

Supramolecular Solid Complexes between Bis-pyridinium-4-oxime and Distinctive Cyanoiron Platforms
 , , , , ,
, , , , ,  and
and 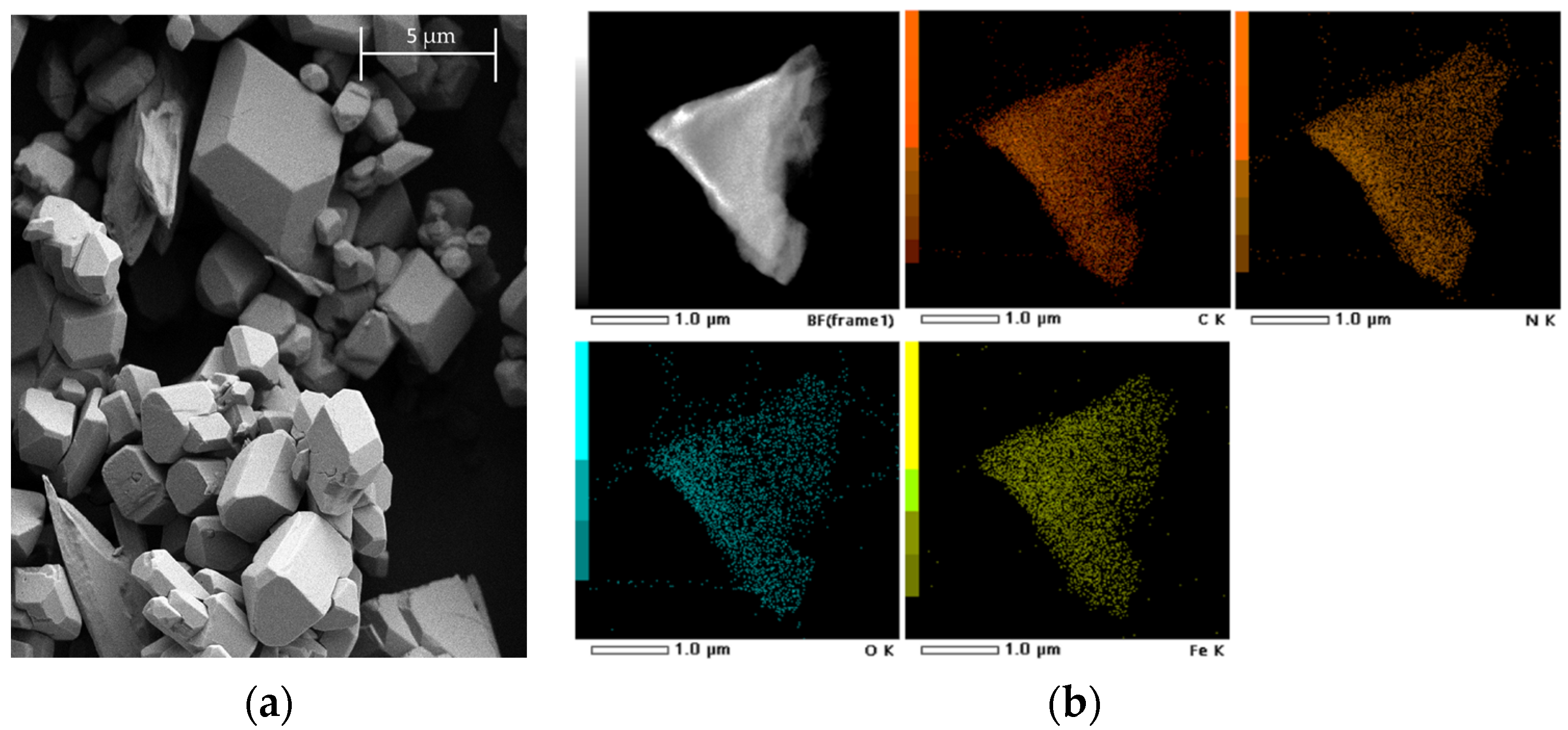{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
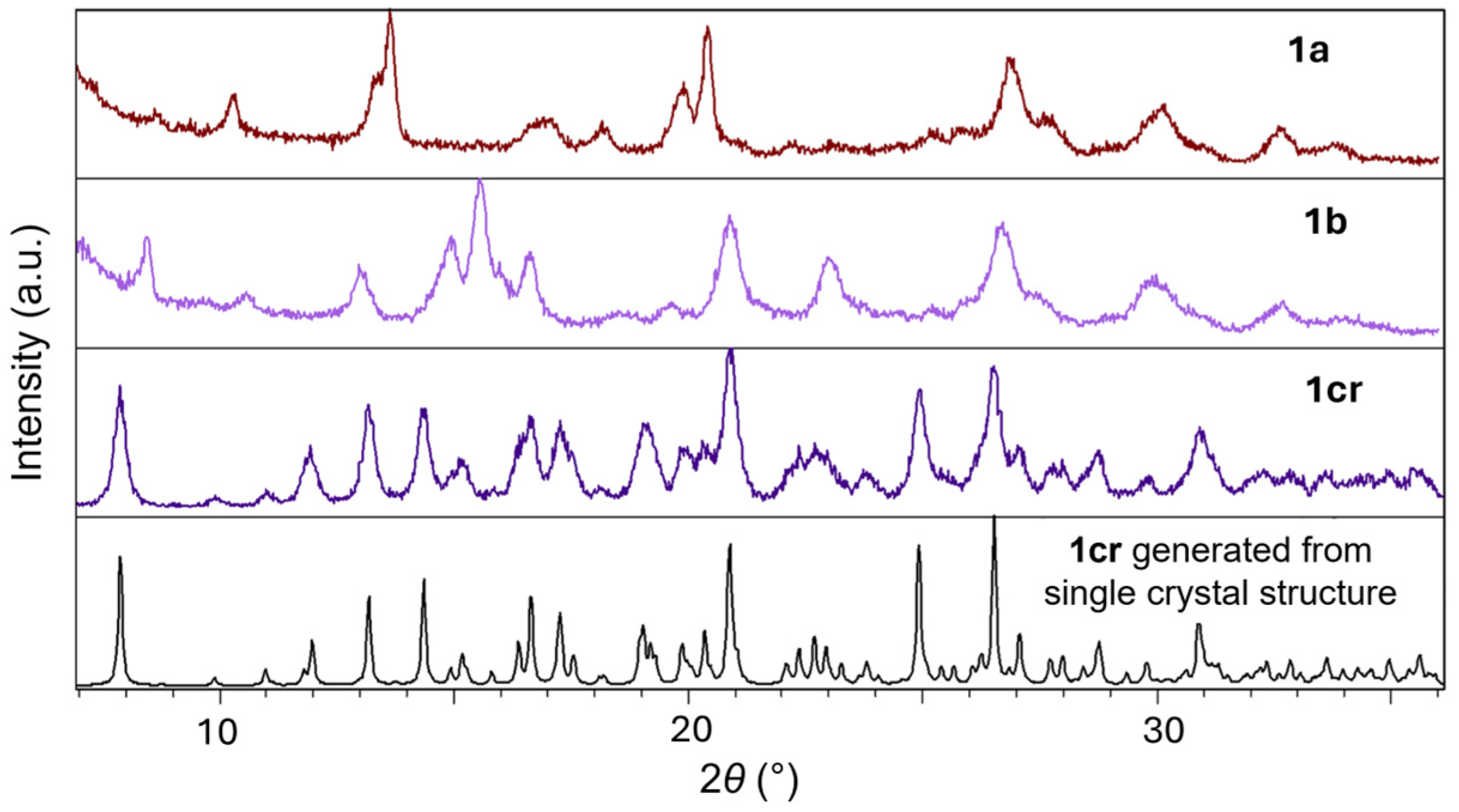
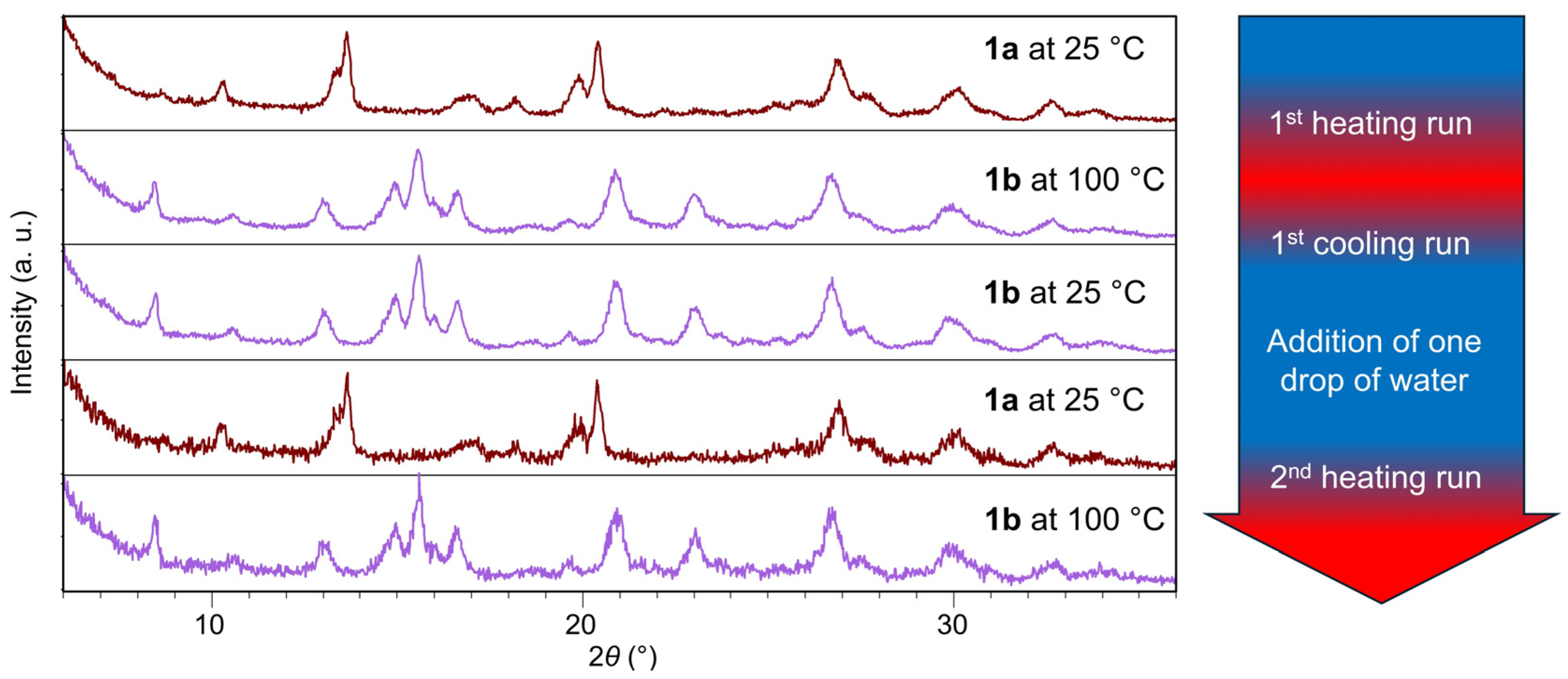
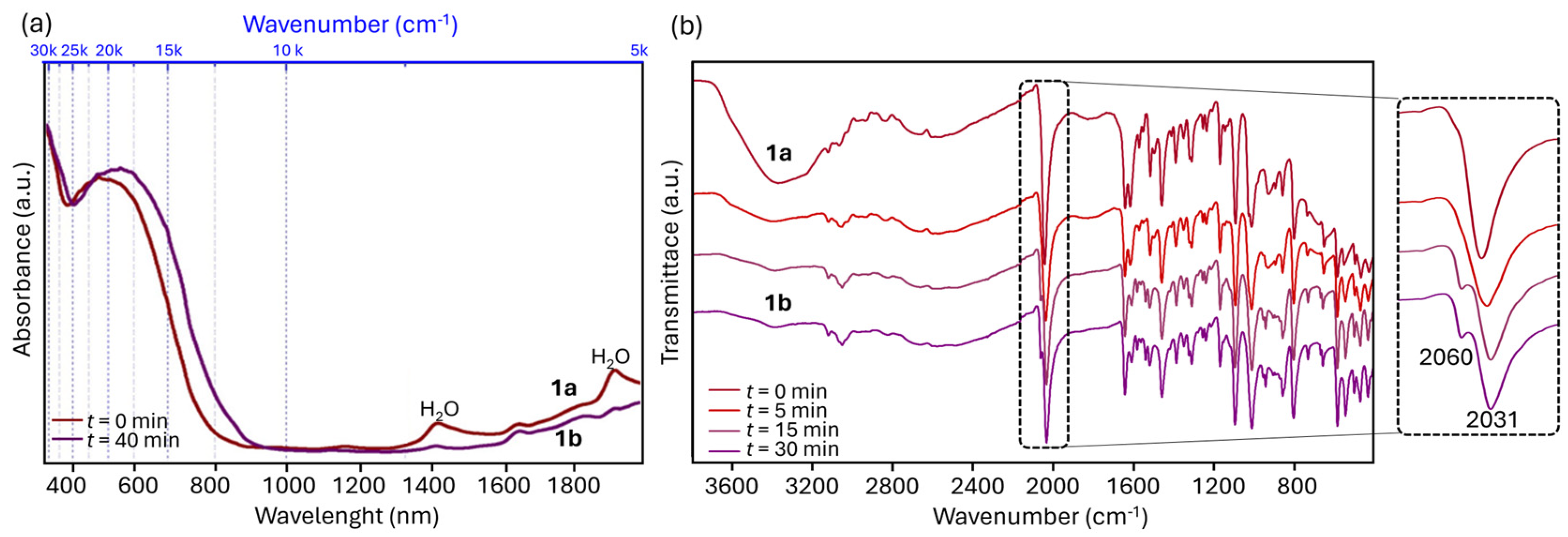
2. Results and Discussion
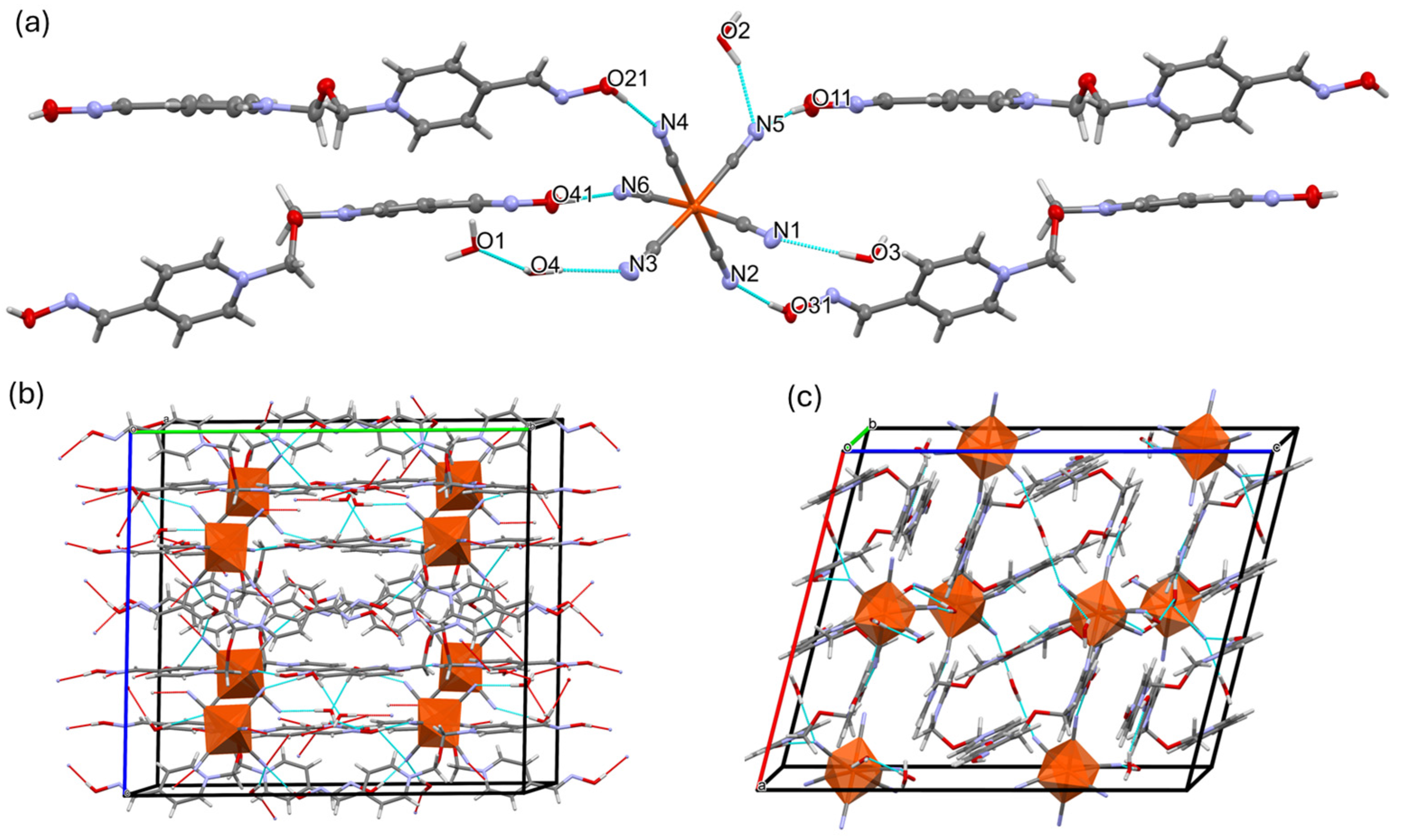
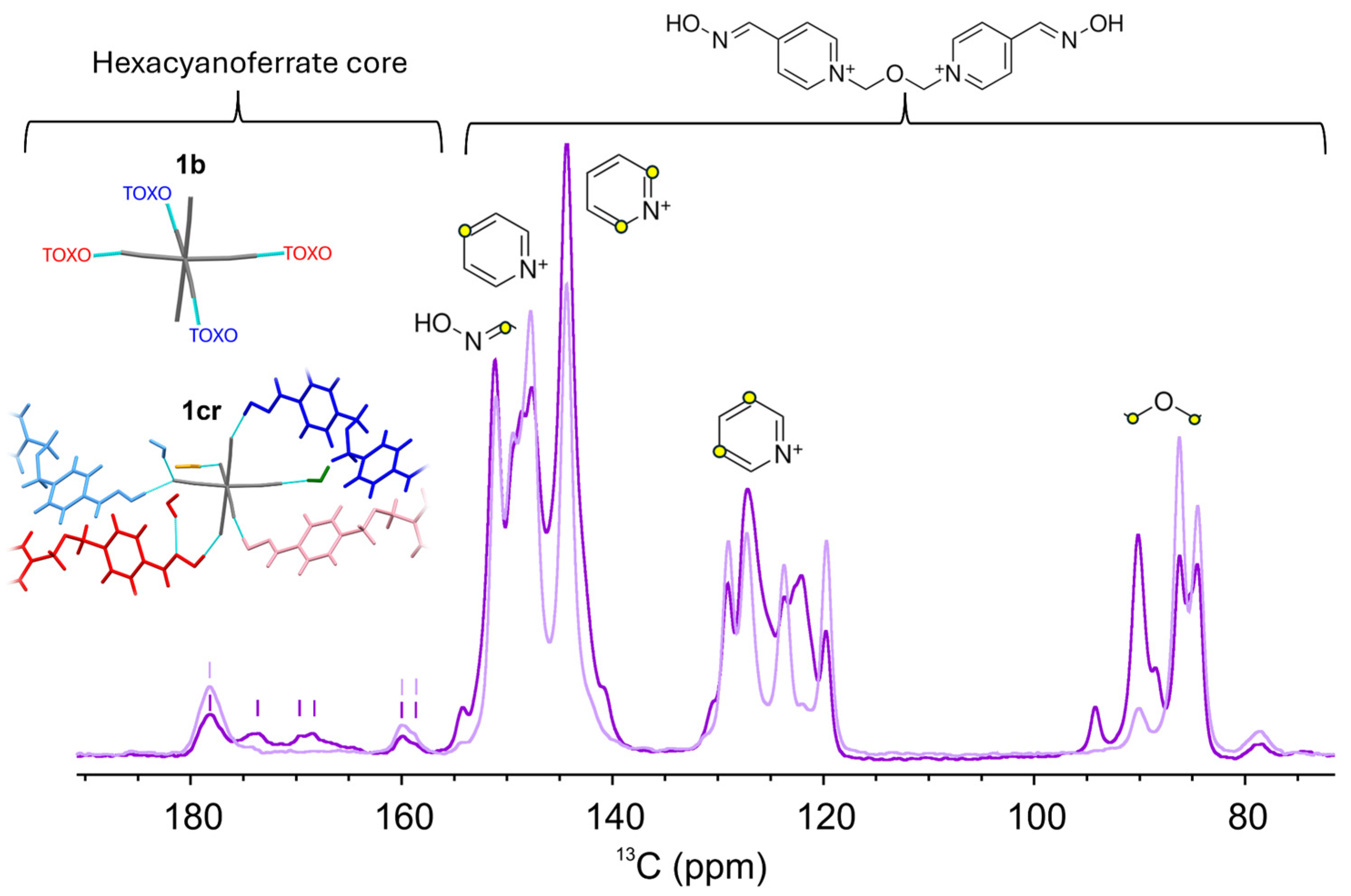
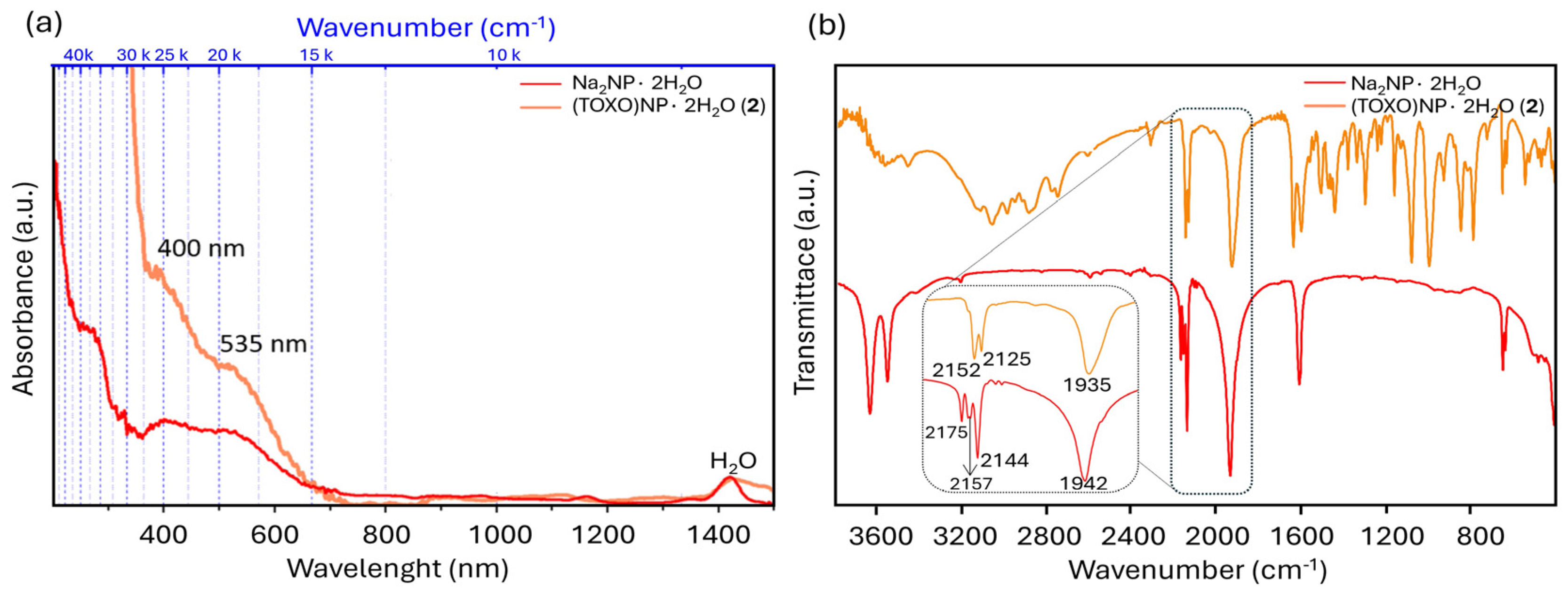
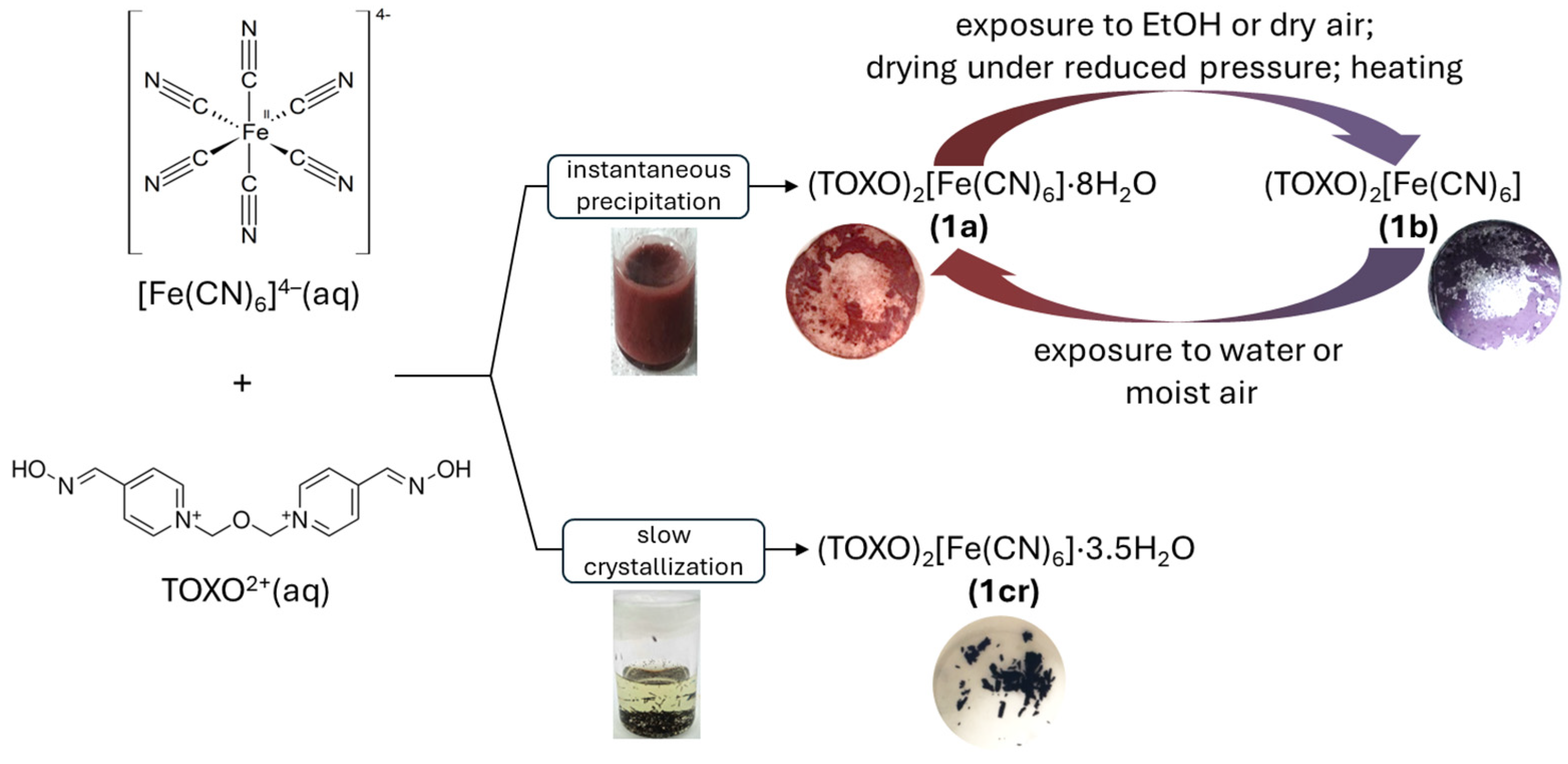
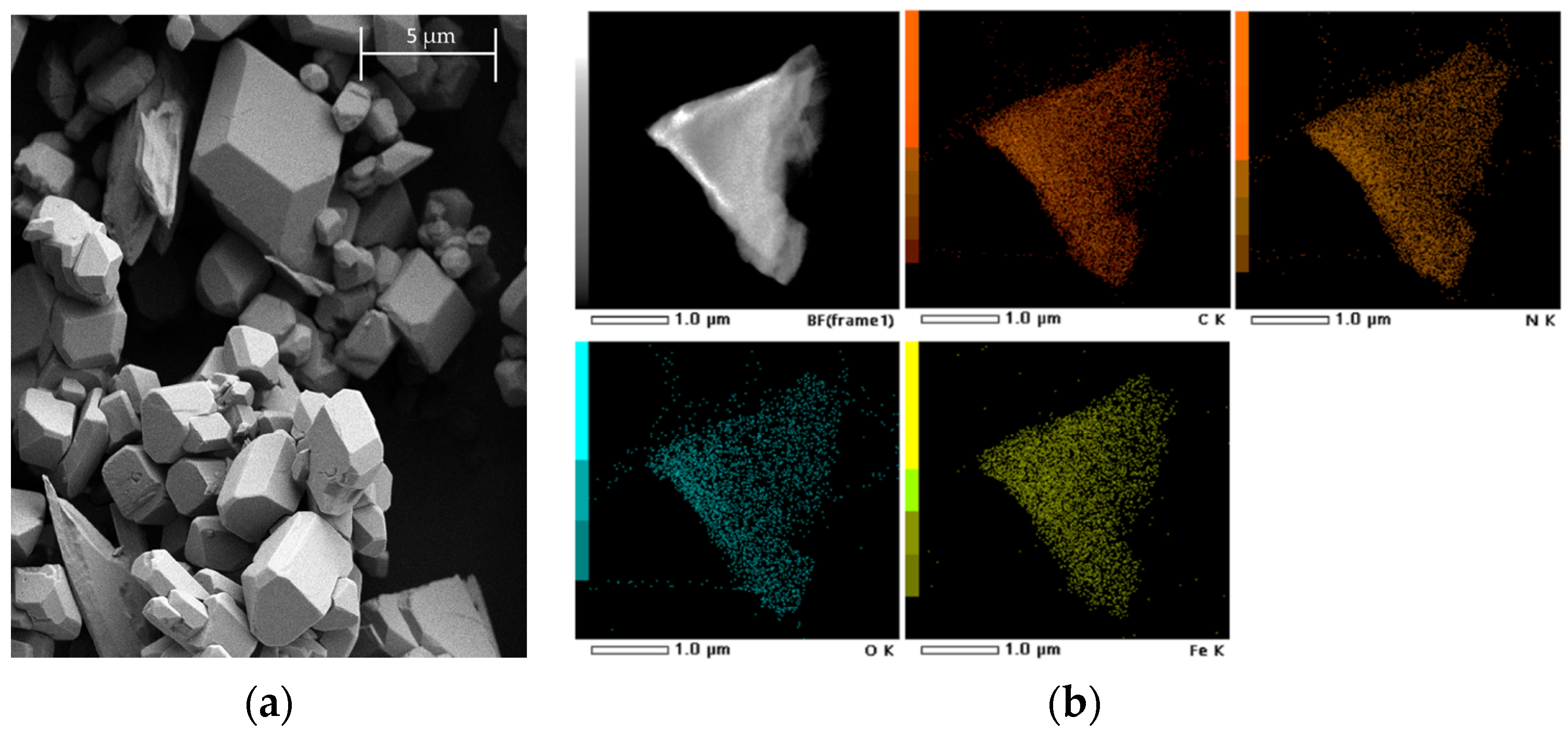
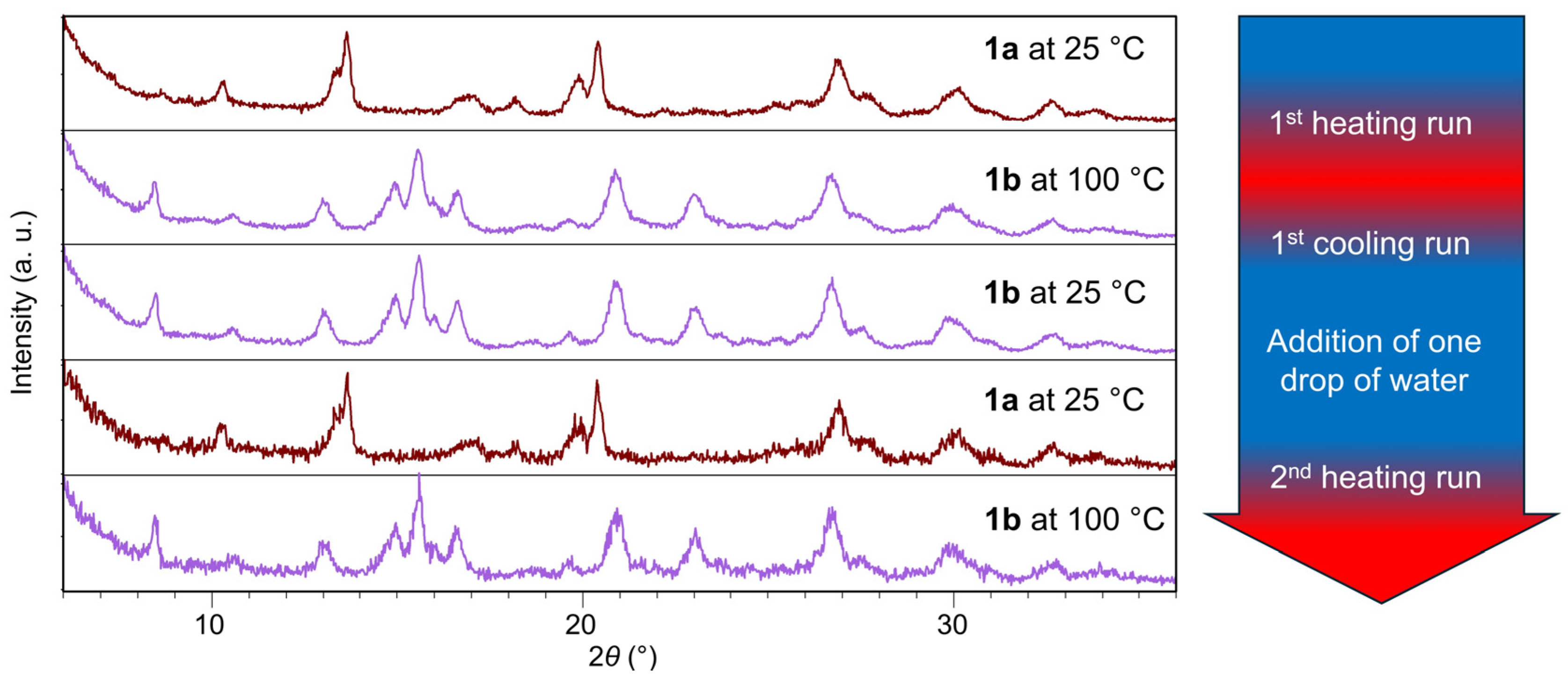
