
A Crucial Role of Proteolysis in the Formation of Intracellular Dinitrosyl Iron Complexes
 , , , and
, , , and
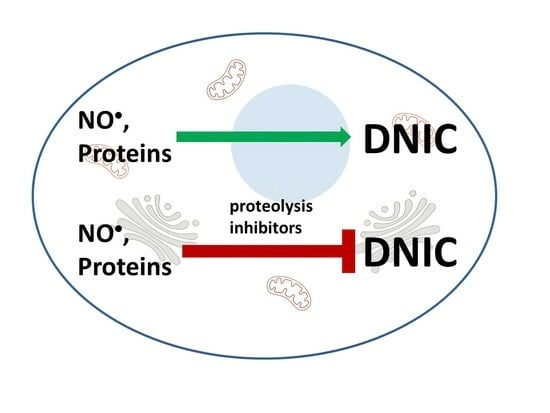
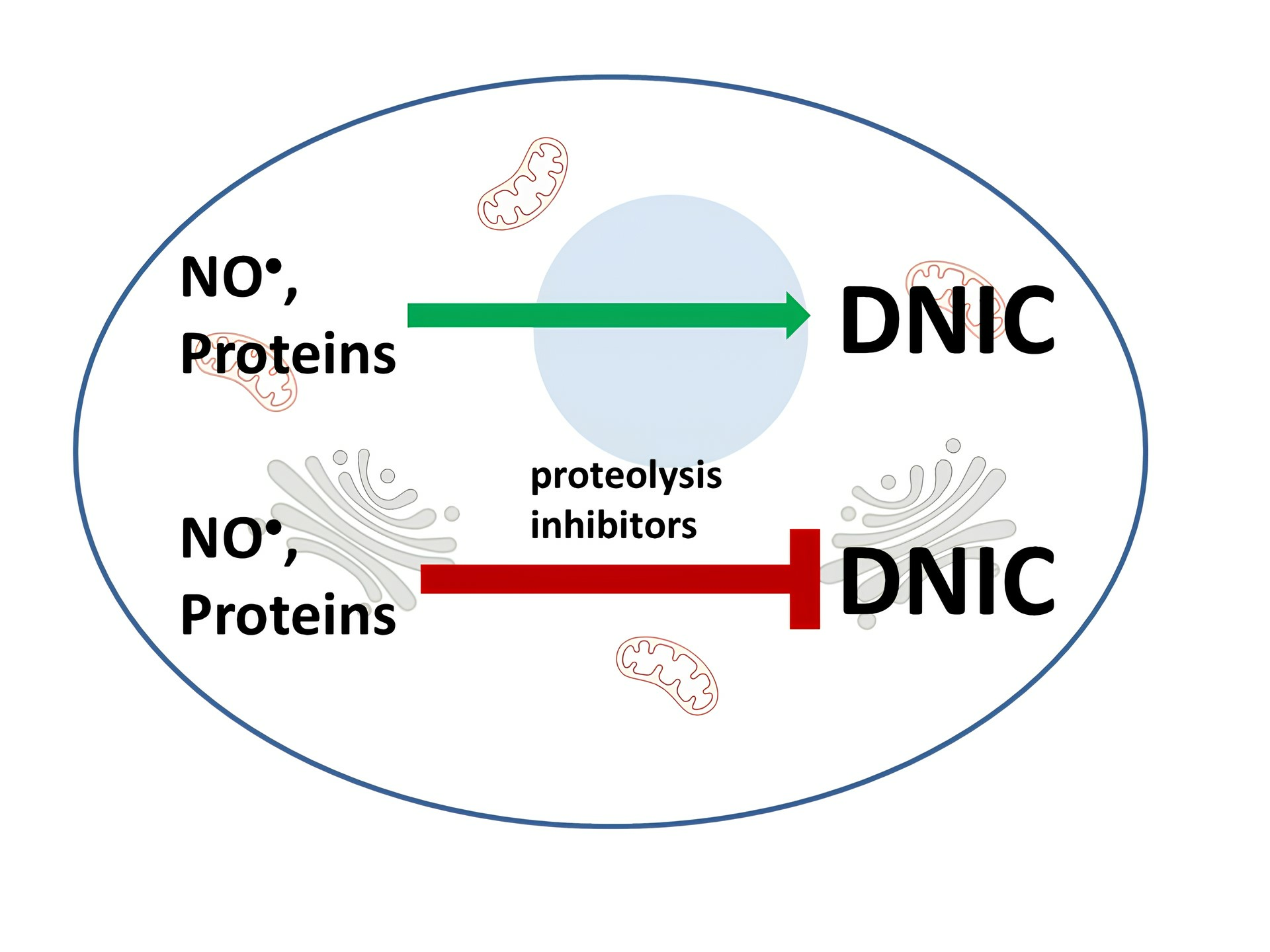
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
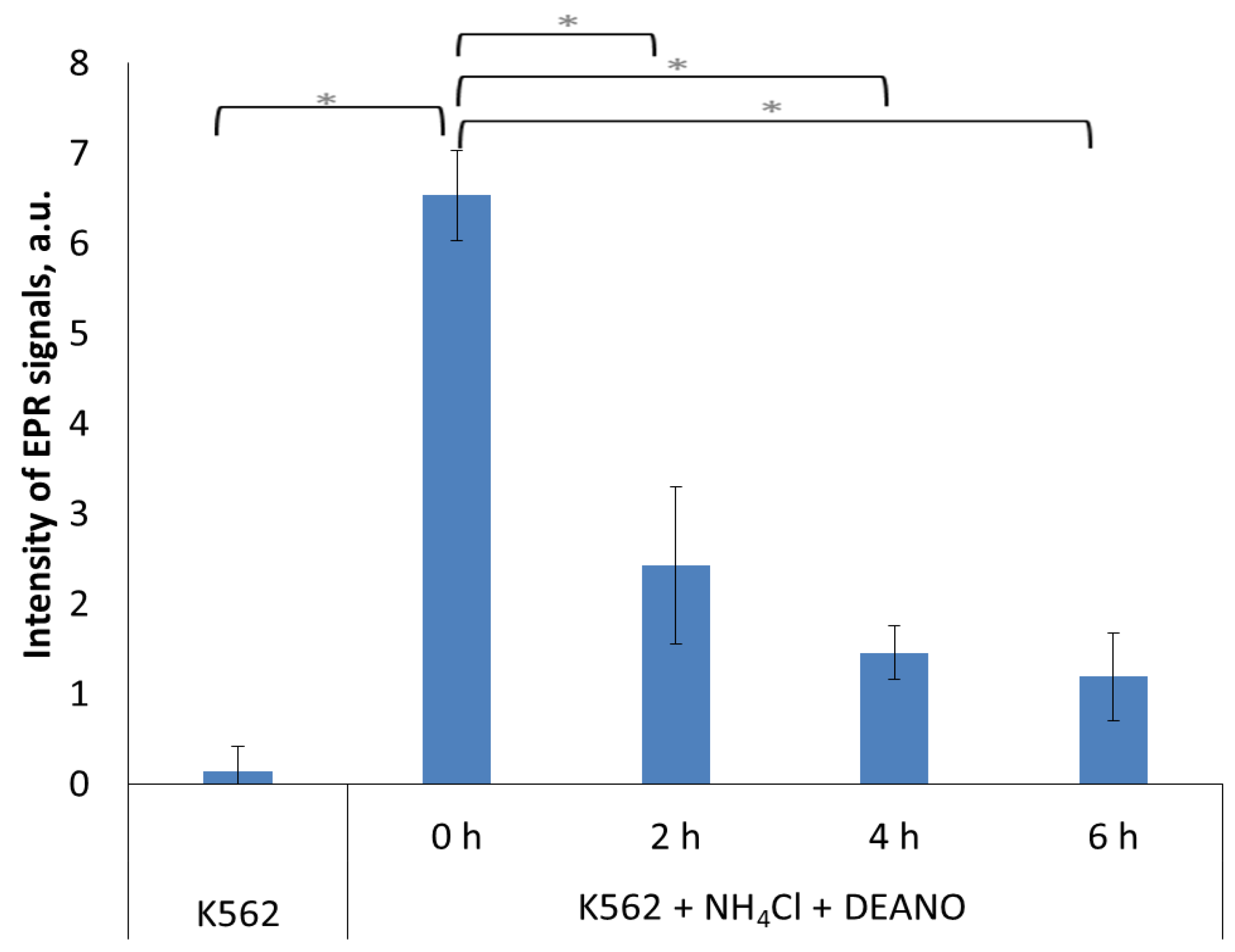
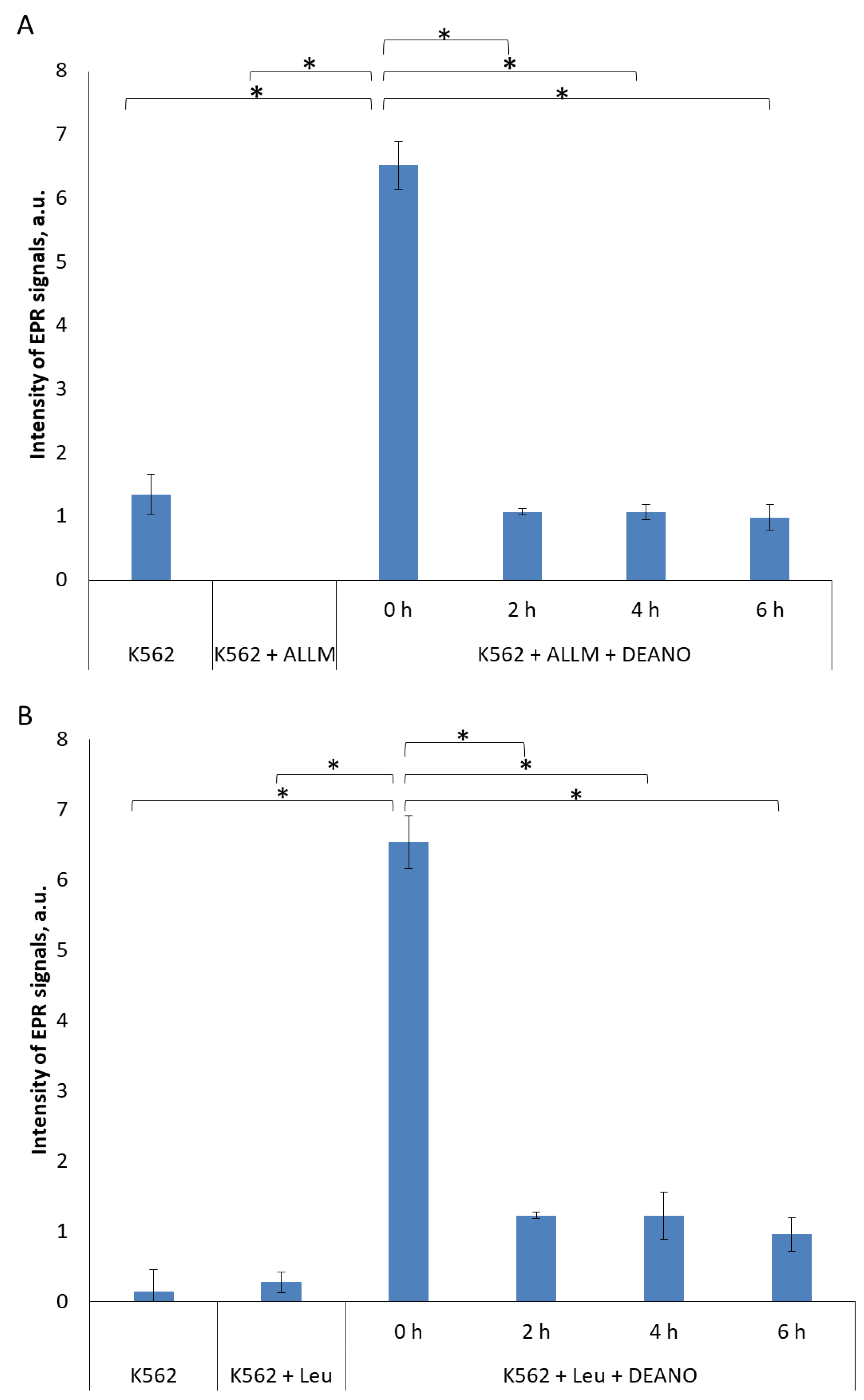
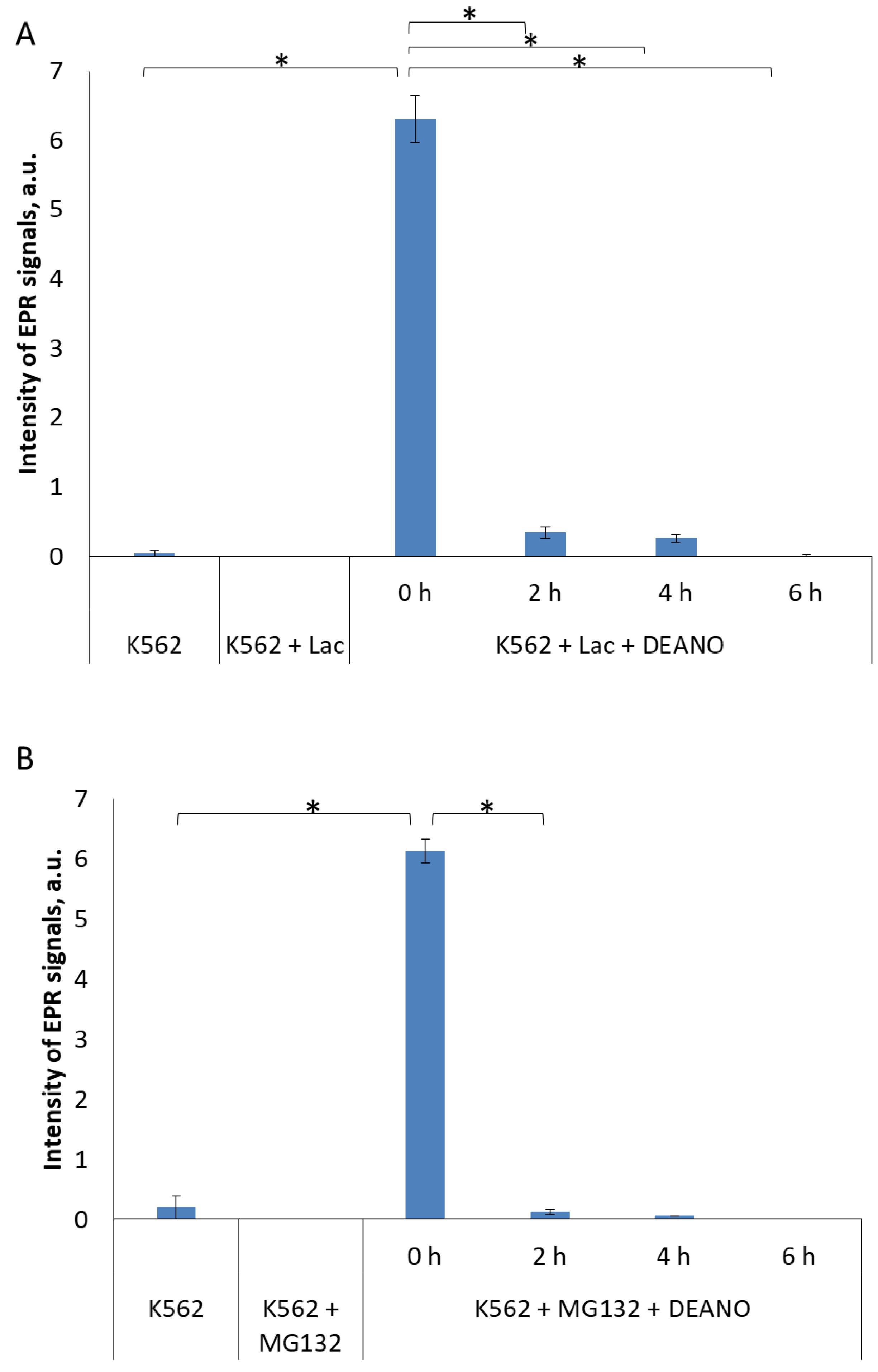
2.1. The Effect of Inhibitors of Lysosomal Proteolysis on DNIC formation
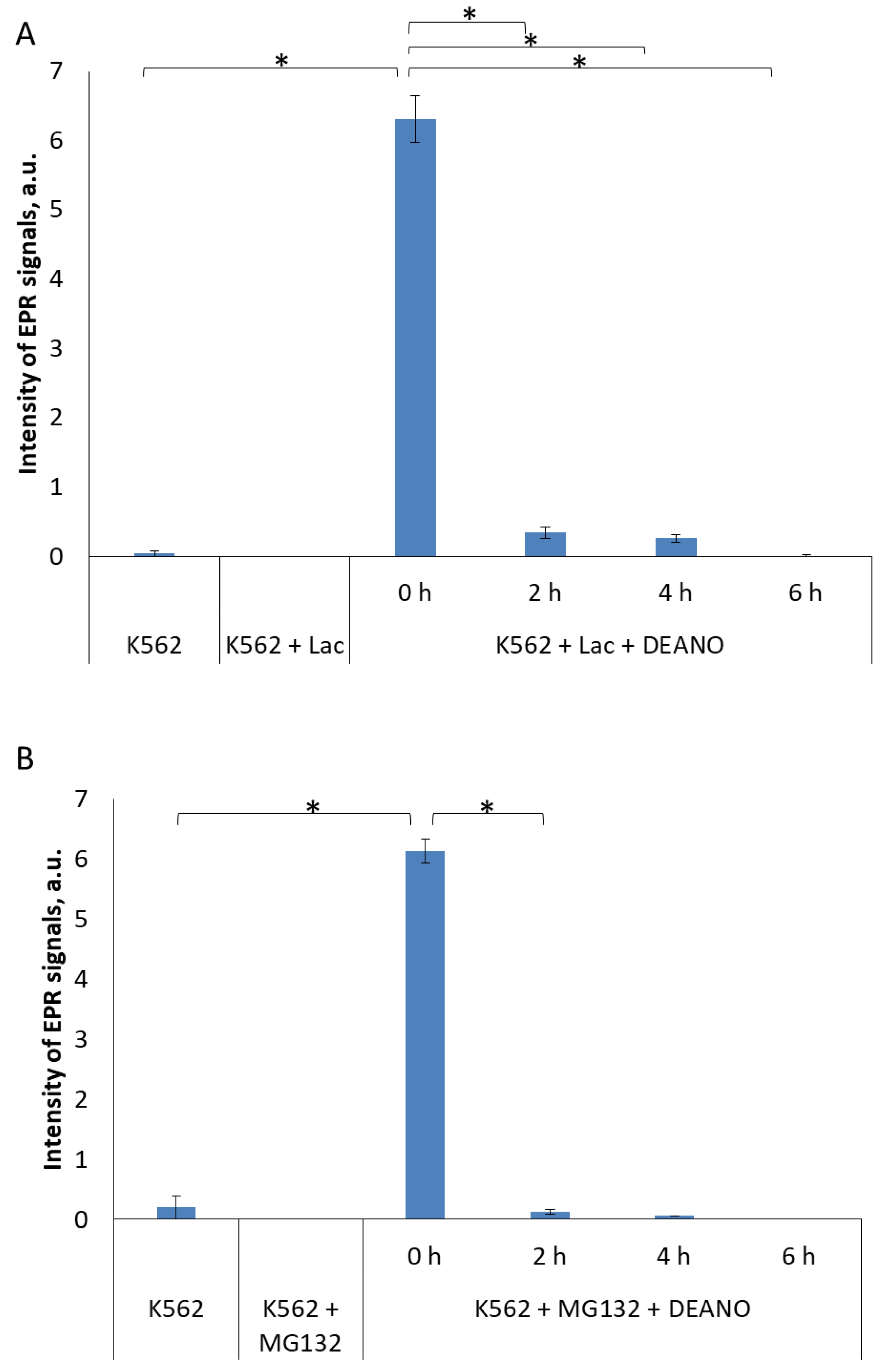
2.2. The Effect of Proteasome Inhibitors on DNIC Synthesis
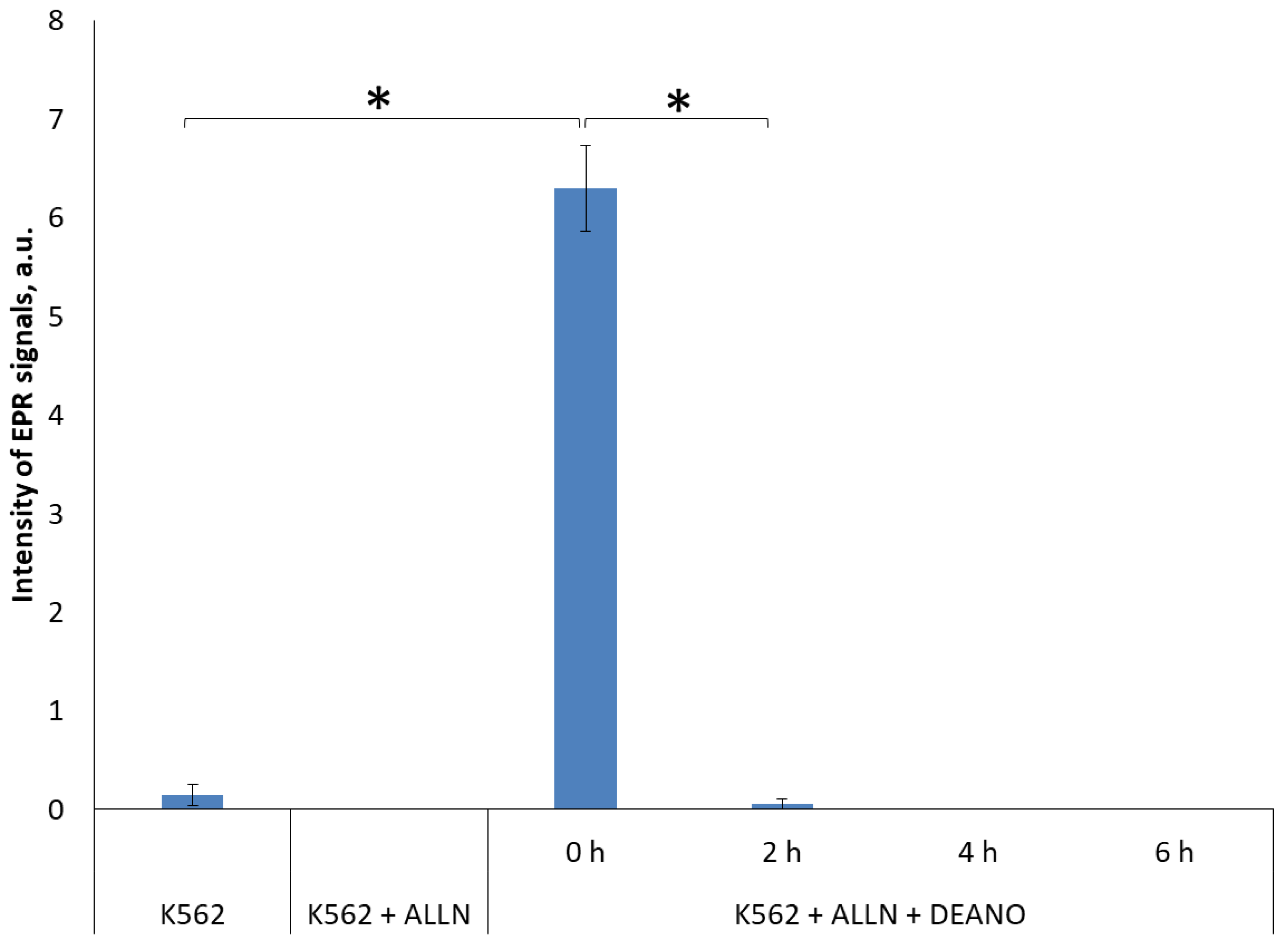
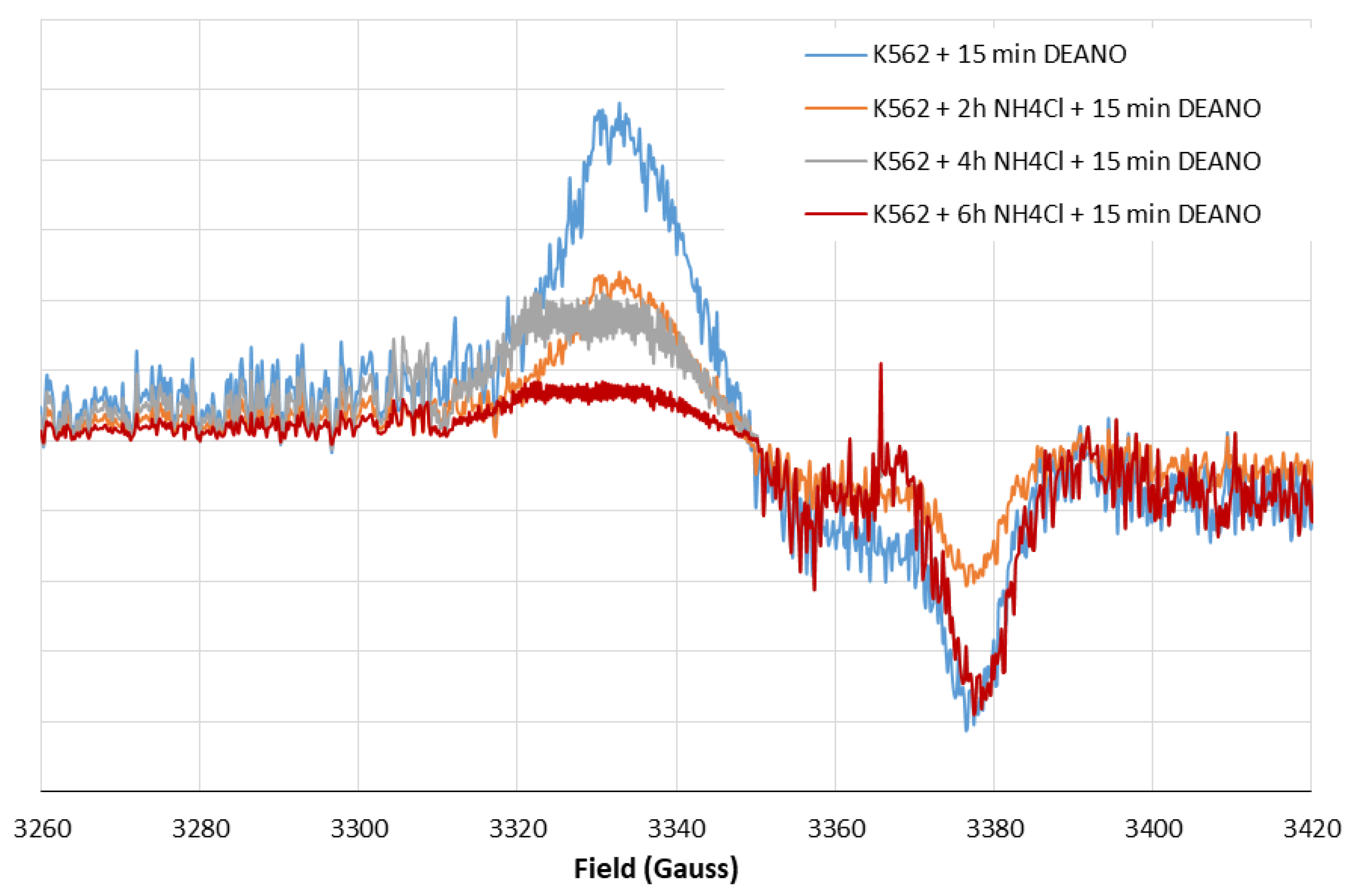
2.3. Effect of the Total Proteolysis INHIBITION on DNIC Synthesis
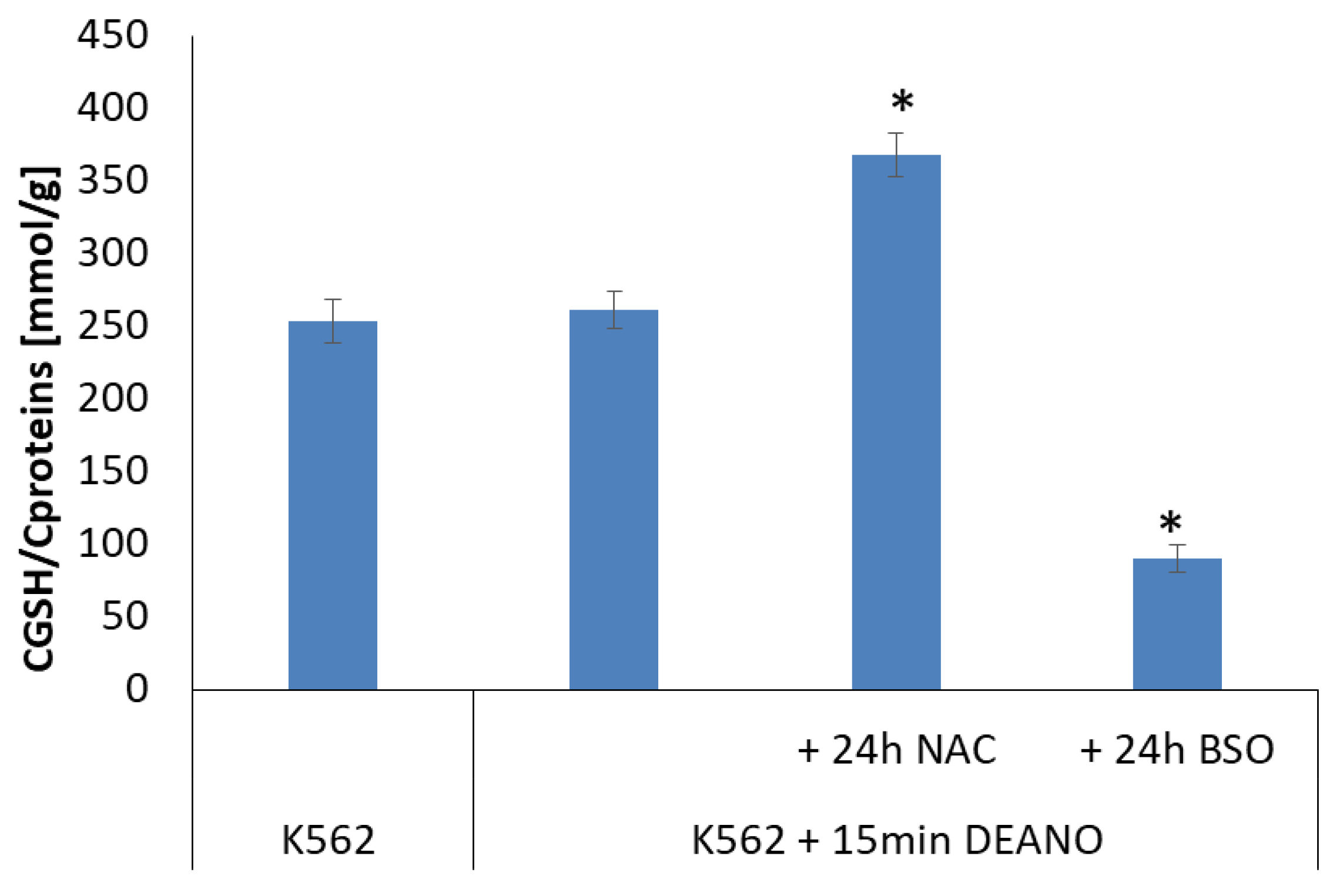
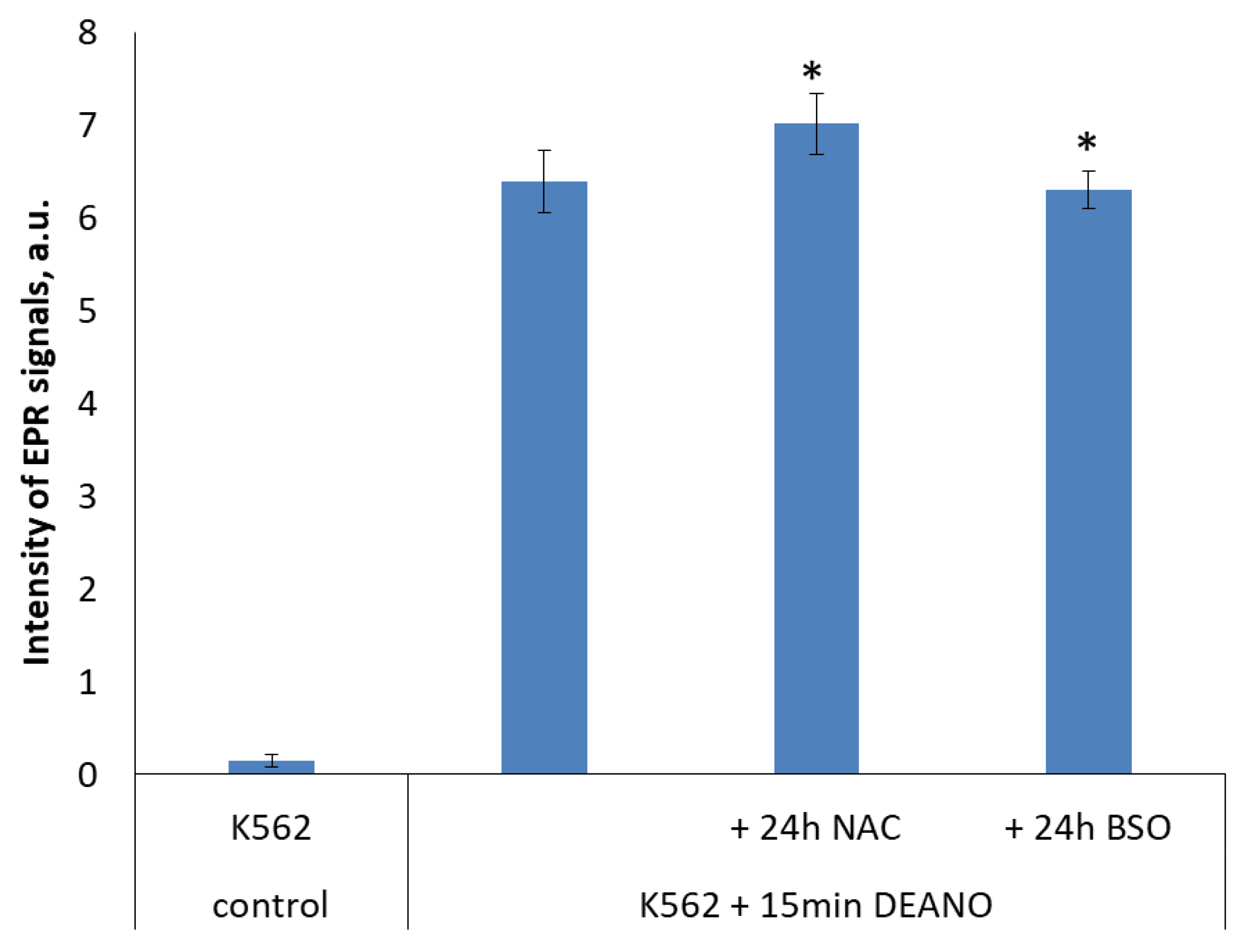
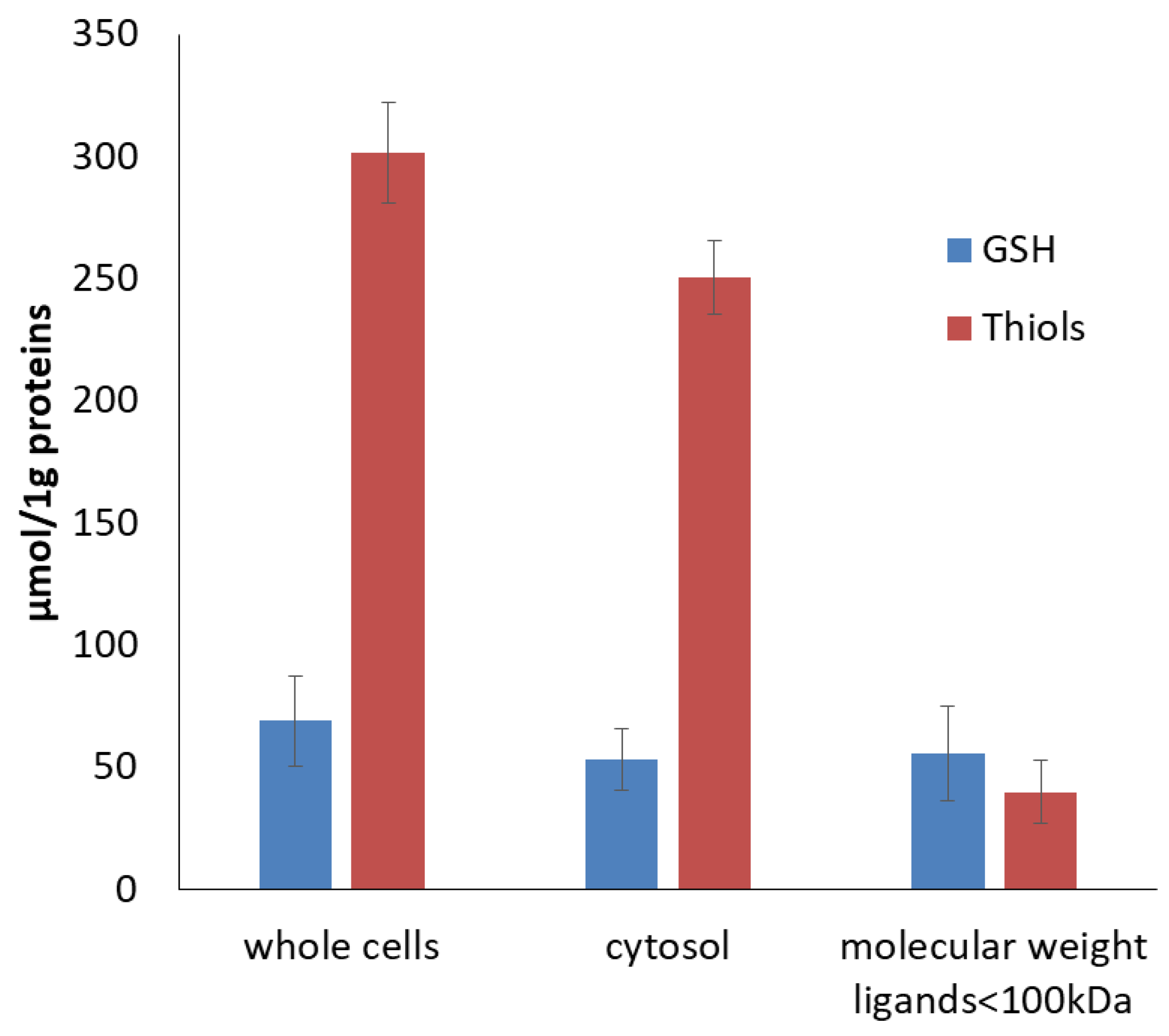
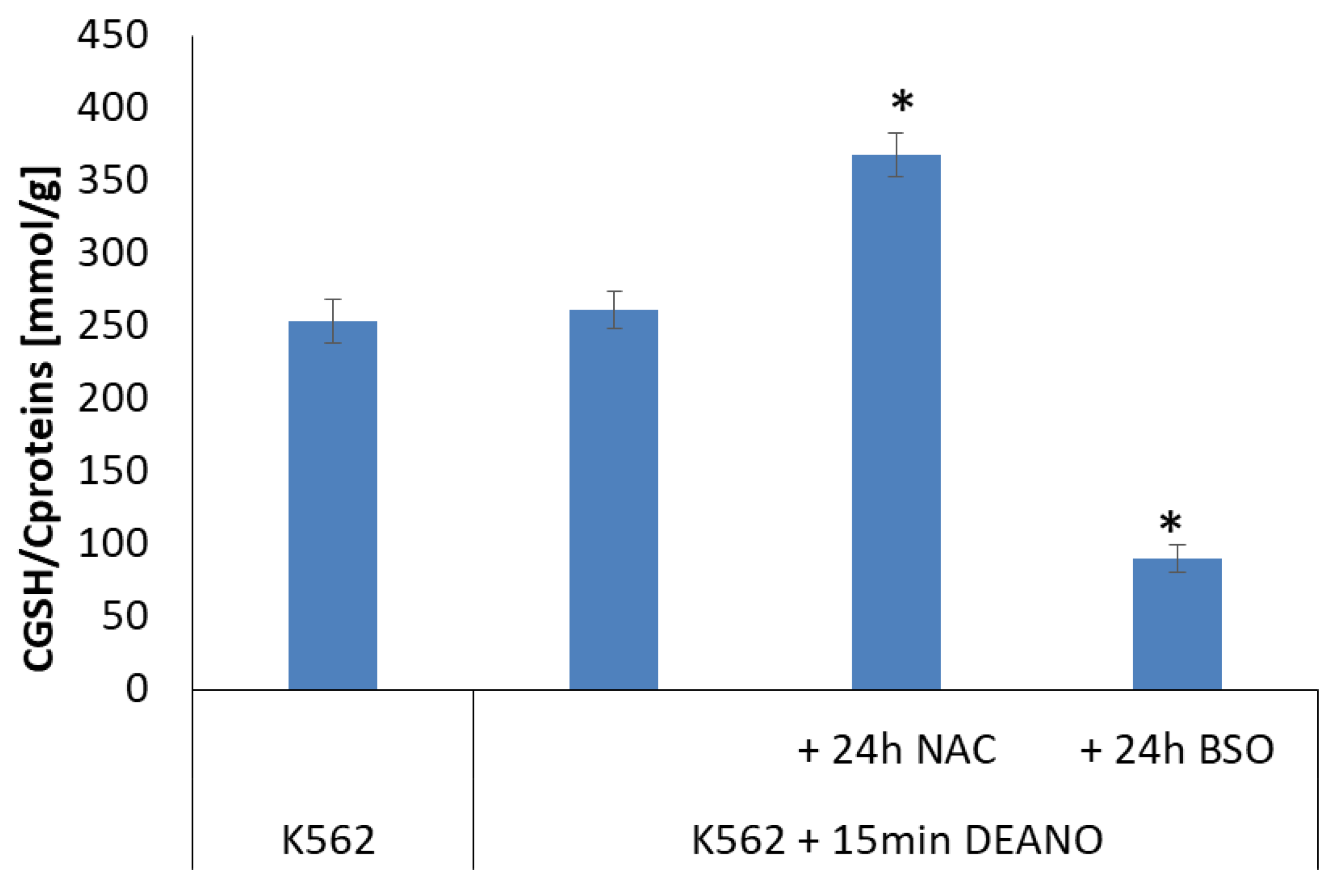
2.4. The Effect of Glutathione Concentration Modulation on DNIC Synthesis
2.5. Cellular Low and High Molecular Ligand Fractions
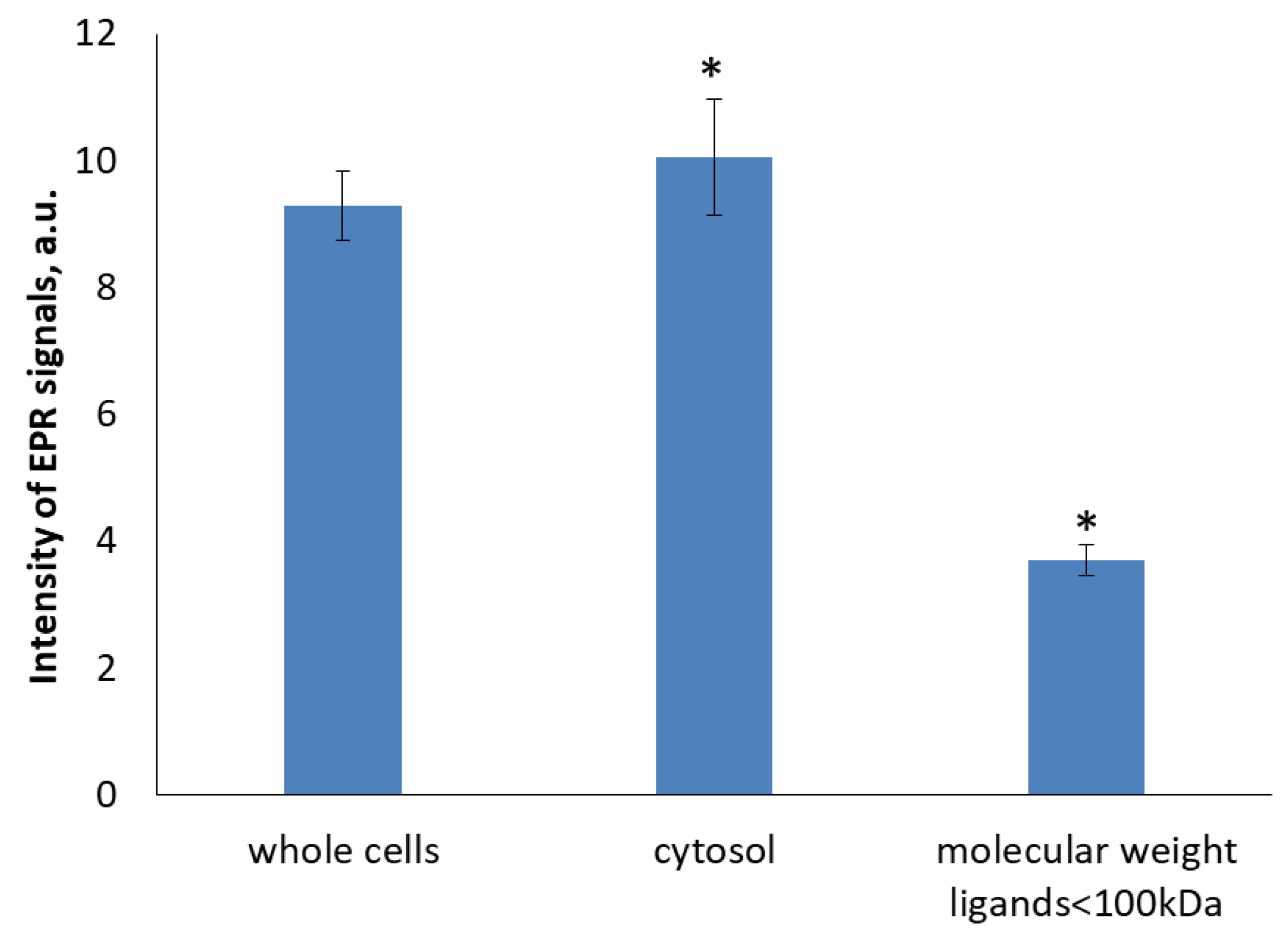
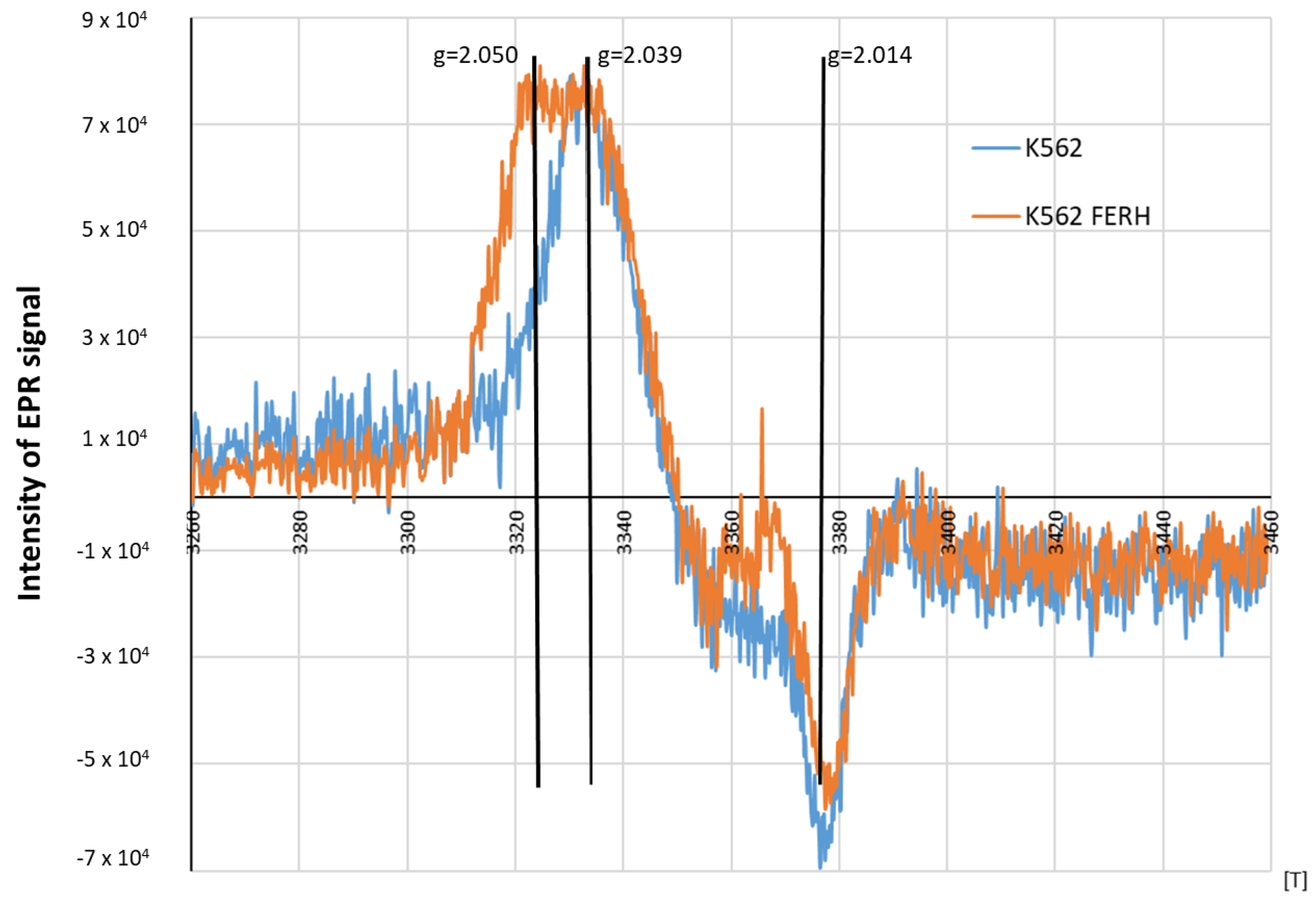
2.6. Characterization of EPR Signal of Protein-Bound DNICs
3. Materials and Methods
3.1. Cell Lines and Chemicals
3.2. Preparation of HFER Transfected K562 Cells
3.3. Inhibition of Proteolysis
3.4. Generation of DNICs
3.5. EPR Measurements
3.6. Preparation of Subcellular Fractions
3.7. Glutathione and Thiol Determination
3.8. Statistical Evaluation
4. Summary and Conclusions
Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALLM | N-Acetyl-L-leucyl-L-leucyl-L-methioninal (lysosome inhibitor) |
| ATPases | class of enzymes that catalyze hydrolysis of adenosine triphosphate |
| BSO | D,L-buthionine [S,R]-sulfoximine |
| DEANO | 2-(N,N-Diethyloamino)-diazenolate-2-oxide |
| DNICs | dinitrosyl iron complexes |
| EDTA | ethylenediaminetetraacetic acid |
| EPR | electron paramagnetic resonance |
| FERH | heavy chain of ferritin |
| GSH | glutathione |
| HMWDNICs | dinitrosyl iron complexes with high-molecular-weight ligands |
| His | histidine |
| HFS | hyperfine structure |
| LMWDNICs | dinitrosyl iron complexes with low-molecular-weight ligands |
| LAC | lactacystin (proteasome inhibitor) |
| LEUPT | leupeptin (lysosome inhibitor) |
| MG-101 | N-Acetyl-L-leucyl-L-leucyl-L-norleucinal (calpain, lysosome and proteasome inhibitor) |
