
Chemoselective Synthesis and Anti-Kinetoplastidal Properties of 2,6-Diaryl-4H-tetrahydro-thiopyran-4-one S-Oxides: Their Interplay in a Cascade of Redox Reactions from Diarylideneacetones

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Results
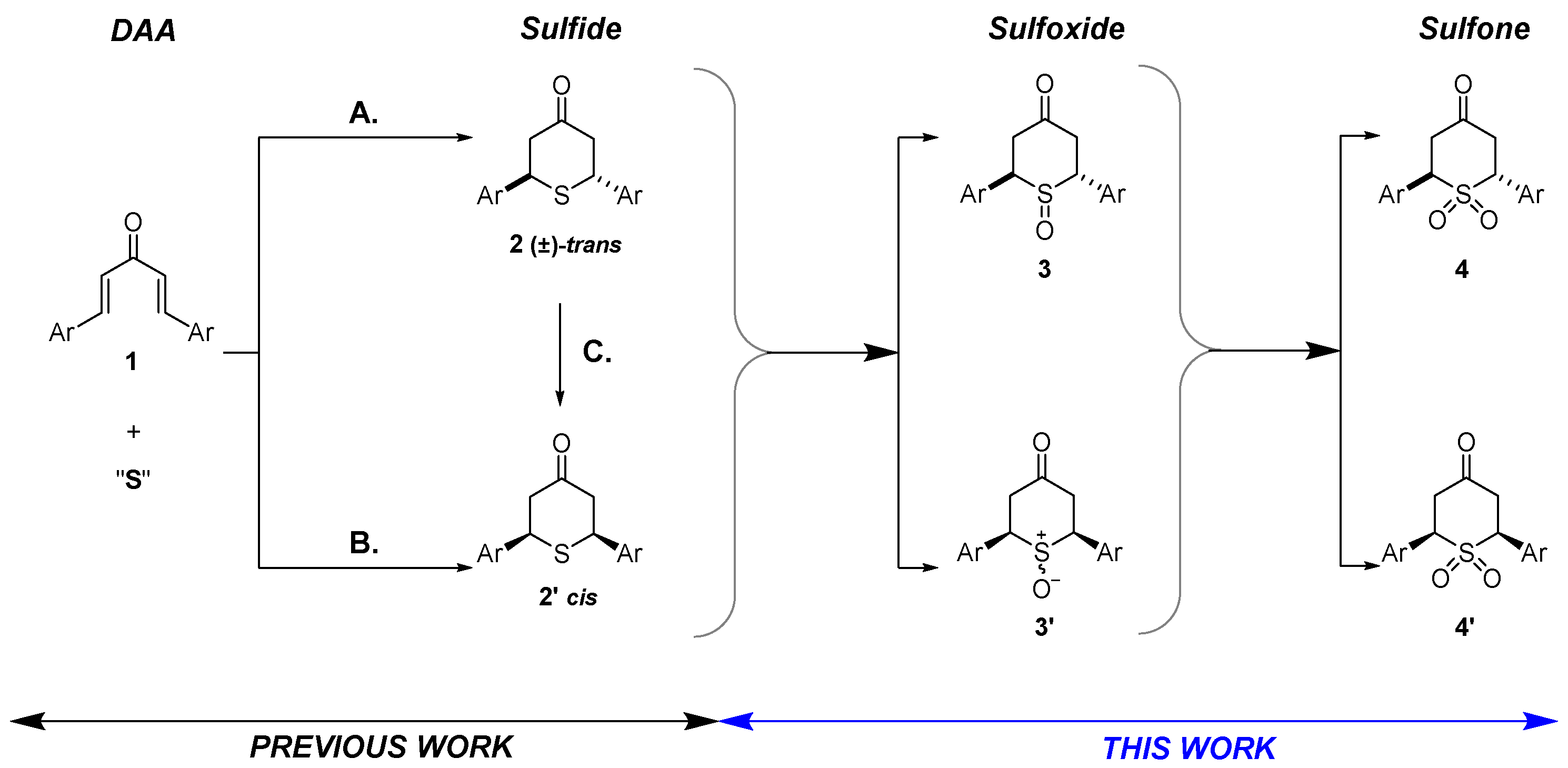
2.1. Optimized Synthesis of Starting Symmetrical and Unsymmetrical Diarylideneacetones 1 and 1′ and Symmetrical (±)-trans and cis-2,6-Diaryl-4H-tetrahydrothiopyran-4-ones 2 and 2′
2.2. Synthesis of 2,6-Diaryl-4H-tetrahydrothiopyran-4-one S-Oxides
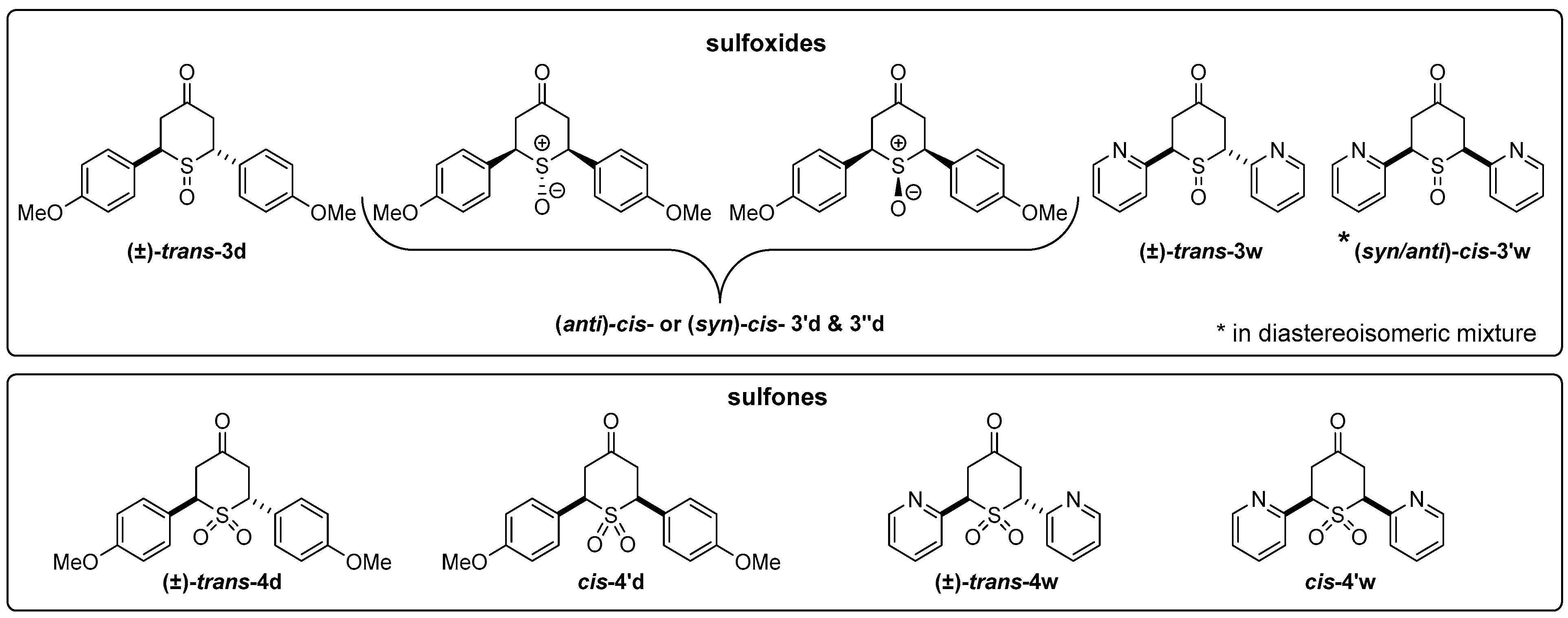
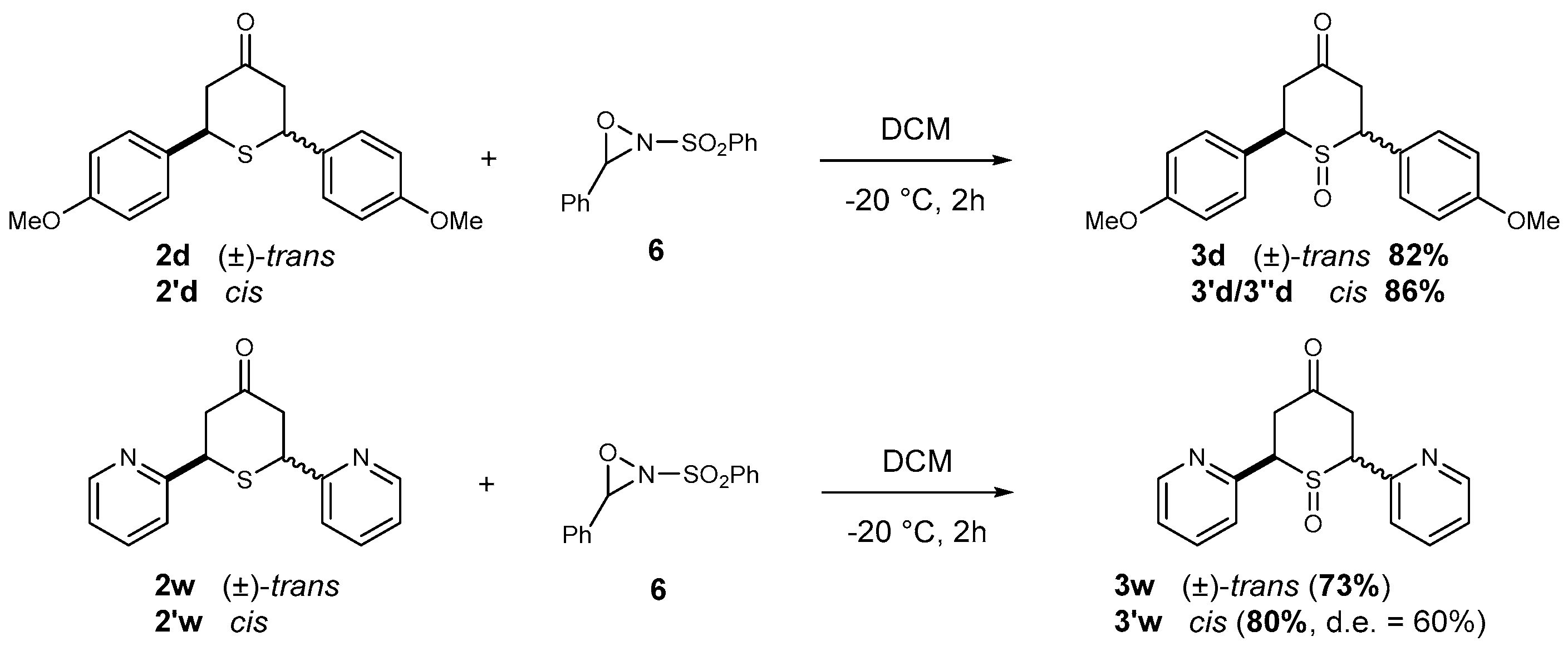
2.2.1. Synthesis of 2,6-DA-4-THTP Sulfoxide Derivatives 3–3′
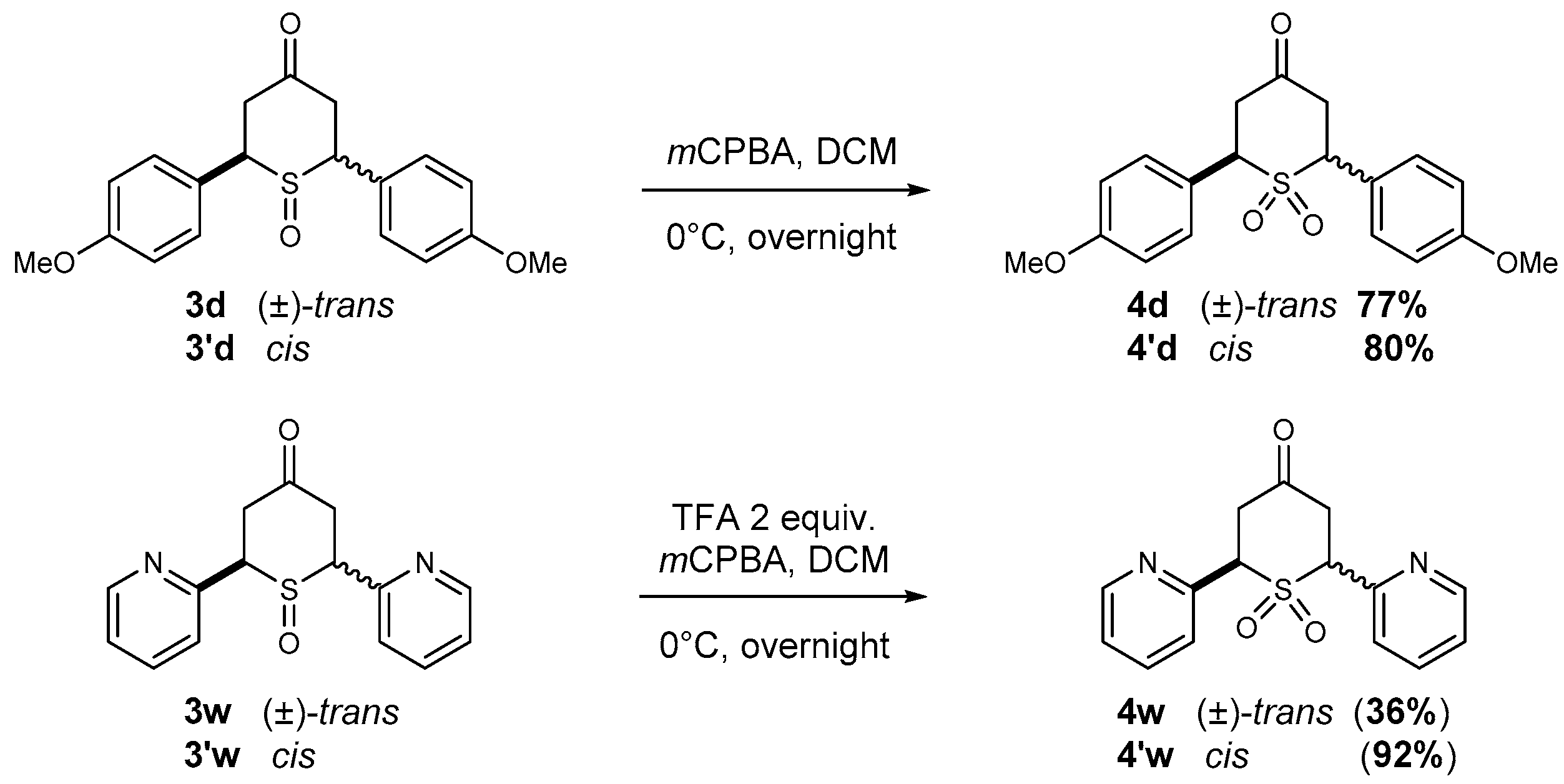
2.2.2. Synthesis of 2,6-DA-4-THTP Sulfone Derivatives 4–4′
2.3. Anti-Kinetoplastid Activities and Cytotoxicity
2.3.1. Primary Evaluation of Diarylideneacetones and 2,6-Diaryltetrahydrothiopyran-4-ones
2.3.2. Primary Evaluation of 2,6-Diaryltetrahydrothiopyran-4-one S-Oxides
