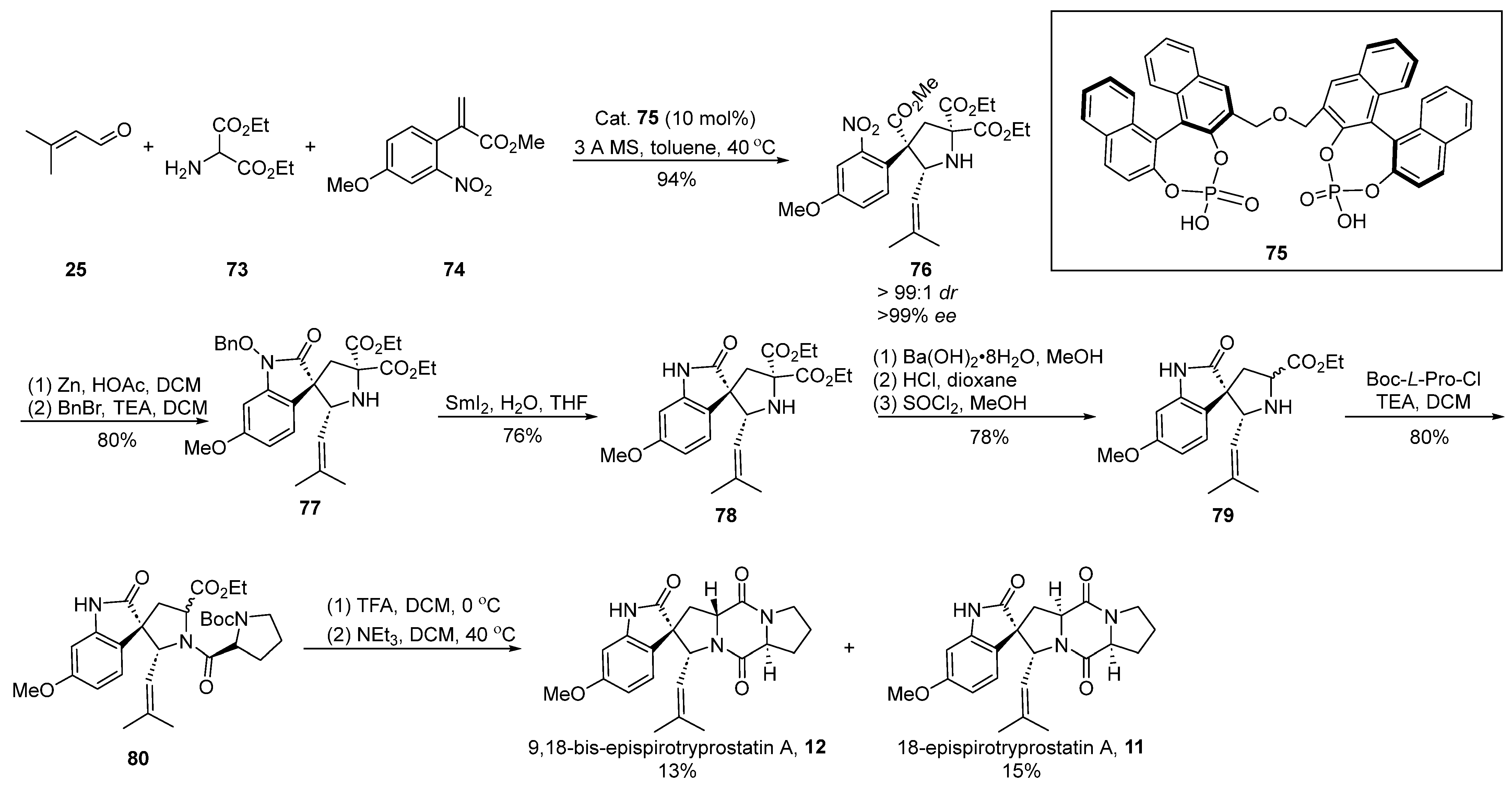
In the same year, Gong [57] introduced a chiral phosphoric acid-catalyzed [3 + 2] cyclization strategy, culminating in the construction of chiral quaternary carbon centers (Scheme 10). This innovative approach facilitated the total synthesis of two distinct enantiomers of spirotryprostatin A. The reaction sequence involved the generation of an imine through the reaction of aldehyde 25 with ethyl aminopropionate 73, followed by a 1,3-dipolar cycloaddition with methyl acrylate derivative 74 under the catalysis of chiral phosphoric acid 75. This process not only facilitated the construction of two chiral carbon centers but also resulted in the formation of intermediate 76. Subsequent zinc powder-mediated cyclization of 76 yielded indole derivative 77. Compound 77 underwent sequential transformations, including deprotection via samarium diiodide, decarboxylation, and hydrolysis, yielding indole spirocyclic pyrrolidine intermediate 79. The further condensation of 79 with proline acyl chloride, Boc removal, and closure of the diketopiperazine ring ultimately afforded the target compounds 9,18-epi-spirotryprostatin A (12) and 18-epi-spirotryprostatin A (11). Upon conducting activity tests on both non-enantiomeric isomers, it was observed that their activities were comparable to that of spirotryprostatin A (1). Consequently, it can be inferred that the configuration at positions C-9 and C-18 in such compounds is not a decisive factor influencing the structure–activity relationship of spirotryprostatin A (1). The synthesis involved a total of 10 steps, culminating in the production of two non-enantiomeric isomers, namely 9,18-epi-spirotryprostatin A (12) and 18-epi-spirotryprostatin A (11), with overall yields of 4.9% and 5.3%, respectively.
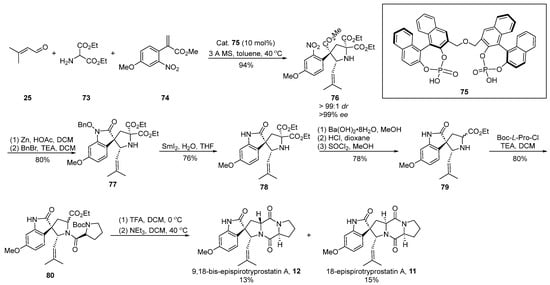
Scheme 10.
Gong’s total synthesis of spirotryprostatin alkaloids via 1,3-dipolar cycloaddition.
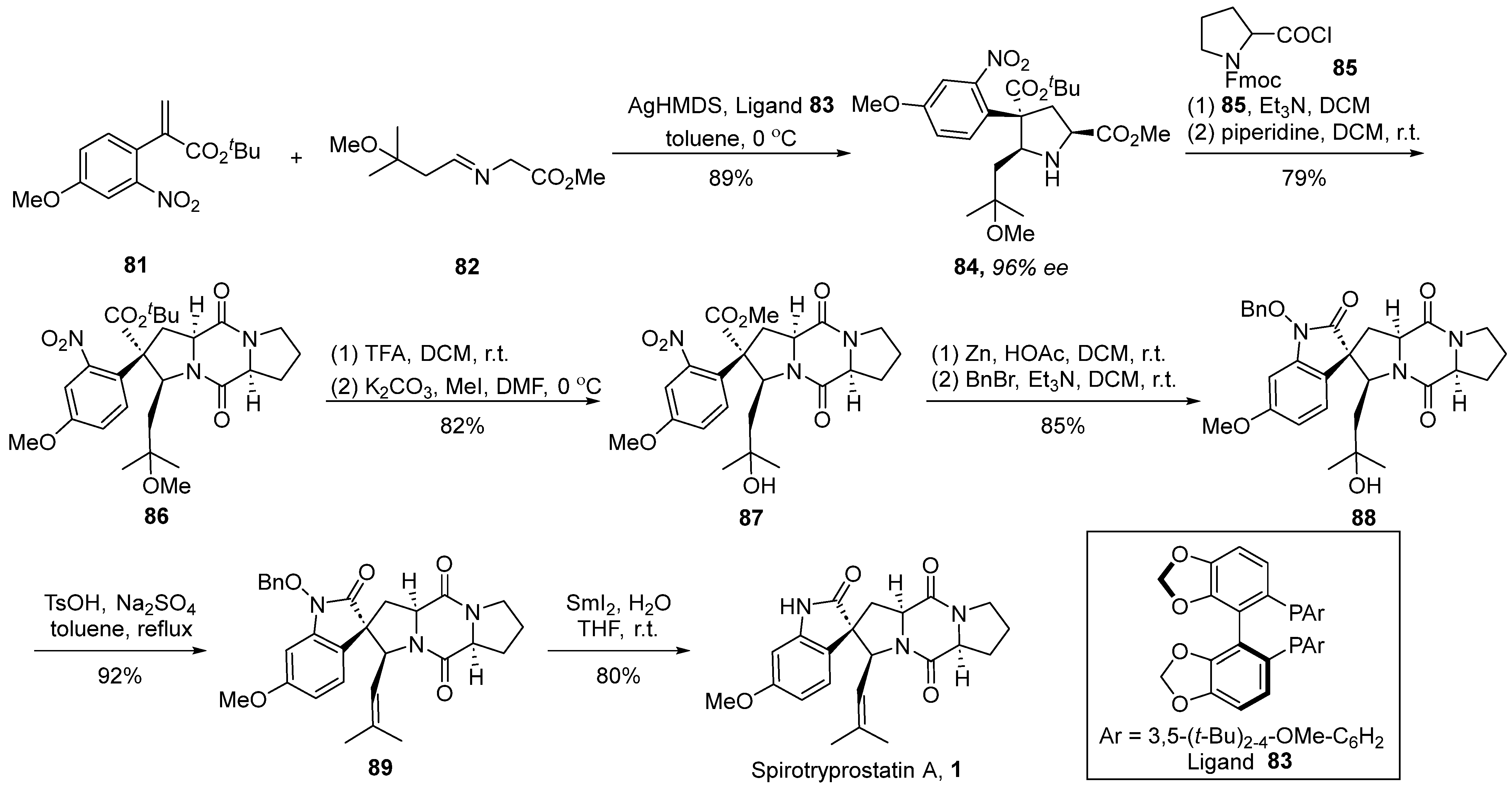
3.4. Wang’s Total Synthesis
In 2023, Wang’s group [58] disclosed an instance of a silver-catalyzed asymmetric [3 + 2] cycloaddition strategy, achieving the total synthesis of spirotryprostatin A (Scheme 11). The authors initiated the synthesis utilizing α-substituted acrylic ester 81 and imine ester 82 as starting materials. Employing AgHMDS and chiral ligand 83 as catalysts facilitated a cyclization process, affording methyl ester derivative 84 with a 96% ee value. Through a coupling reaction with acyl chloride 85, using the methodology established by the Danishefsky group, the formation of fused-ring compound 86 was achieved. Subsequently, intermediate 86 underwent demethoxylation and methylation under the influence of trifluoroacetic acid, yielding intermediate 87. The reduction of the nitro group followed by intramolecular cyclization and benzyl protection afforded pivotal spirocyclic oxindole intermediate 89. Further transformations involving hydroxyl elimination and deprotection culminated in the final product, spirotryprostatin A (1). The synthetic route encompassed a total of six steps with a yield of 36%.