| MG-132 | Z-Leu-Leu-Leu-al (proteasome inhibitor) |
| MG-262 | Z-Leu-Leu-Leu-B(OH)2 (calpain, lysosome and proteasome inhibitor) |
| NAC | N-acetylcysteine |
| PBS | phosphate-buffered saline |
References
- Lukiewicz, S.; Zweier, J.L. (Eds.) Nitric Oxide in Transplant Rejection and Anti-Tumor Defense; Kluwer Academic Publishers: Boston, MA, USA, 1998; ISBN 978-0-7923-8389-5. [Google Scholar]
- Ueno, T.; Yoshimura, T. The Physiological Activity and In Vivo Distribution of Dinitrosyl Dithiolato Iron Complex. Jpn. J. Pharmacol. 2000, 82, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, N.; Kim, E.; Dong, H.T.; Harland, J.B.; Hunt, A.P.; Manickas, E.C.; Oakley, K.M.; Pham, J.; Reed, G.C.; Alfaro, V.S. The Biologically Relevant Coordination Chemistry of Iron and Nitric Oxide: Electronic Structure and Reactivity. Chem. Rev. 2021, 121, 14682–14905. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.C.; Pereira, J.C.M.; Miranda, K.M. Mechanisms of Nitric Oxide Reactions Mediated by Biologically Relevant Metal Centers. Nitrosyl Complexes Inorg. Chem. Biochem. Med. II 2013, 154, 99–135. [Google Scholar]
- Vanin, A.F. Physico-Chemistry of Dinitrosyl Iron Complexes as a Determinant of Their Biological Activity. Int. J. Mol. Sci. 2021, 22, 10356. [Google Scholar] [CrossRef] [PubMed]
- Hickok, J.R.; Sahni, S.; Shen, H.; Arvind, A.; Antoniou, C.; Fung, L.W.M.; Thomas, D.D. Dinitrosyliron Complexes Are the Most Abundant Nitric Oxide-Derived Cellular Adduct: Biological Parameters of Assembly and Disappearance. Free Radic. Biol. Med. 2011, 51, 1558–1566. [Google Scholar] [CrossRef]
- Vanin, A.F.; Malenkova, I.V.; Serezhenkov, V.A. Iron Catalyzes Both Decomposition and Synthesis ofS-Nitrosothiols: Optical and Electron Paramagnetic Resonance Studies. Nitric Oxide 1997, 1, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Corey, C.; Hickok, J.; Wang, Y.; Shiva, S. Differential Mitochondrial Dinitrosyliron Complex Formation by Nitrite and Nitric Oxide. Redox Biol. 2018, 15, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, J.; Kim, E. Synthetic Modeling Chemistry of Iron–Sulfur Clusters in Nitric Oxide Signaling. Acc. Chem. Res. 2015, 48, 2453–2461. [Google Scholar] [CrossRef] [PubMed]