2.3.3. Secondary Antileishmanial Evaluation of Diarylideneacetones, 2,6-Diaryltetrahydrothiopyran-4-ones, and Their Related S-Oxides
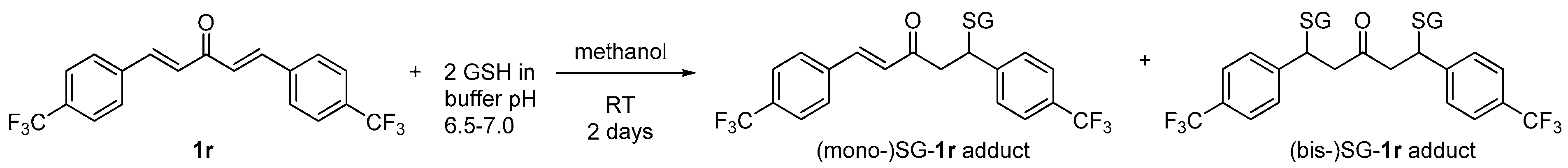
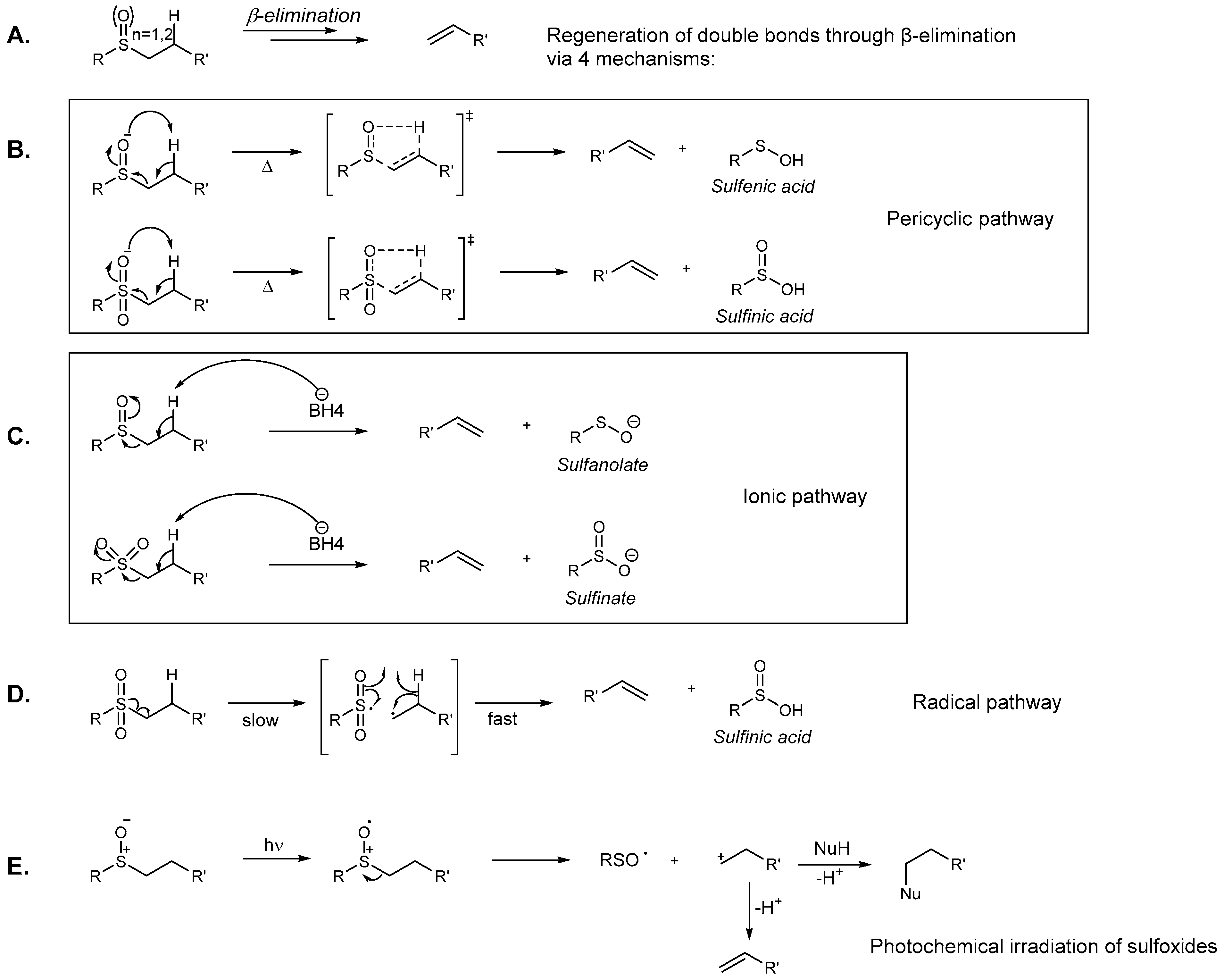
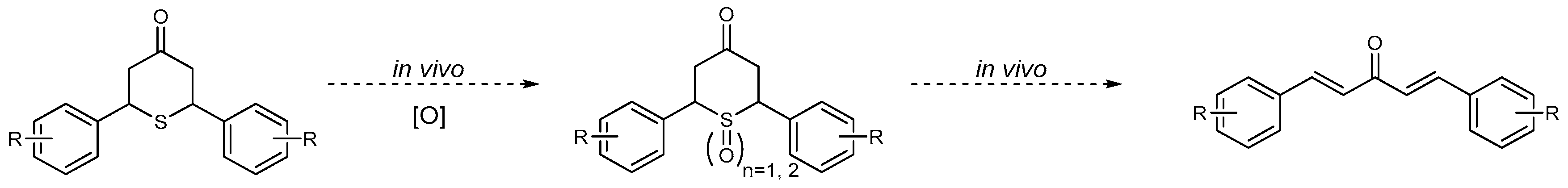
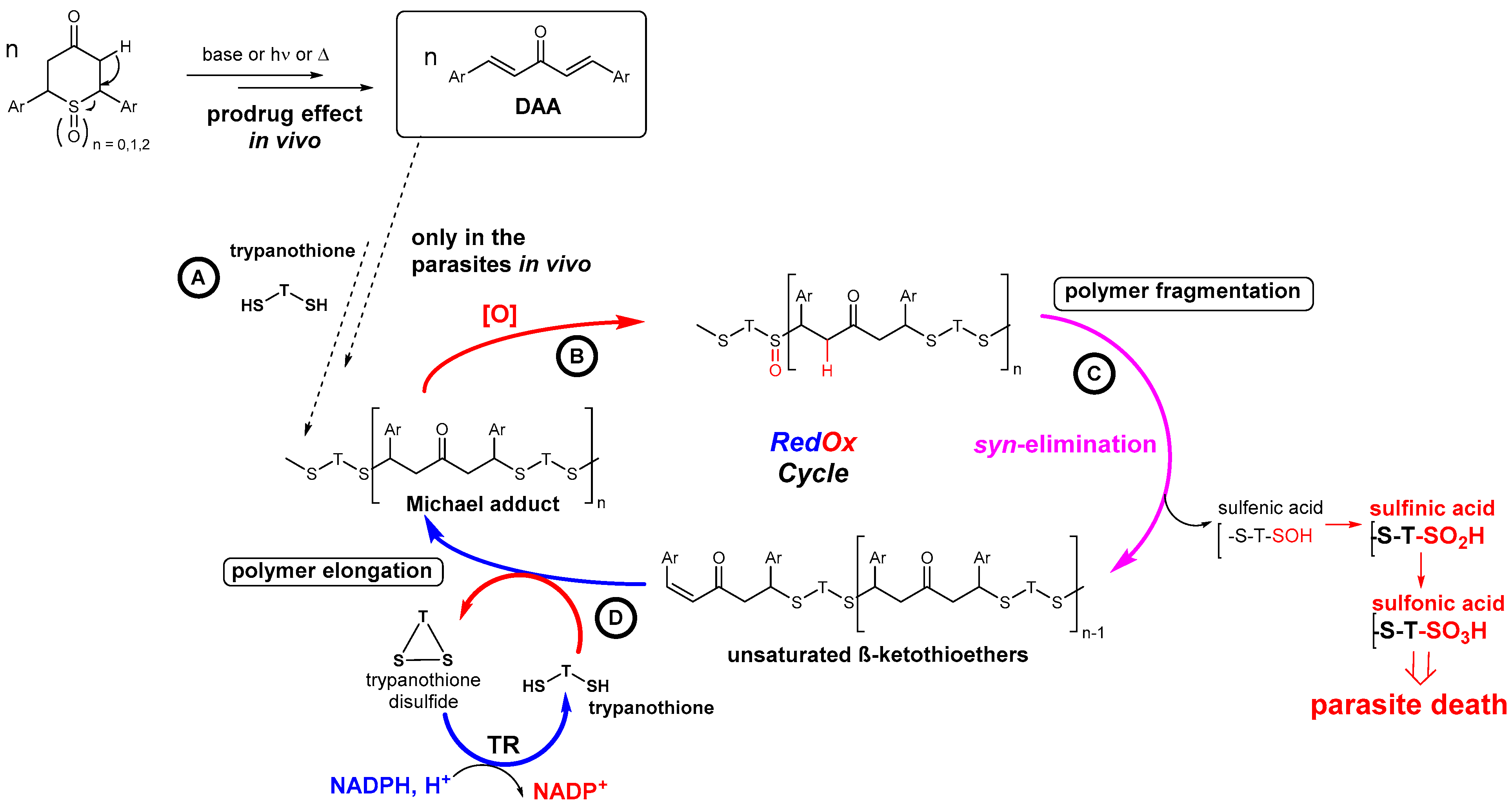
2.3.4. Putative Mode-of-Action through a Cascade of Redox Reactions
3. Discussion
3.1. Dissociation of Toxicity from Trypanocidal Activity
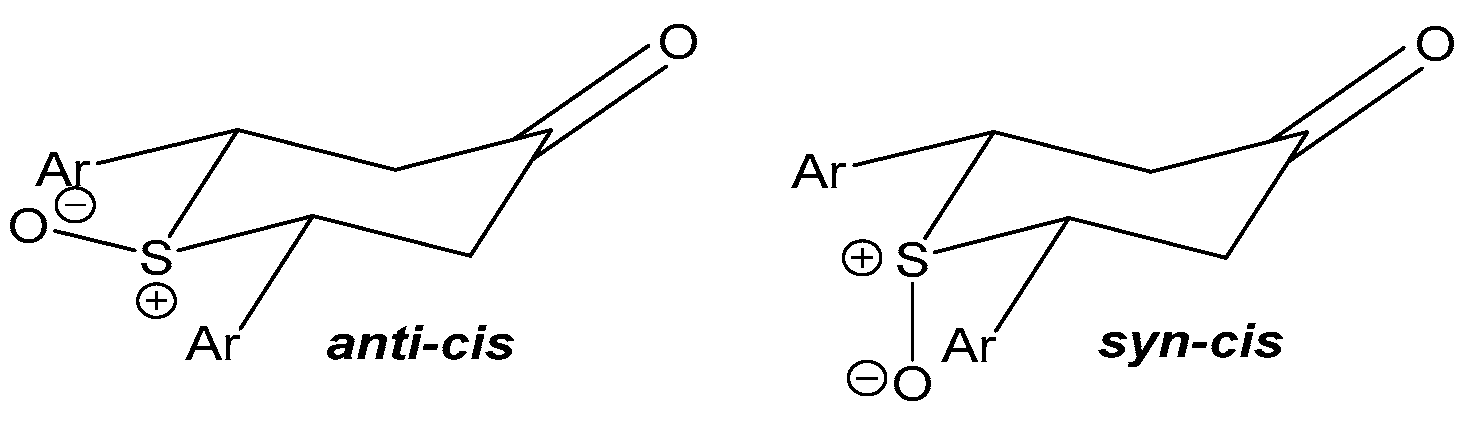
3.2. Effects of the Stereochemical Configuration
3.3. Effects of Sulfur Oxidation
4. Materials and Methods
4.1. Chemistry: General
4.2. Synthesis of 2,6-Diaryl-4H-tetrahydrothiopyran-4-one Sulfoxide and Sulfone Derivatives
4.2.1. Synthesis of “Davis’s Oxaziridine” Reagent
| N-benzylidenebenzenesulfonamide (5) Chemical Formula: C13H11NO2S Molecular Weight: 245.30 g·mol−1 |  |
| 3-Phenyl-2-(phenylsulfonyl)-1,2-oxaziridine (6), the so-called “Davis’s oxaziridine” Chemical Formula: C13H11NO3S Molecular Weight: 261.30 g·mol−1 |  |