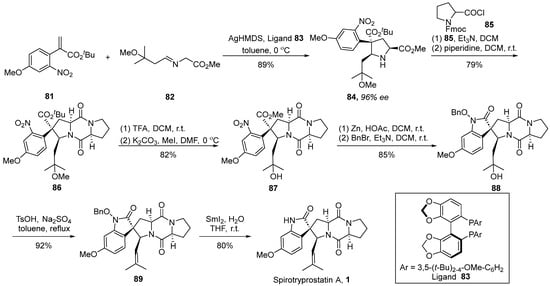
Scheme 11.
Wang’s total synthesis of spirotryprostatin A via 1,3-dipolar cycloaddition.
4. Metal Catalysis
4.1. Heck Coupling
4.1.1. Overman’s Total Synthesis
In the year 2000, Overman et al. [59] achieved the total synthesis of spirotryprostatin B (2) through the strategic implementation of a Heck reaction-mediated cyclization to construct the oxygenated spirocyclic framework (Scheme 12). Diverging from the conventional synthetic approaches to spirotryprostatin-type compounds, which typically involve the initial construction of the spirocyclic oxindole moiety, Overman introduced a pioneering strategy wherein the diketopiperazine scaffold is first assembled, followed by the subsequent synthesis of the spiroatom. A pivotal aspect of this synthetic pathway involves the utilization of an intramolecular asymmetric Heck reaction and the dual capture of a π-allyl palladium intermediate by the adjacent amide nitrogen atom within the diketopiperazine ring. This intermediate is subsequently captured by a trans-selective nucleophilic reagent, concurrently establishing chiral centers at the C3 and C18 positions. The reaction commenced with the commercially available substrate allylic alcohol 90. Initially, an oxygen-induced Claisen rearrangement yielded dienoic ester 91. The subsequent hydrolysis of the two ester groups on 91, followed by TBDPS protection and condensation with ortho-iodoaniline, furnished compound 93. Nitrogen protection of the amide on 93 was accomplished using 2-(trimethylsilyl)ethoxymethyl (SEM), followed by the removal of the TBDPS-protecting group from the hydroxyl moiety. Oxidation with DMP yielded the intermediate aldehyde, which underwent a Horner–Wadsworth–Emmons reaction with phosphonate 94, culminating in the formation of compound 95. The final steps involved a Heck reaction for cyclization, leading to the construction of the oxindole and spirocyclic oxindole compound 96, concurrently generating an equivalent non-enantiomeric isomer. The demethylation of the target configurational intermediate 96 and subsequent steps resulted in the total synthesis of the natural product spirotryprostatin B (2) in 10 steps, achieving a yield of 9%.
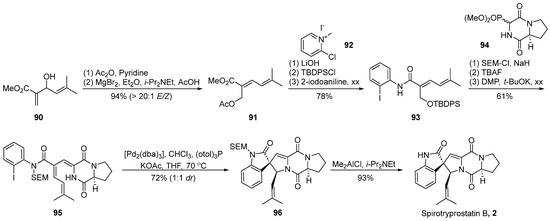
Scheme 12.
Overman’s total synthesis of spirotryprostatin B via Heck coupling.
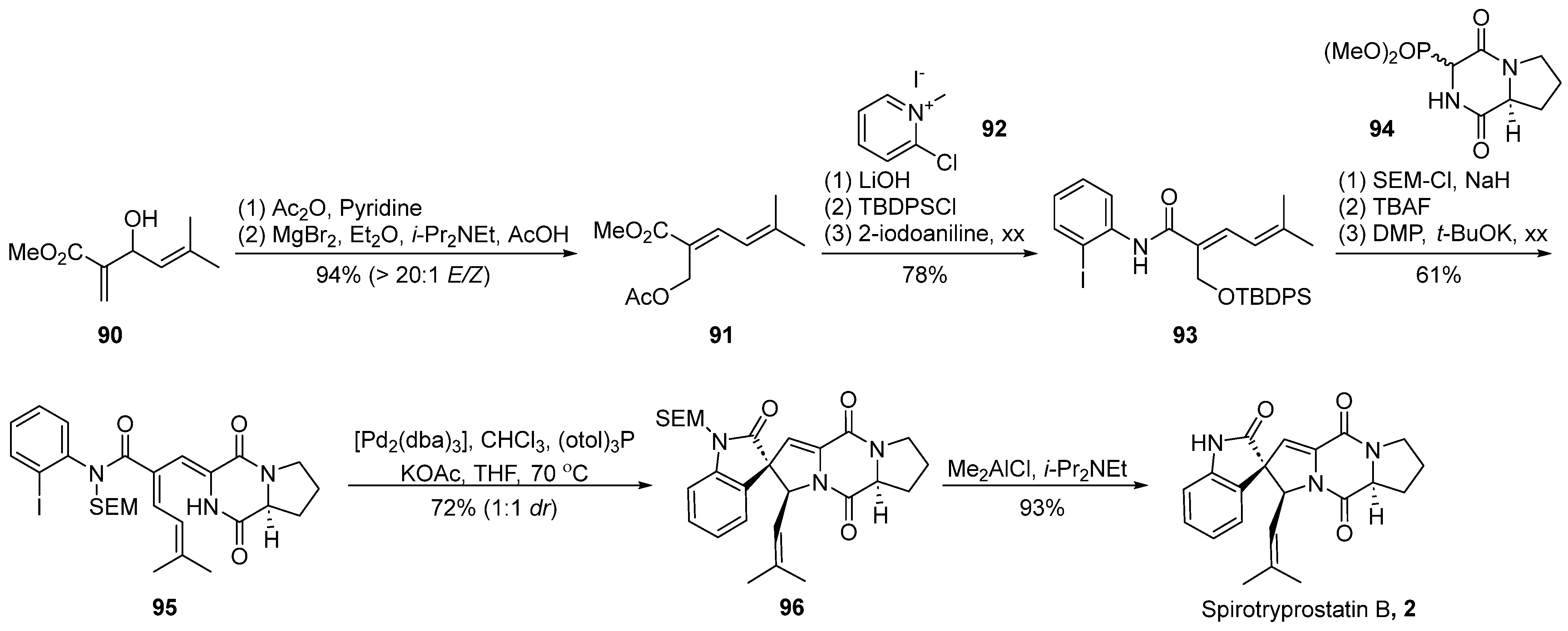
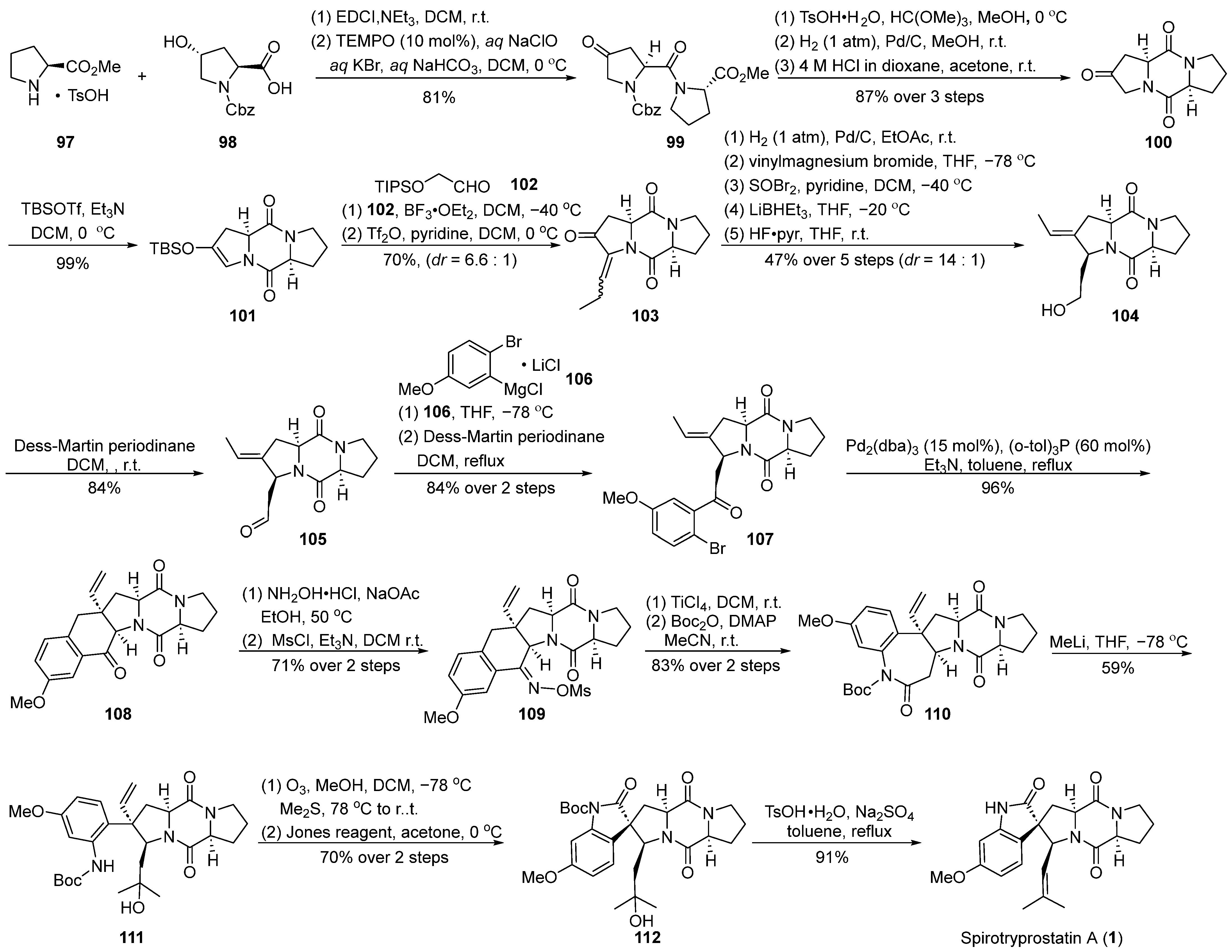
4.1.2. Fukuyama’s Total Synthesis
In 2014, Fukuyama’s group [60] presented a comprehensive synthesis strategy for spirotryprostatin A (1), with a pivotal molecular intramolecular Heck reaction (Scheme 13). Commencing with straightforward L-proline methyl ester salt 97 and 4-hydroxy-L-proline 98, the synthesis initiated with a condensation reaction, followed by oxidation using 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) to yield the highly unstable amide intermediate 99. Recognizing the instability of 99 and its potential impact on yield, a protective measure involved the initial carbonyl protection of the keto group in 99 to form a dimethyl ketal. The subsequent hydrogenation of the formate benzyl ester (Cbz) group triggered an intramolecular cyclization, resulting in the formation of the diketopiperazine intermediate, which, upon acidic hydrolysis, yielded the diketopiperazine intermediate 100. The treatment of ketone 100 with a combination of trifluoromethanesulfonic anhydride (TBSOTf) and triethylamine provided the desired intermediate 101, featuring an unsaturated