- Crack, J.C.; Le Brun, N.E. Biological Iron-Sulfur Clusters: Mechanistic Insights from Mass Spectrometry. Coord. Chem. Rev. 2021, 448, 214171. [Google Scholar] [CrossRef]
- Lewandowska, H.; Kalinowska, M.; Brzoska, K.; Wojciuk, K.; Wojciuk, G.; Kruszewski, M. Nitrosyl Iron Complexes—Synthesis, Structure and Biology. Dalton Trans. 2011, 40, 8273–8289. [Google Scholar] [CrossRef] [PubMed]
- Moënne-Loccoz, P. Spectroscopic Characterization of Heme Iron–Nitrosyl Species and Their Role in NO Reductase Mechanisms in Diiron Proteins. Nat. Prod. Rep. 2007, 24, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.C.; Shiro, Y.; Van Eldik, R. Renaissance in NO Chemistry. Inorg. Chem. 2021, 60, 15831–15834. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.T.; Speelman, A.L.; Kozemchak, C.E.; Sil, D.; Krebs, C.; Lehnert, N. The Fe 2 (NO) 2 Diamond Core: A Unique Structural Motif In Non-Heme Iron-NO Chemistry. Angew. Chem. Int. Ed. 2019, 58, 17695–17699. [Google Scholar] [CrossRef] [PubMed]
- Dong, H. Synthesis and Reactivity of Non-Heme Iron-Nitrosyl Complexes That Model the Active Sites of NO Reductases. Ph.D. Thesis, University of Michigan, Ann Arbor, MI, USA, 2021. [Google Scholar] [CrossRef]
- Medvedeva, V.A.; Ivanova, M.V.; Shumaev, K.B.; Dudylina, A.L.; Ruuge, E.K. Generation of Superoxide Radicals by Heart Mitochondria and the Effects of Dinitrosyl Iron Complexes and Ferritin. Biophysics 2021, 66, 603–609. [Google Scholar] [CrossRef]
- Mukosera, G.T.; Principe, P.; Mata-Greenwood, E.; Liu, T.; Schroeder, H.; Parast, M.; Blood, A.B. Iron Nitrosyl Complexes Are Formed from Nitrite in the Human Placenta. J. Biol. Chem. 2022, 298, 102078. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bhatla, S.C. Heme Oxygenase-Nitric Oxide Crosstalk-Mediated Iron Homeostasis in Plants under Oxidative Stress. Free Radic. Biol. Med. 2022, 182, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Burgova, E.N.; Tkachev, N.A.; Paklina, O.V.; Mikoyan, V.D.; Adamyan, L.V.; Vanin, A.F. The Effect of Dinitrosyl Iron Complexes with Glutathione and S-Nitrosoglutathione on the Development of Experimental Endometriosis in Rats: A Comparative Studies. Eur. J. Pharmacol. 