4.2.2. Synthesis of 2,6-Diaryl-4H-tetrahydrothiopyran-4-one Sulfoxide Derivatives 3–3′
General Procedure I for the Synthesis of Sulfoxide Derivatives
| (±)-trans-2,6-Di-(p-anisyl)-4H-tetrahydrothiopyran-4-one 1-oxide (3d) Chemical Formula: C19H20O4S Molecular Weight: 344.42 g·mol−1 |  |
| (anti)- and (syn)-cis-2,6-Di-(p-anisyl)-4H-tetrahydro-thiopyran-4-one 1-oxides (3′d and 3″d) Chemical Formula: C19H20O4S Molecular Weight: 344.42 g·mol−1 |  |
- Major diastereoisomer:
- Minor diastereoisomer:
| (±)-trans-2,6-Di-(pyridin-2-yl)-4H-tetrahydrothiopyran-4-one 1-oxide (3s) Chemical Formula: C15H14N2O2S Molecular Weight: 286.35 g·mol−1 |  |
| (syn/anti)-cis-2,6-Di-(pyridin-2-yl)-4H-tetra-hydrothiopyran-4-one 1-oxide, as diastereoisomeric mixture (3′s) Chemical Formula: C15H14N2O2S Molecular Weight: 286.35 g.mol−1 |  |
- Major diastereoisomer (80 mol%)
- Minor diastereoisomer (20 mol%)
4.2.3. Synthesis of 2,6-Diaryl-4H-tetrahydrothiopyran-4-ones Sulfone Derivatives 4–4′