pyrrolidine structure. The resulting product exhibited high specificity, achieving a remarkable yield of 99%. To introduce an alkyl side chain at the C-18 position of the final product, the Mukaiyama hydroformylation reaction was employed between 101 and trimethylsilyloxyacetaldehyde 102. Subsequent dehydration with trifluoromethanesulfonyl anhydride resulted in the formation of the unsaturated ketone 103 in a mixture of E-Z isomers (6.6:1). Utilizing palladium catalysis, the hydrogenation of 103 was accomplished, followed by a consecutive three-step reduction–rearrangement process, and deprotection, selectively converting it into intermediate 104. Subsequently, the oxidation of the secondary alcohol to an aldehyde was achieved using a Dess–Martin reagent, and further nucleophilic addition with a Grignard reagent produced an intermediate, which, upon subsequent oxidation with the Dess–Martin reagent, led to the formation of ketone intermediate 107. Under palladium catalysis, intermediate 107 underwent an intramolecular Heck coupling cyclization reaction, resulting in the formation of tetrahydronaphthone 108. The introduction of the nitrogen moiety was achieved through hydroxylamine hydrochloride, generating the E-oxime 109 because of the hinderance of the Ph-ring. Subsequent Beckmann rearrangement, Boc protection, and ring opening with methyl lithium yielded the tertiary alcohol intermediate 111. Ozone decomposition followed by an oxidation step resulted in the formation of the spirocyclic oxindole ketone intermediate 112. Finally, through dehydration and deprotection steps, the ultimate product, spirotryprostatin A (1), was obtained. The synthetic pathway involved a total of 25 steps with an overall yield of 3.4%. Despite the relatively high yields at each step, the extended length of the synthetic route contributed to a lower final yield.
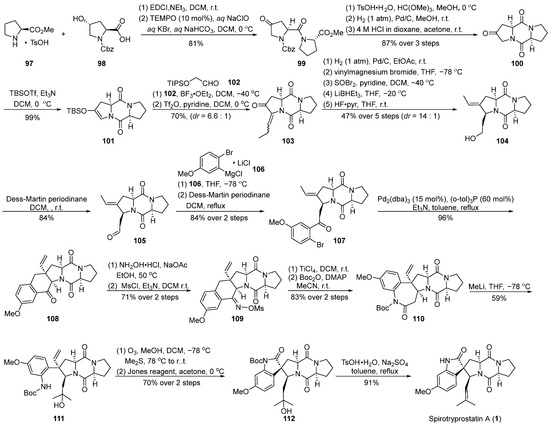
Scheme 13.
Fukuyama’s total synthesis of spirotryprostatin A via Heck coupling.
4.2. Metal Olefination
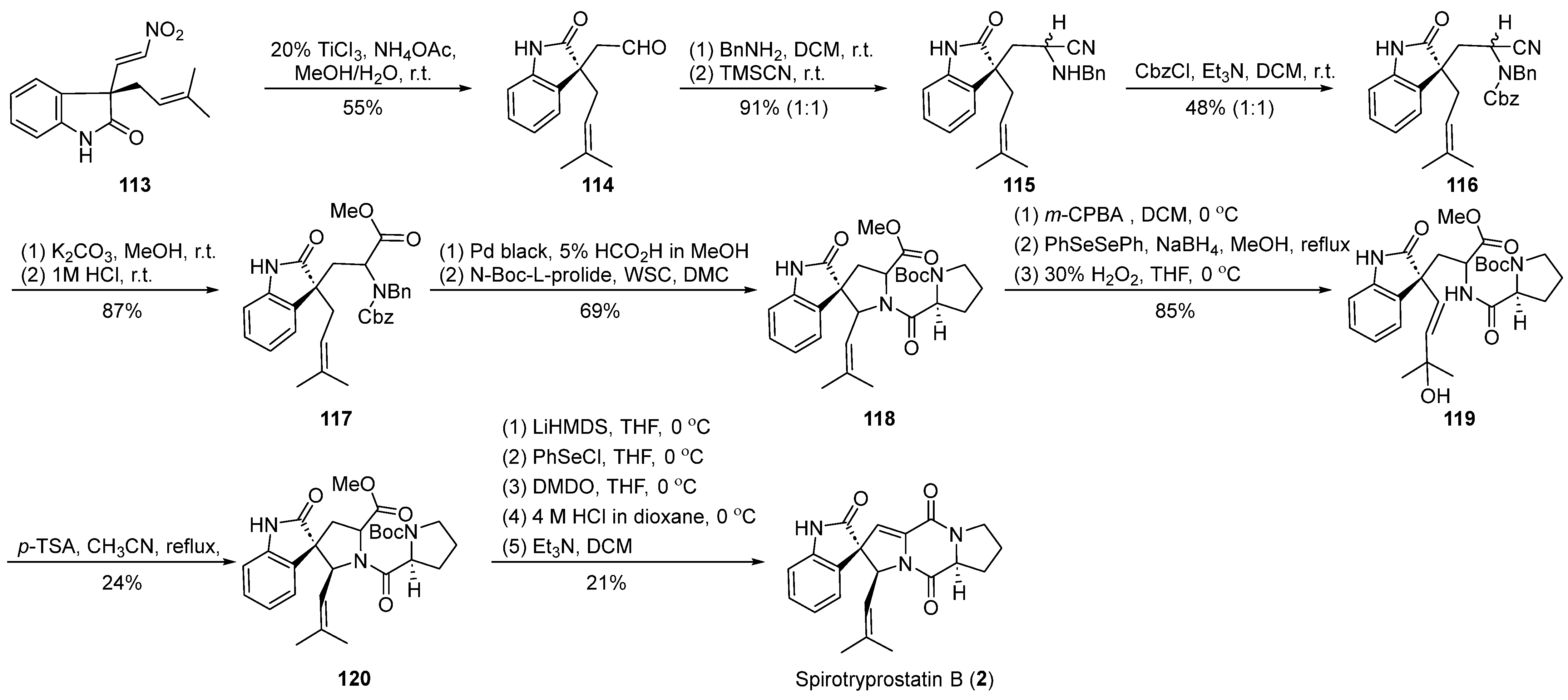
4.2.1. Fuji’s Total Synthesis
In the year 2002, Fuji et al. [61] utilized nitroalkene cyclization as a pivotal step in the synthesis of spirotryprostatin B (2) (Scheme 14). Their strategy for constructing the spirocyclic quaternary carbon was guided by the formation of a chiral auxiliary, whereby the resulting intermediate underwent functional group transformations, introducing distinct fragments and ultimately yielding the cyclization precursor 113. Under the influence of titanium trichloride and ammonium acetate, precursor 113 underwent nitroalkene transformation to yield aldehyde 114. Subsequent steps, including Strecker reaction, Cbz protection, and cyanide hydrolysis, led to the formation of a benzylamine intermediate 117 containing an ester moiety. Compound 117 was further condensed with protected L-proline to generate amide intermediate 118. Successive transformations involving epoxidation, phenylselenol addition, and oxidative cleavage resulted in the conversion to tertiary alcohol intermediate 119. The treatment of intermediate 119 with p-toluenesulfonic acid produced the anticipated spirocyclic oxindole pyrrolidine intermediate 120 with a yield of 24% and the concomitant formation of an isomer. Employing the methodology reported by Danishefsky et al., the treatment of 120 yielded the target product spirotryprostatin B (2), following a 16-step synthetic route with an overall yield of 0.6%, accompanied by a notable presence of byproducts.