2014, 741, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Vladimir, T.; Anatoly, O.; Larisa, I.; Vladimir, P.; Anna, D.; Anna, O. Hypothetical Mechanism of Light Action on Nitric Oxide Physiological Effects. Lasers Med. Sci. 2021, 36, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Vanin, A.F.; Mikoyan, V.D.; Borodulin, R.R.; Burbaev, D.S.; Kubrina, L.N. Dinitrosyl Iron Complexes with Persulfide Ligands: EPR and Optical Studies. Appl. Magn. Reson. 2016, 47, 277–295. [Google Scholar] [CrossRef]
- Vanin, A.F. Dinitrosyl Iron Complexes as a “Working Form” of Nitric Oxide in Living Organisms; Cambridge Scholars Publishing: Newcastle upon Tyne, UK, 2019; ISBN 1-5275-4075-8. [Google Scholar]
- Nalbandyan, R.M.; Vanin, A.F.; Blumenfeld, L.A. EPR Signals of a New Type in Yeast Cells. In Proceedings of the Abstracts of the Meeting Free Radical Processes in Biological Systems, Moscow, Russia; 1965; p. 18. [Google Scholar]
- Chenais, B.; Morjani, H.; Drapier, J.C. Impact of Endogenous Nitric Oxide on Microglial Cell Energy Metabolism and Labile Iron Pool. J. Neurochem. 2002, 81, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Jasid, S.; Simontacchi, M.; Puntarulo, S. Exposure to Nitric Oxide Protects against Oxidative Damage but Increases the Labile Iron Pool in Sorghum Embryonic Axes. J. Exp. Bot. 2008, 59, 3953–3962. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, H.; Sadło, J.; Męczyńska, S.; Stępkowski, T.M.; Wójciuk, G.; Kruszewski, M. Formation of Glutathionyl Dinitrosyl Iron Complexes Protects against Iron Genotoxicity. Dalton Trans. 2015, 44, 12640–12652. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, H.; Męczyńska-Wielgosz, S.; Sikorska, K.; Sadło, J.; Dudek, J.; Kruszewski, M. LDL Dinitrosyl Iron Complex: A New Transferrin-independent Route for Iron Delivery in Hepatocytes. BioFactors 2018, 44, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.A.; Ding, H. L-Cysteine-Mediated Destabilization of Dinitrosyl Iron Complexes in Proteins. J. Biol. Chem. 2001, 276, 30980–30986. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.A.; Eide, L.; Klungland, A.; Ding, H. Reversible Inactivation of E. Coli Endonuclease III via Modification of Its [4Fe-4S] Cluster by Nitric Oxide. DNA Repair 2003, 2, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Tonzetich, Z.J.; Do, L.H.; Lippard, S.J. Dinitrosyl Iron Complexes Relevant to Rieske Cluster Nitrosylation. J. Am. Chem. Soc. 2009, 131, 7964–7965. [Google Scholar] [CrossRef] [PubMed]