General Procedure J for the Synthesis of Sulfone Derivatives
| (±)-trans-2,6-Di-(p-anisyl)-4H-tetrahydrothiopyran-4-one 1,1-dioxide (4d) Chemical Formula: C19H20O5S Molecular Weight: 360.42 g·mol−1 |  |
| cis-2,6-Di-(p-anisyl)-4H-tetrahydrothiopyran-4-one 1,1-dioxide (4′d) Chemical Formula: C19H20O5S Molecular Weight: 360.42 g.mol−1 |  |
| (±)-trans-2,6-Di-(pyridin-2-yl)-4H-tetrahydrothiopyran-4-one 1,1-dioxide (4w) Chemical Formula: C15H14N2O3S Molecular Weight: 302.35 g.mol−1 |  |
| cis-2,6-Di-(pyridin-2-yl)-4H-tetrahydrothiopyran-4-one 1,1-dioxide (4′w) Chemical Formula: C15H14N2O3S Molecular Weight: 302.35 g.mol−1 |  |
4.3. Biological Assays
4.3.1. Assays from the University of Paris-Saclay (Prof. P. Loiseau, BioCIS, UMR 8076 CNRS)
L. donovani (MHOM/ET/67/HU3)—Promastigote Stage
L. donovani (MHOM/ET/67/HU3)—Amastigote Stage
4.3.2. Assays from the University of Antwerp (Prof. L. Maes)
Trypanosoma brucei brucei
Trypanosoma cruzi
Leishmania spp.