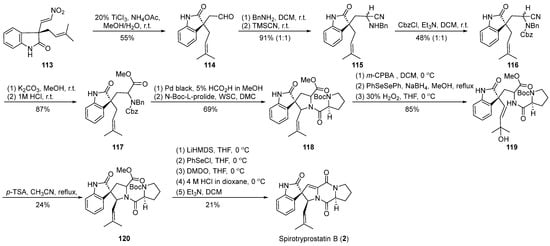
Scheme 14.
Fuji’s total synthesis of spirotryprostatin B via nitroalkene cyclization.
4.2.2. Trost’s Total Synthesis
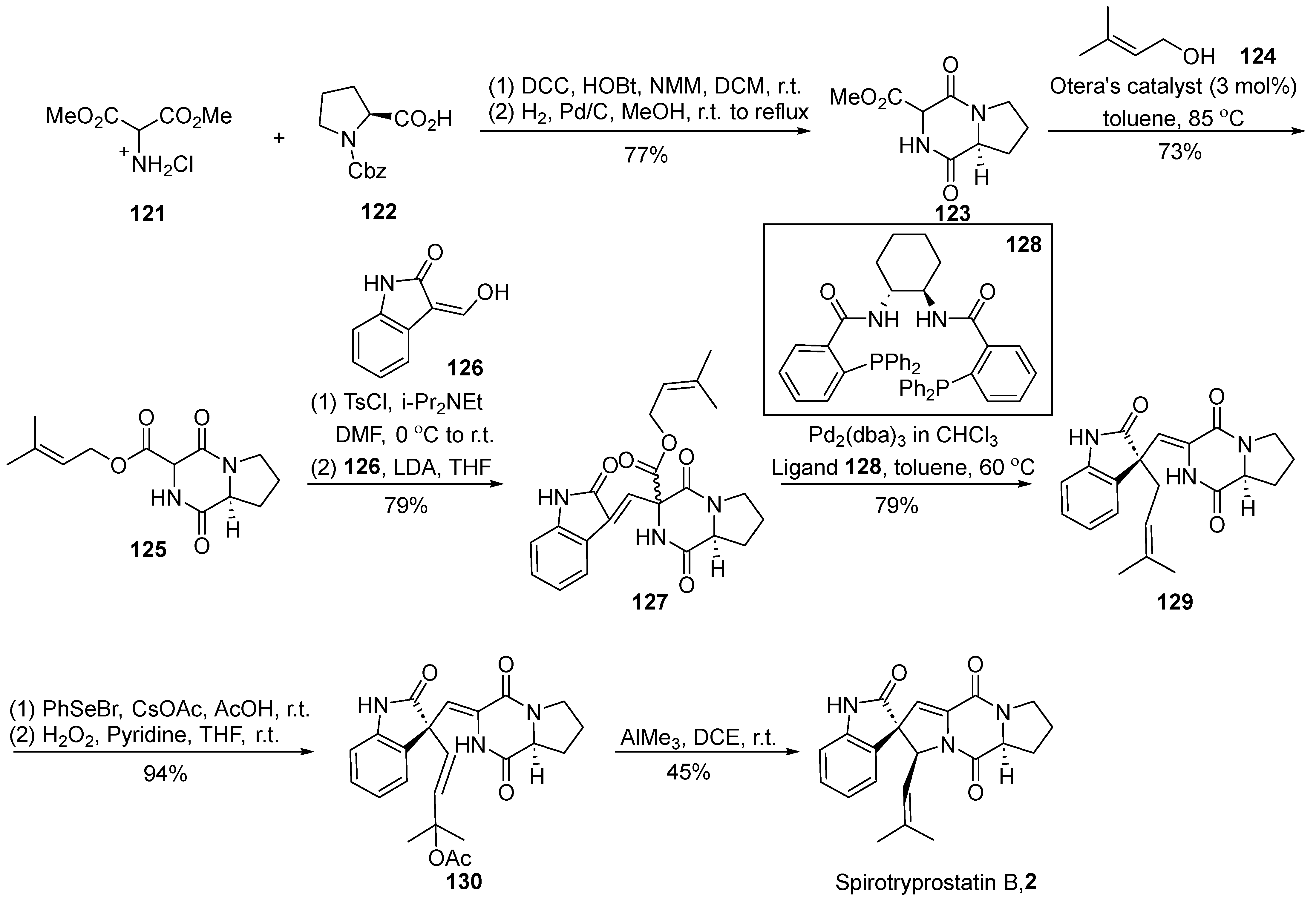
In 2007, Trost et al. [62] disclosed a comprehensive synthesis route for spirotryprostatin B (2) utilizing an efficient palladium-catalyzed asymmetric isoprene functionalization (Scheme 15). A pivotal aspect of this synthetic pathway involves the non-enantioselective isoprenylation reaction under palladium catalysis to construct the spirocyclic carbon stereocenter. Commencing with Cbz-protected L-proline 122 and hydrochloride salt of 2-amino-propane-dicarboxylic acid 121, the reaction involved an initial sequence of condensation, hydrogenation, deprotection, and subsequent condensation to yield intermediate 123. Under the catalysis of the Otera catalyst, intermediate 123 underwent alkylation to generate the diketopiperazine compound 125 bearing an isoprenyl side chain. The coupling of intermediate 125 with oxindole 126, facilitated by a one-pot strategy, produced the nucleophilic precursor compound 127. Subsequent decarboxylative isoprenyl rearrangement occurred under the joint action of a palladium catalyst and ligand 128, yielding the crucial intermediate 129. The treatment of intermediate 129 with PhSeOAc, followed by oxidative elimination using hydrogen peroxide, led to the formation of vinyl acetate 130. Ultimately, the target product was obtained through cyclization under the influence of trimethylaluminum. The entire synthetic route comprised eight steps, achieving the total synthesis of spirotryprostatin B (2) with a yield of 13.7%.
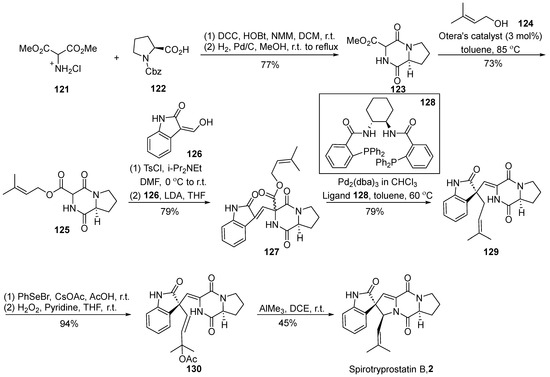
Scheme 15.
Trost’s total synthesis of spirotryprostatin B via palladium-catalyzed asymmetric isoprene functionalization.