- Tonzetich, Z.J.; McQuade, L.E.; Lippard, S.J. Detecting and Understanding the Roles of Nitric Oxide in Biology. Inorg. Chem. 2010, 49, 6338–6348. [Google Scholar] [CrossRef] [PubMed]
- Harrop, T.C.; Tonzetich, Z.J.; Reisner, E.; Lippard, S.J. Reactions of Synthetic [2Fe-2S] and [4Fe-4S] Clusters with Nitric Oxide and Nitrosothiols. J. Am. Chem. Soc. 2008, 130, 15602–15610. [Google Scholar] [CrossRef] [PubMed]
- Tinberg, C.E.; Tonzetich, Z.J.; Wang, H.; Do, L.H.; Yoda, Y.; Cramer, S.P.; Lippard, S.J. Characterization of Iron Dinitrosyl Species Formed in the Reaction of Nitric Oxide with a Biological Rieske Center. J. Am. Chem. Soc. 2010, 132, 18168–18176. [Google Scholar] [CrossRef]
- Vanin, A.F. Dinitrosyl Iron Complexes and S-Nitrosothiols Are Two Possible Forms for Stabilization and Transport of Nitric Oxide in Biological Systems. Biochem. c/c Biokhimiia 1998, 63, 782–793. [Google Scholar]
- Tung, C.-Y.; Tseng, Y.-T.; Lu, T.-T.; Liaw, W.-F. Insight into the Electronic Structure of Biomimetic Dinitrosyliron Complexes (DNICs): Toward the Syntheses of Amido-Bridging Dinuclear DNICs. Inorg. Chem. 2021, 60, 15846–15873. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Suzuki, Y.; Fujii, S.; Vanin, A.F.; Yoshimura, T. In Vivo Distribution and Behavior of Paramagnetic Dinitrosyl Dithiolato Iron Complex in the Abdomen of Mouse. Free Radic. Res. 1999, 31, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Timoshin, A.A.; Vanin, A.F.; Orlova, T.R.; Sanina, N.A.; Ruuge, E.K.; Aldoshin, S.M.; Chazov, E.I. Protein-Bound Dinitrosyl–Iron Complexes Appearing in Blood of Rabbit Added with a Low-Molecular Dinitrosyl–Iron Complex: EPR Studies. Nitric Oxide 2007, 16, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, H.; Męczyńska, S.; Sochanowicz, B.; Sadło, J.; Kruszewski, M. Crucial Role of Lysosomal Iron in the Formation of Dinitrosyl Iron Complexes in vivo. J. Biol. Inorg. Chem. 2007, 12, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Barhoumi, R.; Bailey, R.H.; Burghardt, R.C. Kinetic Analysis of Glutathione in Anchored Cells with Monochlorobimane. Cytometry 1995, 19, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Shumaev, K.B.; Dudylina, A.L.; Ivanova, M.V.; Pugachenko, I.S.; Ruuge, E.K. Dinitrosyl Iron Complexes: Formation and Antiradical Action in Heart Mitochondria. BioFactors 2018, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Mellgren, R.L. Calpain Inhibitors and Serine Protease Inhibitors Can Produce Apoptosis in HL-60 Cells. Arch. Biochem. Biophys. 1996, 334, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Hiwasa, T.; Sawada, T.; Sakiyama, S. Cysteine Proteinase Inhibitors and Ras. Gene Products Share the Same Biological Activities Including Tranforming Activity toward NIH3T3 Mouse Fibroblasts and the Differentiation-Inclucing Activity toward PC12 Rat Pheochromocytoma Cells. Carcinogenesis 1990, 11, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kurinov, I.V.; Harrison, R.W. Two Crystal Structures of the Leupeptin-Trypsin Complex. Protein Sci. 1996, 5, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, L.; Zheng, H.; Mao, C.; Hu, W.; Xiong, K.; Wang, F.; Liu, C. Application and Interpretation of Current Autophagy Inhibitors and Activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Craiu, A.; Gaczynska, M.; Akopian, T.; Gramm, C.F.; Fenteany, G.; Goldberg, A.L.; Rock, K.L. Lactacystin and Clasto-Lactacystin β-Lactone Modify Multiple Proteasome β-Subunits and Inhibit Intracellular Protein Degradation and Major Histocompatibility Complex Class I Antigen Presentation. J. Biol. Chem. 1997, 272, 13437–13445. [Google Scholar] [CrossRef] [PubMed]
- Squier, M.K.T.; Miller, A.C.K.; Malkinson, A.M.; Cohen, J.J. Calpain Activation in Apoptosis. J. Cell. Physiol. 1994, 159, 229–237. [Google Scholar] [CrossRef]
- Goldberg, A.L. Development of Proteasome Inhibitors as Research Tools and Cancer Drugs. J. Cell Biol. 2012, 199, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, G.; De Llano, J.J.M.; Villarroya, A.; Rivett, A.J.; Knecht, E. Changes in the Proteolytic Activities of Proteasomes and Lysosomes in Human Fibroblasts Produced by Serum Withdrawal, Amino-Acid Deprivation and Confluent Conditions. Biochem. J. 2003, 375, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Goldberg, A.L. Proteasome Inhibitors: Valuable New Tools for Cell Biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Kageyama, M.; Ota, T.; Sasaoka, M.; Katsuta, O.; Shinomiya, K. Chemical Proteasome Inhibition as a Novel Animal Model of Inner Retinal Degeneration in Rats. PLoS ONE 2019, 14, e0217945. [Google Scholar] [CrossRef] [PubMed]