Toxicity
5. Conclusions
6. Patent
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2018, 15, 217–231, Erratum in Nat. Rev. Microbiol. 2018, 16, 714. [Google Scholar] [CrossRef] [PubMed]
- Fairlamb, A.H.; Cerami, A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992, 46, 695–729. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.; Lanfranchi, D.A.; Davioud-Charvet, E. Redox-active agents in reactions involving the trypanothione/trypanothione reductase-based system to combat kinetoplastidal parasites. In Trypanosomatid Diseases: Molecular Routes to Drug Discovery; Flohė, L., Jäger, T., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; Volume 4, Chapter 22; pp. 405–429. ISBN 978-3-527-32731-7. [Google Scholar]
- Davioud-Charvet, E.; McLeish, M.J.; Veine, D.; Giegel, D.; Arscott, L.D.; Andricopulo, A.D.; Becker, K.; Müller, S.; Schirmer, R.H.; Williams, C.H.; et al. Mechanism-based inactivation of thioredoxin reductase from Plasmodium falciparum by Mannich bases. Implication for cytotoxicity. Biochemistry 2003, 42, 13319–13330. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Bauer, H.; Melcher, J.; Ruppert, T.; Rattray, L.; Yardley, V.; Davioud-Charvet, E.; Krauth-Siegel, R.L. Irreversible inhibition of trypanothione reductase by unsaturated Mannich bases: A divinylketone as key intermediate. J. Med. Chem. 2005, 48, 7400–7410. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, I.N.; Wong, P.E.; Maes, L.; Müller, T.J.; Krauth-Siegel, R.L.; Barrett, M.P.; Davioud-Charvet, E. Unsaturated Mannich bases active against multidrug-resistant Trypanosoma brucei brucei strains. ChemMedChem 2009, 4, 339–351. [Google Scholar] [CrossRef]
- Martín-Escolano, R.; Moreno-Viguri, E.; Santivañez-Veliz, M.; Martin-Montes, A.; Medina-Carmona, E.; Paucar, R.; Marín, C.; Azqueta, A.; Cirauqui, N.; Pey, A.L.; et al. Second Generation of Mannich Base-Type Derivatives with in Vivo Activity against Trypanosoma cruzi. J. Med. Chem. 2018, 61, 5643–5663. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Roy, A.; Dutta, N.; Majumder, H.K. Reactive oxygen species and imbalance of calcium homeostasis contributes to curcumin induced programmed cell death in Leishmania donovani. Apoptosis 2008, 13, 867–882. [Google Scholar] [CrossRef]
- Lazarin-Bidóia, D.; Desoti, V.C.; Martins, S.C.; Ribeiro, F.M.; Ud Din, Z.; Rodrigues-Filho, E.; Ueda-Nakamura, T.; Nakamura, C.V.; de Oliveira Silva, S. Dibenzylideneacetones are potent trypanocidal compounds that affect the Trypanosoma cruzi redox system. Antimicrob. Agents Chemother. 2015, 60, 890–903. [Google Scholar] [CrossRef]
- Chauhan, I.S.; Rao, G.S.; Shankar, J.; Chauhan, L.K.S.; Kapadia, G.J.; Singh, N. Chemoprevention of Leishmaniasis: In-vitro antiparasitic activity of dibenzalacetone, a synthetic curcumin analog leads to apoptotic cell death in Leishmania donovani. Parasitol. Int. 2018, 67, 627–636. [Google Scholar] [CrossRef]
- de Paula, J.C.; Bakoshi, A.B.K.; Lazarin-Bidóia, D.; Ud Din, Z.; Rodrigues-Filho, E.; Ueda-Nakamura, T.; Nakamura, C.V. Antiproliferative activity of the dibenzylideneacetone derivate (E)-3-ethyl-4-(4-nitrophenyl)but-3-en-2-one in Trypanosoma cruzi. Acta Trop. 2020, 211, 105653. [Google Scholar] [CrossRef]
- Changtam, C.; de Koning, H.P.; Ibrahim, H.; Sajid, M.S.; Gould, M.K.; Suksamrarn, A. Curcuminoid analogs with potent activity against Trypanosoma and Leishmania species. Eur. J. Med. Chem. 2010, 45, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Suarez, J.A.Q.; Maria, D.A.; Rando, D.G.; Martins, C.A.S.; Pardi, P.C.; De, S. Methods to Prepare Penta-1,4-dien-3-ones and Substituted Cyclohexanones and Derivatives with Antitumoral and Antiparasitic Properties, the Compounds and Their Uses. PCT Patent WO2008003155A2, 1 October 2008. [Google Scholar]
- Wenzel, I.N.; Müller, T.J.J.; Hanquet, G.; Lanfranchi, D.A.; Leroux, F.; Gendron, T.; Davioud-Charvet, E. Dibenzylidene- and Heteroarylideneacetone Derivatives as Kinetoplastideae Parasiticides and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Trypanosomiasis and Leishmaniasis. World Intellectual Property Organization. WO2011033115 A2, 24 March 2011. [Google Scholar]
- Alkhaldi, A.A.; Creek, D.J.; Ibrahim, H.; Kim, D.H.; Quashie, N.B.; Burgess, K.E.; Changtam, C.; Barrett, M.P.; Suksamrarn, A.; de Koning, H.P. Potent trypanocidal curcumin analogs bearing a monoenone linker motif act on Trypanosoma brucei by forming an adduct with trypanothione. Mol. Pharmacol. 2015, 87, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Francisco, K.R.; Monti, L.; Yang, W.; Park, H.; Liu, L.J.; Watkins, K.; Amarasinghe, D.K.; Nalli, M.; Roberto Polaquini, C.; Regasini, L.O.; et al. Structure-activity relationship of dibenzylideneacetone analogs against the neglected disease pathogen, Trypanosoma brucei. Bioorganic Med. Chem. Lett. 2023, 81, 129123. [Google Scholar] [CrossRef] [PubMed]
- Wenzel Nicole, I. Synthesis and Mechanism of Antiparasitic Mannich Base Derivatives Affecting the Redox Equilibrium of Trypanosomes and Malaria Parasites. Ph.D. Thesis, Heidelberg University, Heidelberg, Germany, 21 September 2009. [Google Scholar]