4.3. Cascade Reactions
4.3.1. Procter’s Total Synthesis
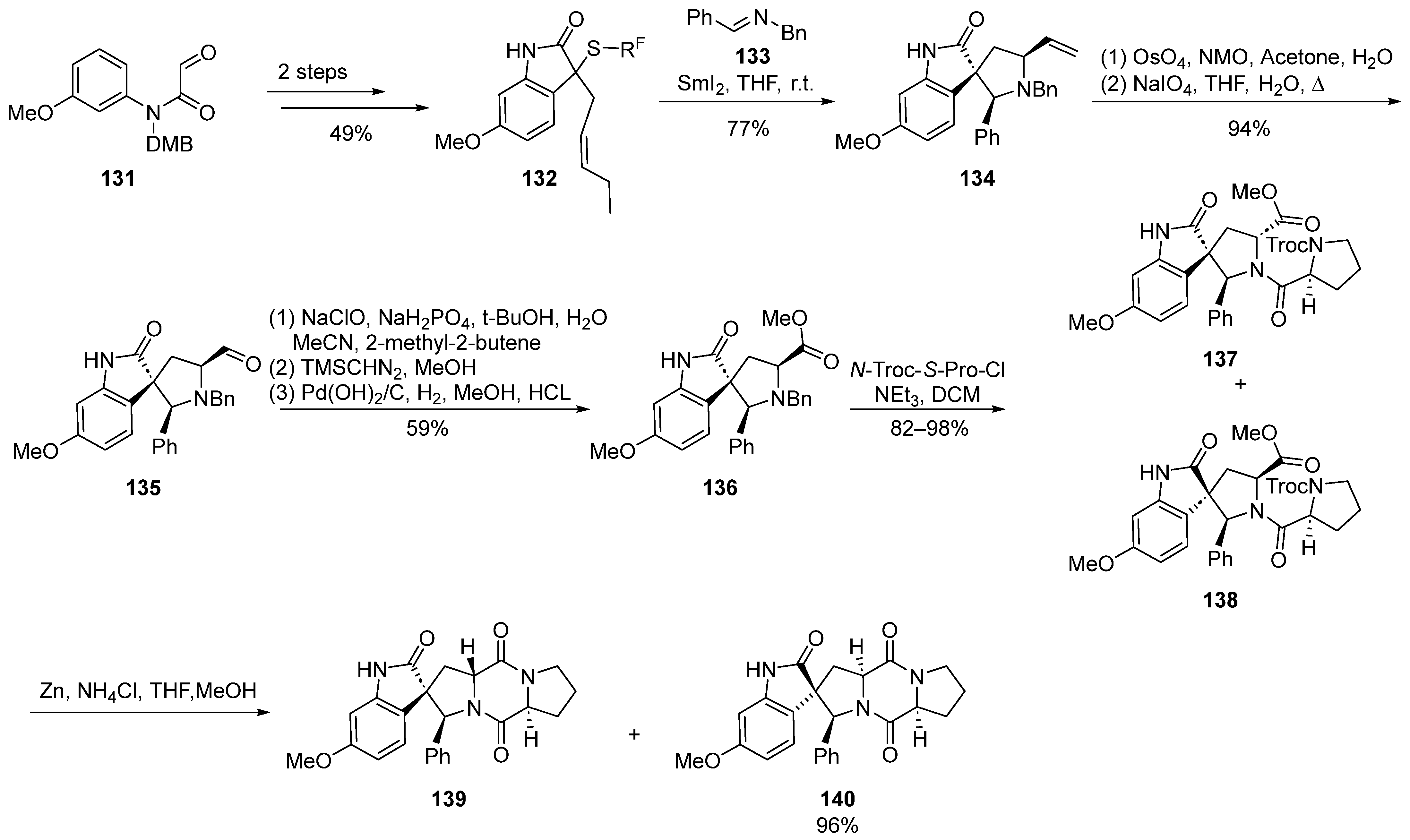
In 2011, Procter et al. [34] documented a cascade reaction triggered by iodine-induced ring formation in the synthesis of spirocyclic indolinone compounds (Scheme 16). Through extensive exploration, they delineated a concise synthetic route for the total synthesis of 18-phenyl-substituted spirotryprostatin A derivatives, spanning a comprehensive sequence of 10 steps with an overall yield ranging between 13.7% and 16.3%. The reaction commenced with amide compound 131 as the starting material, undergoing a two-step transformation to yield the indolone intermediate 132. Subsequently, a tandem reaction employing samarium iodide catalysis and imine 133 led to the formation of the spirocyclic pyrrolidine intermediate 134. The subsequent oxidative cleavage of the terminal alkene in 134 furnished aldehyde intermediate 135. Sequential oxidation, esterification, and debenzylation reactions yielded methyl ester intermediate 136, which, upon condensation with proline acyl chloride, generated the mixture of compounds 137 and 138. The subsequent deprotection of the Troc-protecting group initiated a cyclization reaction, affording two derivatives of spirotryprostatin A, 139 and 140, both featuring an 18-phenyl substitution. In the initial stages of biological activity assessment, the authors observed that compound 140 exhibited a level of bioactivity comparable to that of spirotryprostatin A (1). This finding may provide valuable insights into the structure–activity relationships governing the biological activities of spirotryprostatin-like compounds, thereby serving as a guiding reference for further investigations in this domain.
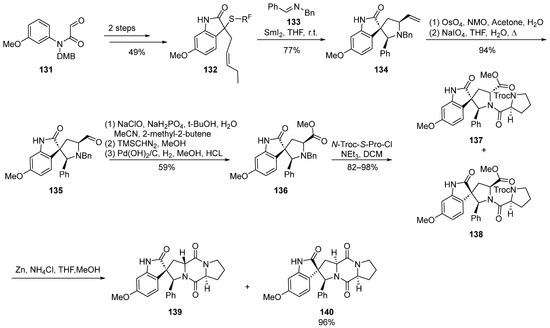
Scheme 16.
Procter’s total synthesis of spirotryprostatin alkaloids via iodine-induced cascade reactions.
4.3.2. Shen’s Total Synthesis
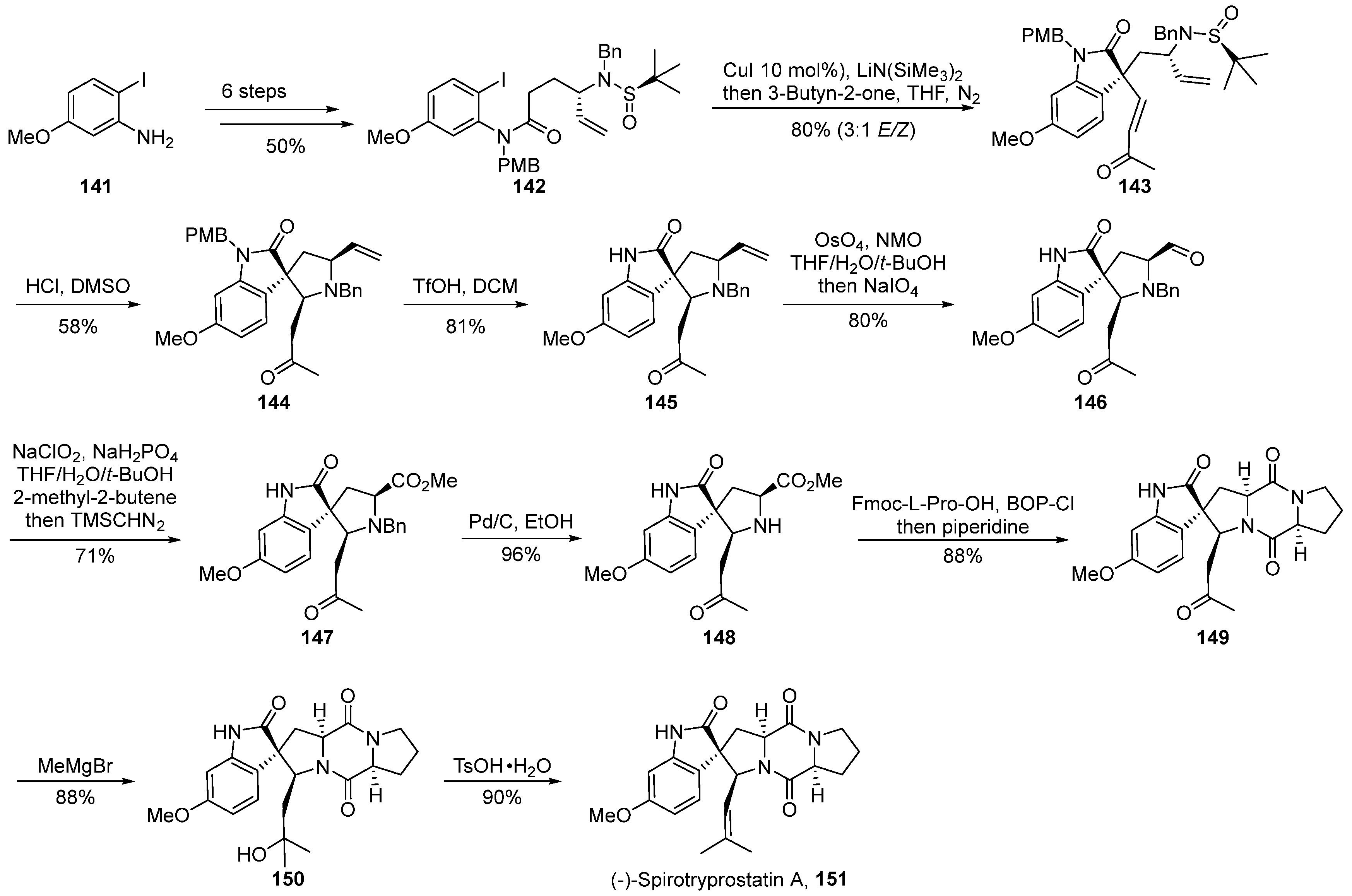
In the year 2022, Shen’s group [63] documented a strategy involving chiral auxiliary-mediated, copper-catalyzed cascade reactions for the introduction of quaternary carbon stereocenters and nitrogen-containing Michael cascade reactions (Scheme 17). This approach facilitated the asymmetric total synthesis of (-)-spirotryprostatin A 151, thereby constructing the spirocyclic pyrrolidine-indole framework. The reaction commenced with 2-iodo-5-methoxyaniline 141 as the initial substrate and underwent a concise six-step transformation to yield the ortho-iodoaniline derivative 142. Subsequently, a copper-catalyzed tandem reaction with an alkyne ketone was employed, introducing a quaternary carbon stereocenter and yielding the pivotal spirocyclic oxidized indole intermediate 143. Following this, under acidic conditions, intermediate 143 underwent removal of the chiral auxiliary and intramolecular cascade aza-Michael reaction, yielding intermediate 144. The treatment of 144 with triflic anhydride removed the protective group, and the subsequent oxidation of the olefin under the influence of osmium tetroxide/sodium periodate produced aldehyde 146. Aldehyde 146 underwent Pinnick oxidation in a buffered solution, followed by esterification with (trimethylsilyl) diazomethane to afford methyl ester intermediate 147. Intermediate 147, under palladium–carbon catalysis, underwent deprotection of the silyl group, followed by a ring-closing reaction using the improved strategy by the Danishefsky research group, leading to intermediate 149. Finally, a formaldehyde reagent addition and subsequent dehydration reactions were employed to achieve the total synthesis of the target compound (-)-spirotryprostatin A 151. The entire synthetic pathway encompassed 15 steps, yielding the final product at an overall yield of 7.4%.