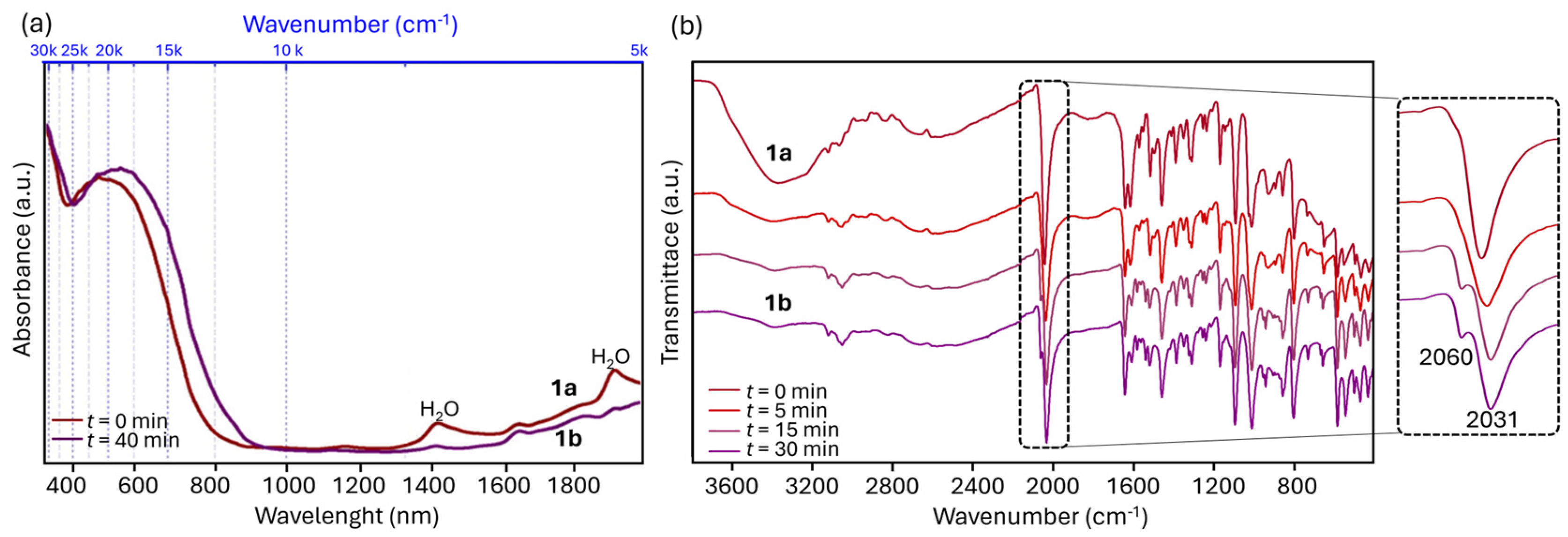
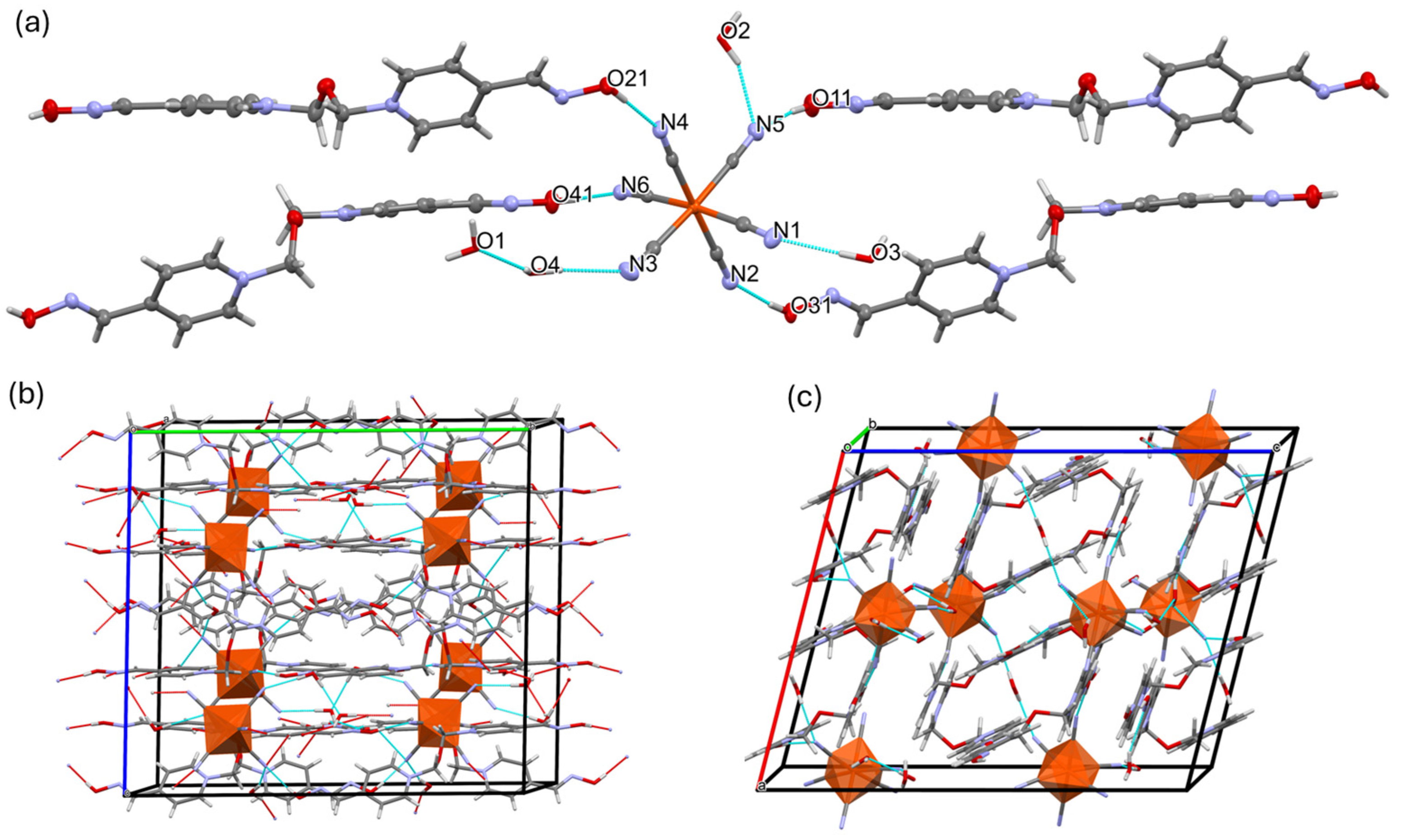
2.1. Inter-Ionic Charge-Transfer Hexacynoferrate(II) Supramolecular Structures
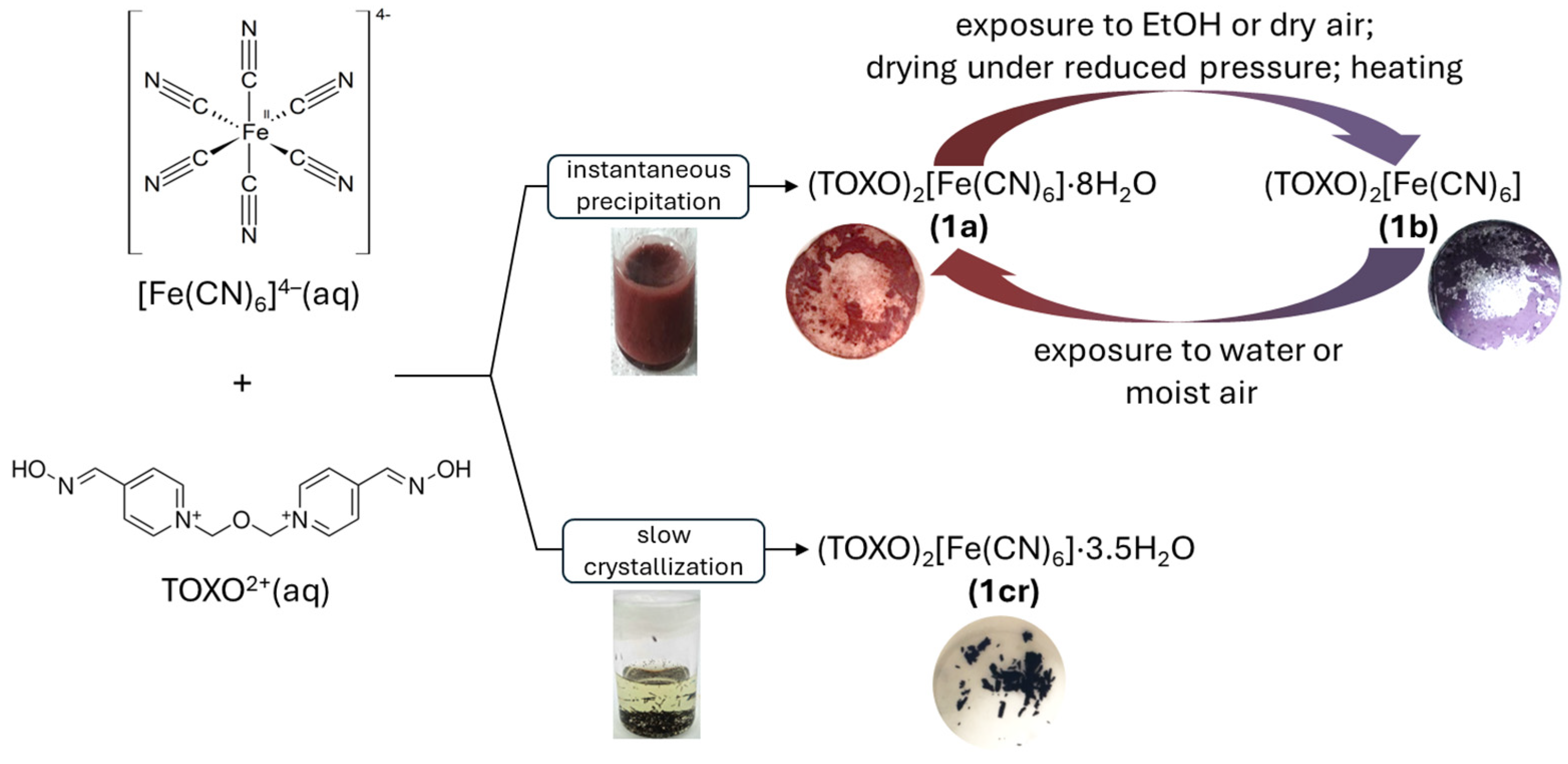
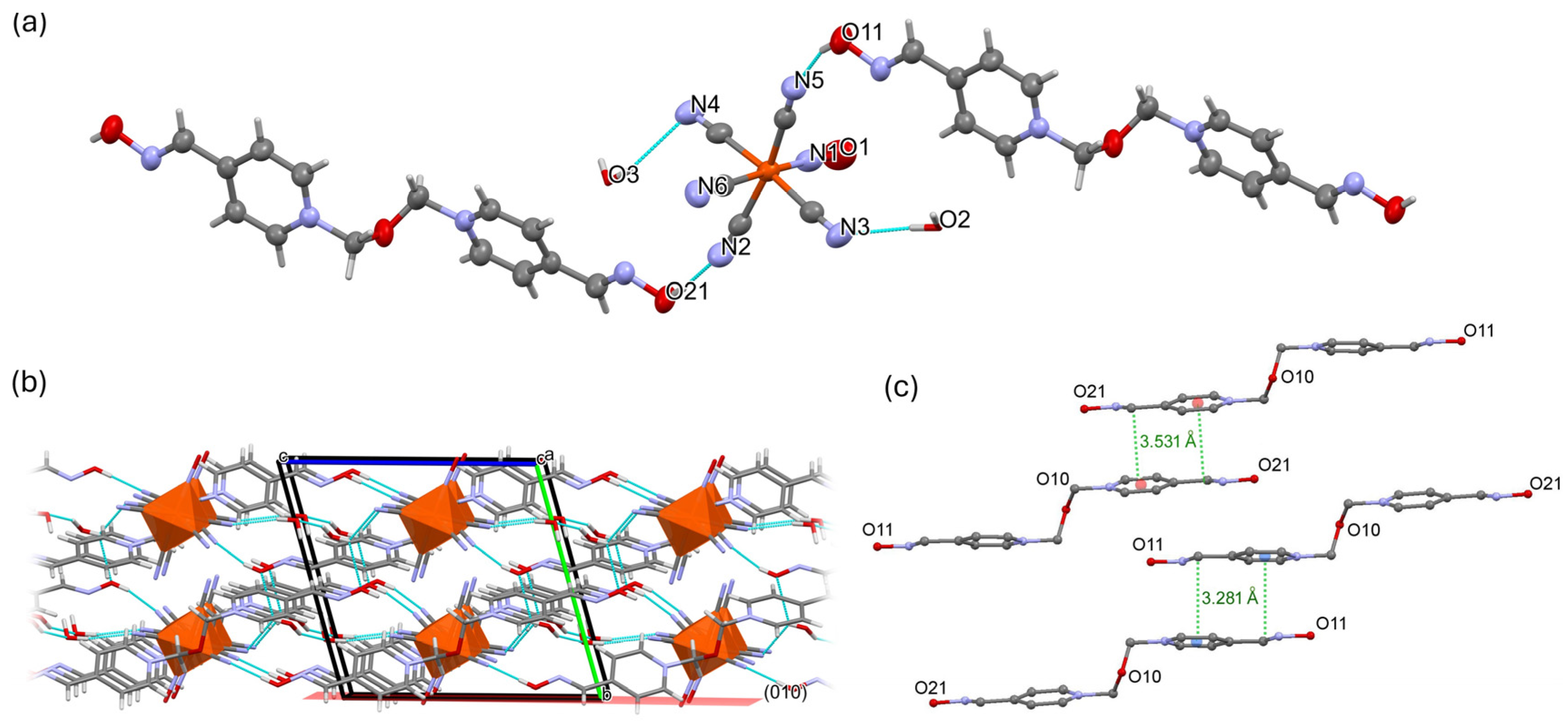
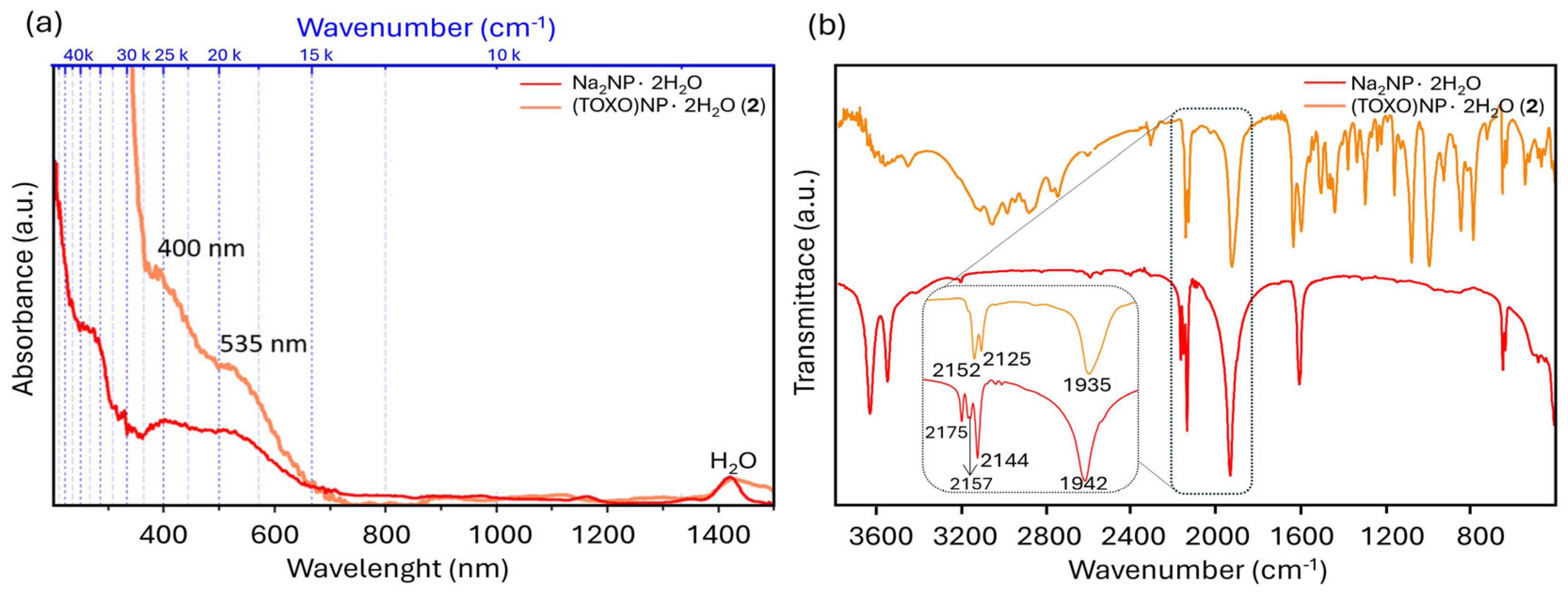
2.2. Inter-Ionic Pentacyanonitrosylferrate Supramolecular Complex
3. Materials and Methods
3.1. Chemicals and Experimental Techniques
3.2. Isolation of Supramolecular Complexes
3.3. SCXRD Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jokanović, M.; Stojiljković, M.P.; Kovač, B.; Ristić, D. Pyridinium oximes