- Berndtsson, M.; Beaujouin, M.; Rickardson, L.; Havelka, A.M.; Larsson, R.; Westman, J.; Liaudet-Coopman, E.; Linder, S. Induction of the Lysosomal Apoptosis Pathway by Inhibitors of the Ubiquitin-proteasome System. Int. J. Cancer 2009, 124, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Frase, H.; Hudak, J.; Lee, I. Identification of the Proteasome Inhibitor MG262 as a Potent ATP-Dependent Inhibitor of the Salmonella Enterica Serovar Typhimurium Lon Protease. Biochemistry 2006, 45, 8264–8274. [Google Scholar] [CrossRef]
- Pujols, L.; Fernández-Bertolín, L.; Fuentes-Prado, M.; Alobid, I.; Roca-Ferrer, J.; Agell, N.; Mullol, J.; Picado, C. Proteasome Inhibition Reduces Proliferation, Collagen Expression, and Inflammatory Cytokine Production in Nasal Mucosa and Polyp Fibroblasts. J. Pharmacol. Exp. Ther. 2012, 343, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, S. Cell-Penetrating Inhibitors of Calpain. Trends Biochem. Sci. 1991, 16, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D. Proteinase and Peptidase Inhibition: Recent Potential Targets for Drug Development. Drug Discov. Today 2002, 7, 1124. [Google Scholar] [CrossRef]
- Vanin, A.F.; Serezhenkov, V.A.; Mikoyan, V.D.; Genkin, M.V. The 2.03 Signal as an Indicator of Dinitrosyl-Iron Complexes with Thiol-Containing Ligands. Nitric Oxide 1998, 2, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, P.; Lewandowska, H.; Drapier, J.C.; Starzynski, R.R.; Bartlomiejczyk, T.; Kruszewski, M. Increase in Labile Iron Pool (LIP) Level and Generation of EPR-Detectable Dinitrosyl-Non-Heme Iron Complexes in L5178Y Cells Exposed to Nitric Oxide. Possible Role of LIP as a Source of Iron for DNIC Formation; Chimie, biologie et therapeutiques; Deregulations du Metabolisme du fer: Gif-sur-Yvette, France, 5 September 2003. [Google Scholar]
- Lee, M.; Arosio, P.; Cozzi, A.; Chasteen, N.D. Identification of the EPR-Active Iron-Nitrosyl Complexes in Mammalian Ferritins. Biochemistry 1994, 33, 3679–3687. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, H.; Stępkowski, T.M.; Sadło, J.; Wójciuk, G.P.; Wójciuk, K.E.; Rodger, A.; Kruszewski, M. Coordination of Iron Ions in The Form of Histidinyl Dinitrosyl Complexes Does Not Prevent Their Genotoxicity. Bioorganic Med. Chem. 2012, 20(22), 6732–6738. [Google Scholar] [CrossRef]