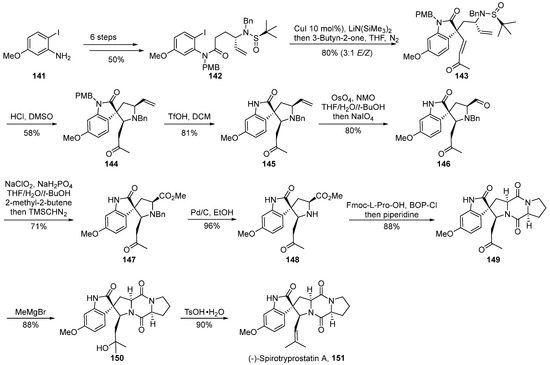
Scheme 17.
Shen’s total synthesis of spirotryprostatin alkaloids via copper-catalyzed cascade reactions.
4.4. MgI2-Catalyzed Ring Expansion
Carreira’s Total Synthesis
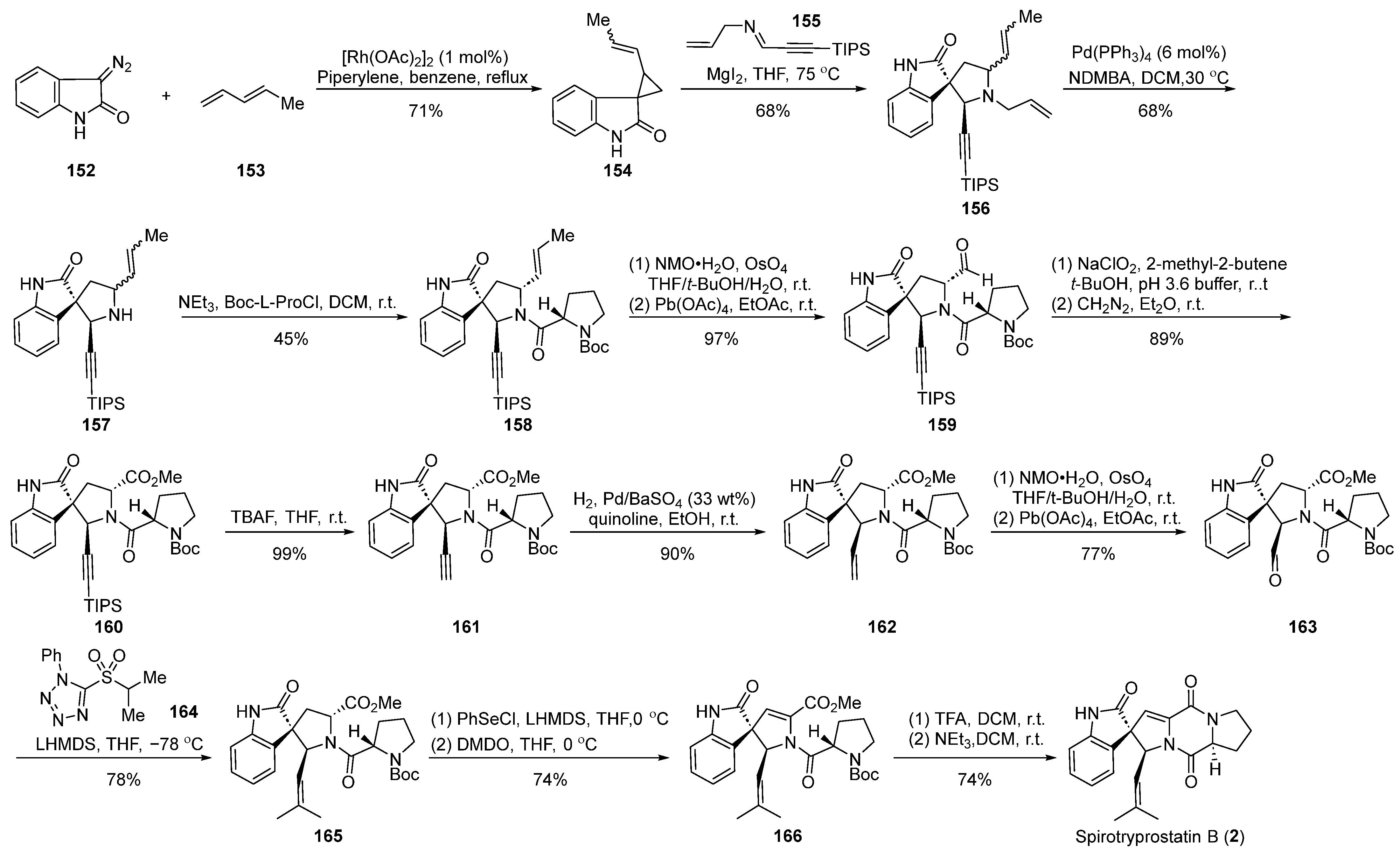
In 2003, the Carreira group reported [64] the application of magnesium iodide-catalyzed cyclization in the total synthesis of spirotryprostatin B (2) (Scheme 18). The reaction commenced with diazoindolone 152 as the starting material, which, under rhodium catalysis, underwent a cyclopropanation reaction with pentadiene 153, affording the indolone cyclopropane intermediate 154. Intermediate 154 and imine 155, through a magnesium iodide-catalyzed [1 + 3] ring addition and ring expansion reaction, yielded the spirocyclic intermediate 156. The subsequent palladium-catalyzed removal of the allyl group produced intermediate 157. A Schotten–Baumann reaction with N-Boc-L-proline acyl chloride introduced the proline five-membered ring, followed by a series of oxidation reactions to obtain aldehyde 159. Aldehyde 159, subjected to Pinnick oxidation, TIPS deprotection, and hydrogenation, yielded compound 162. Further oxidation and olefination led to the spirocyclic pyrrolidine intermediate 165, which, employing Danishefsky’s methodology, underwent a PhSeCl-mediated elimination reaction followed by Boc-deprotection and closure of the diketopiperazine ring to ultimately obtain the target product spirotryprostatin B (2). The synthetic route comprised 19 steps, with intricacies arising from isomer separations and demanding reaction conditions, contributing to a relatively low overall yield of 5.5%.
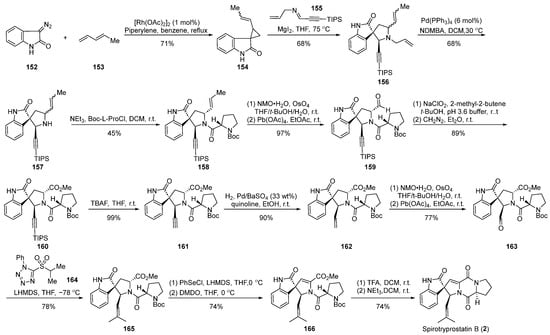
Scheme 18.
Carreira’s total synthesis of spirotryprostatin B via MgI2-catalyzed ring expansion.
5. Conclusions
To date, various synthetic methodologies have been developed for the total synthesis of spirotryprostatin alkaloids, resulting in a diverse array of spirotryprostatin compounds. However, certain approaches, such as oxidative rearrangement, have encountered challenges related to selectivity. Although some methods, such as copper- and silver-catalyzed 1,3-dipolar cycloaddition reactions, show promising potential for asymmetric synthesis, the necessity for the preparation of highly effective chiral ligands to control stereochemical issues poses a significant hurdle to achieving an efficient and concise process, requiring prolonged development efforts. Other approaches utilizing palladium, rhodium, and other metal-catalyzed methods, while capable of stereoselectively synthesizing spirocyclic oxindole diketopiperazines, often involve complex routes, low yields, and the use of expensive noble metals as catalysts, hindering their broader applicability. With the increasing discovery and isolation of spirocyclic oxindole diketopiperazine alkaloids, a continual emergence of highly selective, efficient, and concise synthetic methods is anticipated. Given the notable biological activity of spirotryprostatin compounds, an expanding repertoire of such compounds is being synthesized and applied in clinical practice. It is anticipated that, in the near future, a cohort of new drugs based on the spirotryprostatin scaffold will emerge in the field of medicine, serving as representative products in the development of novel anti-tumor drugs.