in the treatment of poisoning with organophosphorus compounds. In Handbook of Toxicology of Chemical Warfare Agents, 3rd ed.; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2020; Chapter 68; pp. 1145–1159. [Google Scholar]
- Jokanović, M. Structure-Activity Relationship and Efficacy of Pyridinium Oximes in the Treatment of Poisoning with Organophosphorus Compounds: A Review of Recent Data. Curr. Med. Chem. 2012, 12, 1775–1789. [Google Scholar] [CrossRef]
- Kovacic, P. Mechanism of Organophosphates (Nerve Gases and Pesticides) and Antidotes: Electron Transfer and Oxidative Stress. Curr. Med. Chem. 2003, 10, 2705–2709. [Google Scholar] [CrossRef]
- Kovacic, P. Oxidative stress and therapeutic modalities. Med. Hypotheses 2006, 67, 151–156. [Google Scholar] [CrossRef]
- Foretić, B.; Damjanović, V.; Vianello, R.; Picek, I. Novel Insights into the Thioesterolytic Activity of N-Substituted Pyridinium-4-oximes. Molecules 2020, 25, 2385. [Google Scholar] [CrossRef]
- Picek, I.; Vianello, R.; Šket, P.; Plavec, J.; Foretić, B. Tandem β-elimination/hetero-michael addition rearrangement of an N-alkylated pyridinium oxime to an O-alkylated pyridine oxime ether: An experimental and computational study. J. Org. Chem. 2015, 80, 2165–2173. [Google Scholar] [CrossRef]