- Gendron, T. Synthesis and Evaluation of the Antiparasitic Activity of Diarylideneacetones and Their Related Thiopyranone and S-Oxide Prodrugs. Ph.D. Thesis, Strasbourg University, Strasbourg, France, 23 November 2012. [Google Scholar]
- Gendron, T.; Kessedjian, H.; Davioud-Charvet, E.; Lanfranchi, D.A. Diastereoselective synthesis of 2,6-diaryltetrahydrothiopyran-4-ones by phase transfer catalysis. Eur. J. Org. Chem. 2015, 2015, 1790–1796. [Google Scholar] [CrossRef]
- Chaykovsky, M.; Lin, M.; Rosowsky, A.; Modest, E.J. 2,4-Diaminothieno(2,3-d)pyrimidines as antifolates and antimalarials. 2. Synthesis of 2,4-diaminopyrido(4′,3′:4,5)thieno(2,3-d)pyrimidines and 2,4-diamino-8H-thiopyrano(4′,3′:4,5)thieno(2,3-d)pyrimidines. J. Med. Chem. 1973, 16, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Viira, B.; Gendron, T.; Lanfranchi, D.A.; Cojean, S.; Horvath, D.; Marcou, G.; Varnek, A.; Maes, L.; Maran, U.; Loiseau, P.M.; et al. In Silico mining for antimalarial structure-activity knowledge and discovery of novel antimalarial curcuminoids. Molecules 2016, 21, 853. [Google Scholar] [CrossRef]
- Giri, P.; Gupta, L.; Naidu, S.; Joshi, V.; Patel, N.; Giri, S.; Srinivas, N.R. In Vitro Drug-Drug Interaction Potential of Sulfoxide and/or Sulfone Metabolites of Albendazole, Triclabendazole, Aldicarb, Methiocarb, Montelukast and Ziprasidone. Drug Metab. Lett. 2018, 12, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazué, G.; Bray, M.A.; Pécoul, B. Fexinidazole—A new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4, e923. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Bray, M.A.; Cal, M.; Bourdin Trunz, B.; Torreele, E.; Brun, R. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 2011, 55, 5602–5608. [Google Scholar] [CrossRef]
- Guignard, R.F.; Petit, L.; Zard, S.Z. A method for the net contra-thermodynamic isomerization of cyclic trisubstituted alkenes. Org. Lett. 2013, 15, 4178–4181. [Google Scholar] [CrossRef]
- Fox, B.M.; Vroman, J.A.; Fanwick, P.E.; Cushman, M. Preparation and evaluation of sulfide derivatives of the antibiotic brefeldin a as potential prodrug candidates with enhanced aqueous solubilities. J. Med. Chem. 2001, 44, 3915–3924. [Google Scholar] [CrossRef] [PubMed]
- Cubbage, J.W.; Vos, B.W.; Jenks, W.S. Ei Elimination: An Unprecedented Facet of Sulfone Chemistry. J. Am. Chem. Soc. 2000, 122, 20, 4968–4971. [Google Scholar] [CrossRef]
- Cubbage, J.W.; Tetzlaff, T.A.; Groundwater, H.; McCulla, R.D.; Nag, M.; Jenks, W.S. Bimolecular photoreduction of aromatic sulfoxides. J. Org. Chem. 2001, 66, 8621–8628. [Google Scholar] [CrossRef] [PubMed]
- Cubbage, J.W.; Guo, Y.; McCulla, R.D.; Jenks, W.S. Thermolysis of alkyl sulfoxides and derivatives: a comparison of experiment and theory. J. Org. Chem. 2001, 66, 8722–8736. [Google Scholar] [CrossRef] [PubMed]
- Lanfranchi, D.A.; Bour, C.; Hanquet, G. Enantioselective access to key intermediates for salvinorin A and analogues. Eur. J. Org. Chem. 2011, 2011, 2818–2826. [Google Scholar] [CrossRef]
- Marshall, D.R.; Thomas, P.J.; Stirling, C.J.M. Elimination and addition reactions. Part 30. Leaving group abilities in alkene-forming eliminations activated by sulphonyl groups. J. Chem. Soc. Perkin Trans. 2 1977, 14, 1898–1909. [Google Scholar] [CrossRef]
- Gregory, J.D.; Robbins, P.W. Metabolism of Sulfur Compounds (Sulfate Metabolism). Annu. Rev. Biochem. 1960, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Mathew, J.; Balasubramanian, A.S. Mammalian sulfoconjugate metabolism. J. Biosci. 1987, 11, 7–21. [Google Scholar] [CrossRef]
- Carroll, K.S.; Gao, H.; Chen, H.; Stout, C.D.; Leary, J.A.; Bertozzi, C.R. A Conserved Mechanism for Sulfonucleotide Reduction. PLoS Biol. 2005, 3, e250. [Google Scholar] [CrossRef]
- Bauchart-Thevret, C.; Stoll, B.; Burrin, D.G. Intestinal metabolism of sulfur amino acids. Nutr. Res. Rev. 2009, 22, 175–187. [Google Scholar] [CrossRef]
- Block, E. The Organosulfur Chemistry of the GenusAllium—Implications for the Organic Chemistry of Sulfur. Angew. Chem. Int. Ed. Engl. 1992, 31, 1135–1178. [Google Scholar] [CrossRef]
- Oae, S. Organic Sulfur Chemistry: Structure and Mechanism; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Rayner, C.M. Advances in Sulfur Chemistry; JAI Press: Stamford, UK, 2000. [Google Scholar]
- Bordwell, F.G.; Happer, D.A.R.; Cooper, G.D. Concerning driving forces for β-elimination reactions. Tetrahedron Lett. 1972, 13, 2759–2762. [Google Scholar] [CrossRef]
- Redman, R.P.; Thomas, P.J.; Stirling, C.J.M. Elimination and addition reactions. Part 35. Substituent effects on alkene-forming eliminations from carbanions. J. Chem. Soc. Perkin Trans. 2 1978, 11, 1135–1144. [Google Scholar] [CrossRef]
- Baciocchi, E.; Del Giacco, T.; Lanzalunga, O.; Mencarelli, P.; Procacci, B. Photosensitized Oxidation of Alkyl Phenyl Sulfoxides. C−S Bond Cleavage in Alkyl Phenyl Sulfoxide Radical Cations. J. Org. Chem. 2008, 73, 5675–5682. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Stollar, H. The stereochemistry of thiane oxidation: Participation of neighboring groups. Tetrahedron 1974, 30, 2541–2548. [Google Scholar] [CrossRef]