Author Contributions
The initiation of this project was spearheaded by J.-F.W., Z.-X.N. and J.H. The comprehensive gathering and systematic classification of pertinent references were diligently conducted by all authors. It is noteworthy to highlight that each author has made significant contributions to the advancement of this scholarly review. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Joint Construction Project of Henan Provincial Medical Science and Technology Key Program (Nos. LHGJ20210688 and LHGJ20220768).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cui, C.-B.; Kakeya, H.; Osada, H. Novel mammalian cell cycle inhibitors, spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron 1996, 52, 12651–12666. [Google Scholar] [CrossRef]
- Cui, C.B.; Kakeya, H.; Osada, H. Spirotryprostatin B, a Novel Mammalian Cell Cycle Inhibitor Produced by Aspergillus fumigatus. J. Antibiot. 1996, 49, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.M.C.; Neumann, C.S.; Nagayama, S.; Perlstein, E.O.; Schreiber, S.L. A Library of Spirooxindoles Based on a Stereoselective Three-Component Coupling Reaction. J. Am. Chem. Soc. 2004, 126, 16077–16086. [Google Scholar] [CrossRef] [PubMed]
- Antonchick, A.P.; Gerding-Reimers, C.; Catarinella, M.; Schürmann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Highly enantioselective synthesis and cellular evaluation of spirooxindoles inspired by natural products. Nat. Chem. 2010, 2, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. JNCI J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Elderfield, R.C.; Gilman, R.E. Alkaloids of Alstonia Muelleriana. Phytochemistry 1972, 11, 339–343. [Google Scholar] [CrossRef]
- Kornet, M.J.; Thio, A.P. Oxindole-3-spiropyrrolidines and -piperidines. Synthesis and local anesthetic activity. J. Med. Chem. 1976, 19, 892–898. [Google Scholar] [CrossRef]
- Jossang, A.; Jossang, P.; Hadi, H.A.; Sevenet, T.; Bodo, B. Horsfiline, an oxindole alkaloid from Horsfieldia superba. J. Org. Chem. 1991, 56, 6527–6530. [Google Scholar] [CrossRef]
- Pellegrini, C.; Weber, M.; Borschberg, H.-J. Total Synthesis of (+)-Elacomine and (−)-Isoelacomine, Two Hitherto Unnamed Oxindole Alkaloids from Elaeagnus commutata. Helv. Chim. Acta 1996, 79, 151–168. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, X.; Xu, T.; Yang, X.; Liu, Y. Diketopiperazines from Marine Organisms. Chem. Biodivers. 2010, 7, 2809–2829. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Kodato, S.-I.; Hongu, M.; Kawate, T.; Hino, T. Total synthesis of fumitremorgin B. Tetrahedron Lett. 1986, 27, 6217–6220. [Google Scholar] [CrossRef]
- Kodato, S.-I.; Nakagawa, M.; Hongu, M.; Kawate, T.; Hino, T. Total synthesis of (+)-fumitremorgin B, its epimeric isomers, and demethoxy derivatives. Tetrahedron 1988, 44, 359–377. [Google Scholar] [CrossRef]
- Bignan, G.C.; Battista, K.; Connolly, P.J.; Orsini, M.J.; Liu, J.; Middleton, S.A.; Reitz, A.B. Preparation of 3-spirocyclic indolin-2-ones as ligands for the ORL-1 receptor. Bioorg. Med. Chem. Lett. 2005, 15, 5022–5026. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.M.M. Recent advances in the synthesis of biologically active spirooxindoles. Tetrahedron 2014, 70, 9735–9757. [Google Scholar] [CrossRef]
- Yu, B.; Yu, D.-Q.; Liu, H.-M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, W.-L.; Fang, Y.-C.; Zhu, T.-J.; Gu, Q.-Q.; Zhu, W.-M. Cytotoxic Alkaloids and Antibiotic Nordammarane Triterpenoids from the Marine-Derived Fungus Aspergillus sydowi. J. Nat. Prod. 2008, 71, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fang, Y.; Zhu, T.; Zhang, M.; Lin, A.; Gu, Q.; Zhu, W. Seven new prenylated indole diketopiperazine alkaloids from holothurian-derived fungus Aspergillus fumigatus. Tetrahedron 2008, 64, 7986–7991. [Google Scholar] [CrossRef]
- Afiyatullov, S.S.; Zhuravleva, O.I.; Chaikina, E.L.; Anisimov, M.M. A new spirotryprostatin from the marine isolate of the fungus Aspergillus fumigatus. Chem. Nat. Compd. 2012, 48, 95–98. [Google Scholar] [CrossRef]
- Shi, Y.-S.; Zhang, Y.; Chen, X.-Z.; Zhang, N.; Liu, Y.-B. Metabolites Produced by the Endophytic Fungus Aspergillus fumigatus from the Stem of Erythrophloeum fordii Oliv. Molecules 2015, 20, 10793–10799. [Google Scholar] [CrossRef]
- Gao, N.; Shang, Z.-C.; Yu, P.; Luo, J.; Jian, K.-L.; Kong, L.-Y.; Yang, M.-H. Alkaloids from the endophytic fungus Penicillium brefeldianum and their cytotoxic activities. Chin. Chem. Lett. 2017, 28, 1194–1199. [Google Scholar] [CrossRef]
- Rajanarendar, E.; Ramakrishna, S.; Govardhan Reddy, K.; Nagaraju, D.; Reddy, Y.N. A facile synthesis, anti-inflammatory and analgesic activity of isoxazolyl-2,3-dihydrospiro[benzo[f]isoindole-1,3′-indoline]-2′,4,9-triones. Bioorg. Med. Chem. Lett. 2013, 23, 3954–3958. [Google Scholar] [CrossRef] [PubMed]
- Arun, Y.; Bhaskar, G.; Balachandran, C.; Ignacimuthu, S.; Perumal, P.T. Facile one-pot synthesis of novel dispirooxindole-pyrrolidine derivatives and their antimicrobial and anticancer activity against A549 human lung adenocarcinoma cancer cell line. Bioorg. Med. Chem. Lett. 2013, 23, 1839–1845. [Google Scholar] [CrossRef]
- Yang, Y.-T.; Zhu, J.-F.; Liao, G.; Xu, H.-J.; Yu, B. The Development of Biologically Important Spirooxindoles as New Antimicrobial Agents. Curr. Med. Chem. 2018, 25, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.R.; Chandrasekhara Rao, L.; Bangade, V.M.; Shirsat, P.K.; George, S.A.; Jagadeesh babu, N.; Meshram, H.M. A convenient and rapid microwave-assisted synthesis of spirooxindoles in aqueous medium and their antimicrobial activities. New J. Chem. 2016, 40, 2225–2232. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, L.; Sun, W.; Lu, J.; McEachern, D.; Li, X.; Yu, S.; Bernard, D.; Ochsenbein, P.; Ferey, V.; et al. Diastereomeric Spirooxindoles as Highly Potent and Efficacious MDM2 Inhibitors. J. Am. Chem. Soc. 2013, 135, 7223–7234. [Google Scholar] [CrossRef]
- O′Malley, G.J.; Cava, M.P. Tremorgenic mycotoxins: Synthesis of 6-demethoxyfumitremorgin C. Tetrahedron Lett. 1987, 28, 1131–1134. [Google Scholar] [CrossRef]
- Bertamino, A.; Aquino, C.; Sala, M.; Simone, N.d.; Mattia, C.A.; Erra, L.; Musella, S.; Iannelli, P.; Carotenuto, A.; Grieco, P.; et al. Design and synthesis of spirotryprostatin-inspired diketopiperazine systems from prolyl spirooxoindolethiazolidine derivatives. Bioorg. Med. Chem. 2010, 18, 4328–4337. [Google Scholar] [CrossRef]
- Wang, L.; Shi, X.-M.; Dong, W.-P.; Zhu, L.-P.; Wang, R. Efficient construction of highly functionalized spiro[γ-butyrolactone-pyrrolidin-3,3′-oxindole] tricyclic skeletons via an organocatalytic 1,3-dipolar cycloaddition. Chem. Commun. 2013, 49, 3458–3460. [Google Scholar] [CrossRef]
- Boddy, A.J.; Bull, J.A. Stereoselective synthesis and applications of spirocyclic oxindoles. Org. Chem. Front. 2021, 8, 1026–1084. [Google Scholar] [CrossRef]
- Mei, G.-J.; Shi, F. Catalytic asymmetric synthesis of spirooxindoles: Recent developments. Chem. Commun. 2018, 54, 6607–6621. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cobo, A.A.; Franz, A.K. Recent advances in organocatalytic asymmetric multicomponent cascade reactions for enantioselective synthesis of spirooxindoles. Org. Chem. Front. 2021, 8, 4315–4348. [Google Scholar] [CrossRef]