- Cesareo, E.; Parker, L.J.; Pedersen, J.Z.; Nuccetelli, M.; Mazzetti, A.P.; Pastore, A.; Federici, G.; Caccuri, A.M.; Ricci, G.; Adams, J.J.; et al. Nitrosylation of Human Glutathione Transferase P1-1 with Dinitrosyl Diglutathionyl Iron Complex in vitro and in vivo. J. Biol. Chem. 2005, 280, 42172–42180. [Google Scholar] [CrossRef] [PubMed]
- De Maria, F.; Pedersen, J.Z.; Caccuri, A.M.; Antonini, G.; Turella, P.; Stella, L.; Lo Bello, M.; Federici, G.; Ricci, G. The Specific Interaction of Dinitrosyl-Diglutathionyl-Iron Complex, a Natural NO Carrier, with the Glutathione Transferase Superfamily: Suggestion for an Evolutionary Pressure in the Direction of the Storage of Nitric Oxide. J. Biol. Chem. 2003, 278, 42283–42293. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.C.; Bosworth, C.A.; Hennon, S.W.; Mahtani, H.A.; Bergonia, H.A.; Lancaster, J.R. Nitric Oxide-Induced Conversion of Cellular Chelatable Iron into Macromolecule-Bound Paramagnetic Dinitrosyliron Complexes. J. Biol. Chem. 2008, 283, 28926–28933. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Salerno, L.; Di Giacomo, C.; Acquaviva, R.; Siracusa, M.A.; Vanella, A. Imidazole Derivatives as Antioxidants and Selective Inhibitors of nNOS. Nitric Oxide 2006, 14, 45–50. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wójciuk, K.E.; Sadło, J.; Lewandowska, H.; Brzóska, K.; Kruszewski, M. A Crucial Role of Proteolysis in the Formation of Intracellular Dinitrosyl Iron Complexes. Molecules 2024, 29, 1630. https://doi.org/10.3390/molecules29071630
Wójciuk KE, Sadło J, Lewandowska H, Brzóska K, Kruszewski M. A Crucial Role of Proteolysis in the Formation of Intracellular Dinitrosyl Iron Complexes. Molecules. 2024; 29(7):1630. https://doi.org/10.3390/molecules29071630
Chicago/Turabian StyleWójciuk, Karolina E., Jarosław Sadło, Hanna Lewandowska, Kamil Brzóska, and Marcin Kruszewski. 2024. "A Crucial Role of Proteolysis in the Formation of Intracellular Dinitrosyl Iron Complexes" Molecules 29, no. 7: 1630. https://doi.org/10.3390/molecules29071630