- Thiruvalluvar, A.; Balamurugan, S.; Butcher, R.J.; Pandiarajan, K.; Devanathan, D. 2-(4-fluorophenyl)-6-phenyltetrahydro-2H-thiopyran-4-one 1-oxide. Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, o4486. [Google Scholar] [CrossRef]
- Thiruvalluvar, A.; Balamurugan, S.; Butcher, R.J.; Pandiarajan, K.; Devanathan, D. 2-r-(4-Chloro-phen-yl)-6-c-phenyl-3,4,5,6-tetra-hydro-2H-thio-pyran-4-one 1-oxide. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, o2367. [Google Scholar] [CrossRef] [PubMed]
- Wojaczyńska, E.; Wojaczyński, J. Enantioselective Synthesis of Sulfoxides: 2000–2009. Chem. Rev. 2010, 110, 4303–4356. [Google Scholar] [PubMed]
- Brunel, J.M.; Kagan, H.B. Catalytic asymmetric oxidation of sulfides with high enantioselectivities. Synlett 1996, 1996, 404–406. [Google Scholar] [CrossRef]
- Song, Z.J.; King, A.O.; Waters, M.S.; Lang, F.; Zewge, D.; Bio, M.; Leazer Jr, J.L.; Javadi, G.; Kassim, A.; Tschaen, D.M.; et al. An efficient asymmetric synthesis of an estrogen receptor modulator by sulfoxide-directed borane reduction. Proc. Natl. Acad. Sci. USA 2004, 101, 5776–5781. [Google Scholar] [CrossRef]
- Davis, F.A.; Jenkins, R.; Yocklovich, S.G. 2-Arenesulfonyl-3-aryloxaziridines: A new class of aprotic oxidizing agents (oxidation of organic sulfur compounds). Tetrahedron Lett. 1978, 19, 5171–5174. [Google Scholar] [CrossRef]
- Davis, F.A.; Chattopadhyay, S.; Towson, J.C.; Lal, S.; Reddy, T. Chemistry of oxaziridines. 9. Synthesis of 2-sulfonyl- and 2-sulfamyloxaziridines using potassium peroxymonosulfate (oxone). J. Org. Chem. 1988, 53, 2087–2089. [Google Scholar] [CrossRef]
- Foster, A.B.; Inch, T.D.; Qadir, M.H.; Webber, J.M. Assignment of sulphoxide configuration by the nuclear magnetic resonance method. Chem. Commun. 1968, 18, 1086–1089. [Google Scholar] [CrossRef]
- Devanathan, D.; Pandiarajan, K. 1H and 13C NMR spectral study of some 2r-aryl-6c-phenylthian-4-ones, their 1-oxides and 1,1-dioxides. Spectrosc. Lett. 2009, 42, 147–155. [Google Scholar] [CrossRef]
- Chen, C.H.; Reynolds, G.A.; Luss, H.R.; Perlstein, J.H. Chemistry of 1,1-dioxothiopyrans. 1. Syntheses and reactions of 2,6-diphenyl-4H-thiopyran-4-one 1,1-dioxide and 4H-thioflaven-4-one 1,1-dioxide. J. Org. Chem. 1986, 51, 3282–3289. [Google Scholar] [CrossRef]
- Rule, N.G.; Detty, M.R.; Kaeding, J.E.; Sinicropi, J.A. Syntheses of 4H-thiopyran-4-one 1,1-dioxides as precursors to sulfone-containing analogs of tetracyanoquinodimethane. J. Org. Chem. 1995, 60, 1665–1673. [Google Scholar] [CrossRef]
- Williams, D.R.; Fu, L. General Methodology for the Preparation of 2,5-Disubstituted-1,3-oxazoles. Org. Lett. 2010, 12, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.H.; Maciá, B.; Fernández-Ibáñez, M.Á.; Minnaard, A.J.; Feringa, B.L. Catalytic asymmetric conjugate addition of dialkylzinc reagents to α,β-unsaturated sulfones. Org. Biomol. Chem. 2010, 8, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.; Davioud-Charvet, E.; Müller, T.J.J. Versatile synthesis of dissymmetric diarylideneacetones via Pd-catalyzed coupling-isomerization reaction. Synthesis 2012, 44, 3829–3835, Erratum in Synthesis 2013, 45, 1270. [Google Scholar]
- Angiolini, L.; Ghedini, N.; Tramontini, M. The Mannich bases in polymer synthesis. 10. Synthesis of poly (β-ketothioethers) and their behaviour towards hydroperoxide reagents. Polym. Commun. 1985, 26, 218–221. [Google Scholar]
- Sokolova, A.Y.; Wyllie, S.; Patterson, S.; Oza, S.L.; Read, K.D.; Fairlamb, A.H. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob. Agents Chemother. 2010, 54, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Patterson, S.; Stojanovski, L.; Simeons, F.R.C.; Norval, S.; Kime, R.; Read, K.D.; Fairlamb, A.H. The antitrypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci. Transl. Med. 2012, 4, 119re1. [Google Scholar] [CrossRef]
- Ortiz, C.; Breuning, M.; Robledo, S.; Echeverri, F.; Vargas, E.; Quiñones, W. Biological activities of 4H-thiochromen-4-one 1,1-dioxide derivatives against tropical disease parasites: A target-based drug design approach. Heliyon 2023, 9, e17801. [Google Scholar] [CrossRef]
- Sharko, A.; Spitzbarth, B.; Hermans, T.M.; Eelkema, R. Redox-controlled shunts in a synthetic chemical reaction cycle. J. Am. Chem. Soc. 2023, 145, 9672–9678. [Google Scholar] [CrossRef]
- Conard, C.R.; Dolliver, M.A. Dibenzalacetone. Org. Synth. 1932, 12, 22. [Google Scholar]
- Sehnal, P.; Taghzouti, H.; Fairlamb, I.J.S.; Jutand, A.; Lee, A.F.; Whitwood, A.C. Heteroaromatic analogues of dibenzylideneacetone (dba) and Pd2(het-dba)3 complexes: Effect of a thienyl moiety on the reactivity of Pd(η2-th[n]-dba)(PPh3)2/Pd(PPh3)2 (n=1 or 2) and Pd(η2-th2 -dba)(dppe)/Pd(dppe) in oxidative addition reactions with iodobenzene. Organometallics 2009, 28, 824–829. [Google Scholar]
- Corbel, B.; Medinger, L.; Haelters, J.P.; Sturtz, G. An efficient synthesis of dialkyl 2-oxoalkanephosphonates and diphenyl-2-oxoalkylphosphine oxides from 1-chloralkyl ketones. Synthesis 1985, 1985, 1048–1051. [Google Scholar] [CrossRef]
- Müller, T.J.J.; Ansorge, M.; Aktah, D. An Unexpected coupling-isomerization sequence as an entry to novel three-component-pyrazoline syntheses. Angew. Chem. Int. Ed. 2000, 39, 1253–1256. [Google Scholar] [CrossRef]