- Yu, Q.; Guo, P.; Jian, J.; Chen, Y.; Xu, J. Nine-step total synthesis of (−)-strychnofoline. Chem. Commun. 2018, 54, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Coote, S.C.; Quenum, S.; Procter, D.J. Exploiting Sm(ii) and Sm(iii) in SmI2-initiated reaction cascades: Application in a tag removal–cyclisation approach to spirooxindole scaffolds. Org. Biomol. Chem. 2011, 9, 5104–5108. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, L.; Liu, X.; Li, D.; Zhu, H.; Wang, P.; Liu, Y.; Yang, D.; Wang, R. Development of Biligands Magnesium Catalysis in Asymmetric Conjugate Reactions of C3-Pyrrolyl-Oxindoles. Org. Lett. 2017, 19, 4351–4354. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, S.; Danishefsky, S.J.; Sepp-Lorenzino, L.; Rosen, N. Total Synthesis of Spirotryprostatin A, Leading to the Discovery of Some Biologically Promising Analogues. J. Am. Chem. Soc. 1999, 121, 2147–2155. [Google Scholar] [CrossRef]
- Wang, H.; Ganesan, A. Concise synthesis of the cell cycle inhibitor demethoxyfumitremorgin C. Tetrahedron Lett. 1997, 38, 4327–4328. [Google Scholar] [CrossRef]
- Wang, H.; Ganesan, A. The N-Acyliminium Pictet−Spengler Condensation as a Multicomponent Combinatorial Reaction on Solid Phase and Its Application to the Synthesis of Demethoxyfumitremorgin C Analogues. Org. Lett. 1999, 1, 1647–1649. [Google Scholar] [CrossRef]
- Wang, H.; Ganesan, A. A Biomimetic Total Synthesis of (−)-Spirotryprostatin B and Related Studies. J. Org. Chem. 2000, 65, 4685–4693. [Google Scholar] [CrossRef]
- Xi, Y.K.; Zhang, H.; Li, R.X.; Kang, S.Y.; Li, J.; Li, Y. Total Synthesis of Spirotryprostatins through Organomediated Intramolecular Umpolung Cyclization. Chemistry 2019, 25, 3005–3009. [Google Scholar] [CrossRef]
- von Nussbaum, F.; Danishefsky, S.J. A Rapid Total Synthesis of Spirotryprostatin B: Proof of Its Relative and Absolute Stereochemistry. Angew. Chem. Int. Ed. 2000, 39, 2175–2178. [Google Scholar] [CrossRef]
- Miyake, F.Y.; Yakushijin, K.; Horne, D.A. Preparation and synthetic applications of 2-halotryptophan methyl esters: Synthesis of spirotryprostatin B. Angew. Chem. Int. Ed. 2004, 43, 5357–5360. [Google Scholar] [CrossRef] [PubMed]
- Miyake, F.Y.; Yakushijin, K.; Horne, D.A. A Concise Synthesis of Spirotryprostatin A. Org. Lett. 2004, 6, 4249–4251. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Cramer, N.; Silverman, S.M. Enantioselective Construction of Spirocyclic Oxindolic Cyclopentanes by Palladium-Catalyzed Trimethylenemethane-[3 + 2]-Cycloaddition. J. Am. Chem. Soc. 2007, 129, 12396–12397. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-H.; Zhang, W.-Q.; Gong, L.-Z. Asymmetric Organocatalytic Three-Component 1,3-Dipolar Cycloaddition: Control of Stereochemistry via a Chiral Brønsted Acid Activated Dipole. J. Am. Chem. Soc. 2008, 130, 5652–5653. [Google Scholar] [CrossRef]
- Chen, X.-H.; Wei, Q.; Luo, S.-W.; Xiao, H.; Gong, L.-Z. Organocatalytic Synthesis of Spiro[pyrrolidin-3,3′-oxindoles] with High Enantiopurity and Structural Diversity. J. Am. Chem. Soc. 2009, 131, 13819–13825. [Google Scholar] [CrossRef]
- Liu, T.-L.; Xue, Z.-Y.; Tao, H.-Y.; Wang, C.-J. Catalytic asymmetric 1,3-dipolar cycloaddition of N-unprotected 2-oxoindolin-3-ylidene derivatives and azomethine ylides for the construction of spirooxindole-pyrrolidines. Org. Biomol. Chem. 2011, 9, 1980–1986. [Google Scholar] [CrossRef]
- Fejes, I.; Nyerges, M.; Szöllõsy, Á.; Blaskó, G.; Tõke, L. 2-Oxoindolin-3-ylidene derivatives as 2π components in 1,3-dipolar cycloadditions of azomethine ylides. Tetrahedron 2001, 57, 1129–1137. [Google Scholar] [CrossRef]
- Ding, K.; Wang, G.; Deschamps, J.R.; Parrish, D.A.; Wang, S. Synthesis of spirooxindoles via asymmetric 1,3-dipolar cycloaddition. Tetrahedron Lett. 2005, 46, 5949–5951. [Google Scholar] [CrossRef]
- Zubia, A.; Mendoza, L.; Vivanco, S.; Aldaba, E.; Carrascal, T.; Lecea, B.; Arrieta, A.; Zimmerman, T.; Vidal-Vanaclocha, F.; Cossío, F.P. Application of Stereocontrolled Stepwise [3 + 2] Cycloadditions to the Preparation of Inhibitors of α4β1-Integrin-Mediated Hepatic Melanoma Metastasis. Angew. Chem. Int. Ed. 2005, 44, 2903–2907. [Google Scholar] [CrossRef]
- Yang, Q.-L.; Xie, M.-S.; Xia, C.; Sun, H.-L.; Zhang, D.-J.; Huang, K.-X.; Guo, Z.; Qu, G.-R.; Guo, H.-M. A rapid and divergent access to chiral azacyclic nucleoside analogues via highly enantioselective 1,3-dipolar cycloaddition of β-nucleobase substituted acrylates. Chem. Commun. 2014, 50, 14809–14812. [Google Scholar] [CrossRef] [PubMed]
- Sebahar, P.R.; Williams, R.M. The Asymmetric Total Synthesis of (+)- and (−)-Spirotryprostatin B. J. Am. Chem. Soc. 2000, 122, 5666–5667. [Google Scholar] [CrossRef]
- Sebahar, P.R.; Osada, H.; Usui, T.; Williams, R.M. Asymmetric, stereocontrolled total synthesis of (+) and (−)-spirotryprostatin B via a diastereoselective azomethine ylide [1,3]-dipolar cycloaddition reaction. Tetrahedron 2002, 58, 6311–6322. [Google Scholar] [CrossRef]
- Onishi, T.; Sebahar, P.R.; Williams, R.M. Concise, Asymmetric Total Synthesis of Spirotryprostatin A. Org. Lett. 2003, 5, 3135–3137. [Google Scholar] [CrossRef]
- Onishi, T.; Sebahar, P.R.; Williams, R.M. Concise, asymmetric total synthesis of spirotryprostatin A. Tetrahedron 2004, 60, 9503–9515. [Google Scholar] [CrossRef]
- Antonchick, A.P.; Schuster, H.; Bruss, H.; Schürmann, M.; Preut, H.; Rauh, D.; Waldmann, H. Enantioselective synthesis of the spirotryprostatin A scaffold. Tetrahedron 2011, 67, 10195–10202. [Google Scholar] [CrossRef]
- Cheng, M.-N.; Wang, H.; Gong, L.-Z. Asymmetic Organocatalytic 1,3-Dipolar Cycloaddition of Azomethine Ylide to Methyl 2-(2-Nitrophenyl)acrylate for the Synthesis of Diastereoisomers of Spirotryprostatin A. Org. Lett. 2011, 13, 2418–2421. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, W.; Liu, S.; Zou, T.; Zhang, K.; Gong, C.; Guo, W.; Kong, F.; Nie, L.; Hu, S.; et al. Highly Stereodivergent Synthesis of Chiral C4-Ester-Quaternary Pyrrolidines: A Strategy for the Total Synthesis of Spirotryprostatin A. Org. Lett. 2023, 25, 3391–3396. [Google Scholar] [CrossRef]
- Overman, L.E.; Rosen, M.D. Total Synthesis of (-)-Spirotryprostatin B and Three Stereoisomers. Angew. Chem. Int. Ed. 2000, 39, 4596–4599. [Google Scholar] [CrossRef]
- Kitahara, K.; Shimokawa, J.; Fukuyama, T. Stereoselective synthesis of spirotryprostatin A. Chem. Sci. 2014, 5, 904–907. [Google Scholar] [CrossRef]
- Bagul, T.D.; Lakshmaiah, G.; Kawabata, T.; Fuji, K. Total Synthesis of Spirotryprostatin B via Asymmetric Nitroolefination. Org. Lett. 2002, 4, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Stiles, D.T. Total Synthesis of Spirotryprostatin B via Diastereoselective Prenylation. Org. Lett. 2007, 9, 2763–2766. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Liu, T.; Zhao, J.; Dong, J.; Zhao, Y.; Yang, Y.; Yan, X.; Xu, W.; Shen, X. Enantioselective Total Synthesis of Spirotryprostatin A. J. Org. Chem. 2022, 87, 16743–16754. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Carreira, E.M. Total Synthesis of (−)-Spirotryprostatin B. Angew. Chem. Int. Ed. 2003, 42, 694–696. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).