- Braun, R.U.; Ansorge, M.; Müller, T.J.J. The coupling-isomerization synthesis of chalcones. Chem. Eur. J. 2006, 12, 9081–9094. [Google Scholar] [CrossRef]
- Kuo, P.-C.; Damu, A.G.; Cherng, C.-Y.; Jeng, J.-F.; Teng, C.-M.; Lee, E.-J.; Wu, T.-S. Isolation of a natural antioxidant, dehydrozingerone from zingiber officinale and synthesis of its analogues for recognition of effective antioxidant and antityrosinase agents. Arch. Pharm. Res. 2005, 28, 518–528. [Google Scholar] [CrossRef]
- Weber, W.M.; Hunsaker, L.A.; Roybal, C.N.; Bobrovnikova-Marjon, E.V.; Abcouwer, S.F.; Royer, R.E.; Deck, L.M.; Vander Jagt, D.L. Activation of NFkappaB is inhibited by curcumin and related enones. Bioorg. Med. Chem. 2006, 14, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Agbaje, O.C.; Fadeyi, O.O.; Okoro, C.O. Lewis acid mediated diastereoselective synthesis of fused fluorinated spiroketal as potential biologically active compounds. Tetrahedron Lett. 2011, 52, 5297–5300. [Google Scholar] [CrossRef]
- Lin, H.; Hu, G.-X.; Guo, J.; Ge, Y.; Liang, G.; Lian, Q.-Q.; Chu, Y.; Yuan, X.; Huang, P.; Ge, R.-S. Mono-carbonyl curcumin analogues as 11β-hydroxysteroid dehydrogenase 1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4362–4366. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Singh, S.K. Room-Temperature Total Hydrogenation of Biomass-Derived Furans and Furan/Acetone Aldol Adducts over a Ni-Pd Alloy Catalyst. ACS Sustain. Chem. Eng. 2018, 6, 4793–4800. [Google Scholar] [CrossRef]
- Liu, N.; Zhu, W.; Yao, J.; Yin, L.; Lu, T.; Dou, X. Catalyst-Controlled Chemodivergent Synthesis of Spirochromans from Diarylideneacetones and Organoboronic Acids. ACS Catal. 2020, 10, 2596–2602. [Google Scholar] [CrossRef]
- Jahan, N.; Das, A.; Ansary, I. Synthesis of Dibenzo-Fused 15-Membered Dioxa-ketone Macrocycles through Ring-Closing Metathesis Reaction. ChemistrySelect 2022, 7, e202201831. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vitro Assays—CC50 or IC50 (µM) a | |||||
|---|---|---|---|---|---|
| Cmpd | Structure | hMRC-5 | T. cruzi | T. b. brucei | L. infantum |
| 1p |  | 1.0 | ≤0.3 | ≤0.3 | 1.0 |
| 1r |  | 26.2 | 1.6 | 0.2 | >64 |
| 1s |  | 32.8 | 1.8 | 0.5 | >64.0 |
| 1w |  | 1.9 | 0.3 | 0.03 | 0.4 |
| 1x |  | 1.1 | 0.9 | ≤0.3 | 1.0 |
| 1y |  | 1.1 | ≤0.3 | ≤0.3 | 1.0 |
 | |||||
| 2′d | (syn-cis)-3″d | (anti-cis)-3‴d | |||
| Proton | δ (ppm) | δ (ppm) | Δδ (ppm) | δ (ppm) | Δδ (ppm) |
| HX(ax) | 4.28 | 4.07 | −0.21 | 4.12 | −0.16 |
| HA(ax) | 3.01 | 3.73 | +0.72 | 3.24 | +0.23 |
| HB(eq) | 2.93 | 2.67 | −0.26 | 3.01 | +0.08 |
| CC50 or IC50 (µM) a |  2w |  2′w |  3w |  3′w d |  4w |  4′w |
|---|---|---|---|---|---|---|
| hMRC-5 | ≥64 | ≥64 | 8.0 | 8.1 | 7.9 | 7.5 |
| T. cruzib | ≥64 | ≥64 | 0.8 | 0.5 | 0.4 | 0.4 |
| T. b. bruceic | 26.1 | 29.1 | 0.1 | 0.1 | 0.5 | 0.1 |
| L. infantumb | ≥64 | ≥64 | 2.0 | 2.0 | 2.0 | 2.0 |
| L. donovanib | >100 | >100 | 25.0 | 2.5 | 5.0 | 1.6 |
| CC50 or IC50 (µM) a |  2d/2′d |  3d/3′d/3″d |  4d/4′d d |  2w/2′w |  3w/3′w e |  4w/4′w |
|---|---|---|---|---|---|---|
| hMRC-5 | ≥64/≥64 | 32.2 b/≥64/32.0 | ≥64 b/≥ 64 | ≥64/≥64 | 8.0/8.1 | 7.9/7.5 |
| T. cruzib | ≥64/≥64 | 8.9 b/≥64/8.5 | ≥64 b/34.4 | ≥64/≥64 | 0.8/0.5 | 0.4/0.4 |
| T. b. bruceic | ≥64/≥64 | 3.1 b/2.3/2.4 | 43.3 b/2.1 | 26.1/29.1 | 0.1/0.1 | 0.5/0.1 |
| L. infantumb | ≥64/≥64 | ≥64/≥64/27.3 | ≥64/20.3 | ≥64/≥64 | 2.0/2.0 | 2.0/2.0 |
| L. donovanib | nd/>100 | 50.2/>100/35.0 | >100/nd | >100/>100 | 25.0/2.5 | 5.0/1.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gendron, T.; Lanfranchi, D.A.; Wenzel, N.I.; Kessedjian, H.; Jannack, B.; Maes, L.; Cojean, S.; Müller, T.J.J.; Loiseau, P.M.; Davioud-Charvet, E. Chemoselective Synthesis and Anti-Kinetoplastidal Properties of 2,6-Diaryl-4H-tetrahydro-thiopyran-4-one S-Oxides: Their Interplay in a Cascade of Redox Reactions from Diarylideneacetones. Molecules 2024, 29, 1620. https://doi.org/10.3390/molecules29071620
Gendron T, Lanfranchi DA, Wenzel NI, Kessedjian H, Jannack B, Maes L, Cojean S, Müller TJJ, Loiseau PM, Davioud-Charvet E. Chemoselective Synthesis and Anti-Kinetoplastidal Properties of 2,6-Diaryl-4H-tetrahydro-thiopyran-4-one S-Oxides: Their Interplay in a Cascade of Redox Reactions from Diarylideneacetones. Molecules. 2024; 29(7):1620. https://doi.org/10.3390/molecules29071620
Chicago/Turabian StyleGendron, Thibault, Don Antoine Lanfranchi, Nicole I. Wenzel, Hripsimée Kessedjian, Beate Jannack, Louis Maes, Sandrine Cojean, Thomas J. J. Müller, Philippe M. Loiseau, and Elisabeth Davioud-Charvet. 2024. "Chemoselective Synthesis and Anti-Kinetoplastidal Properties of 2,6-Diaryl-4H-tetrahydro-thiopyran-4-one S-Oxides: Their Interplay in a Cascade of Redox Reactions from Diarylideneacetones" Molecules 29, no. 7: 1620. https://doi.org/10.3390/molecules29071620
APA StyleGendron, T., Lanfranchi, D. A., Wenzel, N. I., Kessedjian, H., Jannack, B., Maes, L., Cojean, S., Müller, T. J. J., Loiseau, P. M., & Davioud-Charvet, E. (2024). Chemoselective Synthesis and Anti-Kinetoplastidal Properties of 2,6-Diaryl-4H-tetrahydro-thiopyran-4-one S-Oxides: Their Interplay in a Cascade of Redox Reactions from Diarylideneacetones. Molecules, 29(7), 1620. https://doi.org/10.3390/molecules29071620