- Zorbaz, T.; Kovarik, Z. Neuropharmacology of oxime antidotes. Period. Biol. 2020, 121–122, 35–54. [Google Scholar] [CrossRef]
- Foretić, B.; Vianello, R.; Matković-Čalogović, D.; Jadreško, D.; Picek, I. Supramolecular inter-ionic charge-transfer complexes between derivatives of pyridinium-4-oxime cations and hexacyanoferrate(II) anions. New J.Chem. 2018, 42, 16115–16126. [Google Scholar] [CrossRef]
- Papadakis, R. Mono- and Di-Quaternized 4,4’-Bipyridine Derivatives as Key Building Blocks for Medium- and Environment-Responsive Compounds and Materials. Molecules 2020, 25, 1. [Google Scholar] [CrossRef]
- Paolella, A.; Faure, C.; Timoshevskii, V.; Marras, S.; Bertoni, G.; Guerfi, A.; Vijh, A.; Armande, M.; Zaghib, K. A review on hexacyanoferrate-based materials for energy storage and smart windows: Challenges and perspectives. J. Mater. Chem. A 2017, 5, 18919–18932. [Google Scholar] [CrossRef]
- Reguera, L.; Avila, Y.; Reguera, E. Transition metal nitroprussides: Crystal and electronic structure, and related properties. Coord. Chem. Rev. 2021, 434, 213764. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, S.; Yin, J.J.; He, W.; Lu, W.; Ma, M.; Gu, N.; Zhang, Y. Prussian Blue Nanoparticles as Multienzyme Mimetics and Reactive Oxygen Species Scavengers. J. Am. Chem. Soc. 2016, 138, 5860–5865. [Google Scholar] [CrossRef]
- Boxhoorn, G.; Moolhuysen, J.; Coolegem, J.P.G.; van Santen, R.A. Cyanometallates: An underestimated class of molecular sieves. J. Chem. Soc. Chem. Commun. 1985, 19, 1305–1307. [Google Scholar] [CrossRef]
- González, M.; Osiry, H.; Martínez, M.; Rodríguez-Hernández, J.; Lemus-Santana, A.A.; Reguera, E. Magnetic interaction in a 2D solid through hydrogen bonds and π-π stacking. J. Magn. Magn. Mater. 2019, 471, 70–76. [Google Scholar] [CrossRef]
- Wang, L.; Tricard, S.; Yue, P.; Zhao, J.; Fang, J.; Shen, W. Polypyrrole and graphene quantum dots @ Prussian Blue hybrid film on graphite felt electrodes: Application for amperometric determination of l-cysteine. Biosens. Bioelectron. 2016, 77, 1112–1118. [Google Scholar] [CrossRef]
- Culp, J.T.; Madden, C.; Kauffman, K.; Shi, F.; Matranga, C. Screening Hofmann Compounds as CO2 Sorbents: Nontraditional Synthetic Route to Over 40 Different Pore-Functionalized and Flexible Pillared Cyanonickelates. Inorg. Chem. 2013, 52, 4205–4216. [Google Scholar] [CrossRef]
- Stepanenko, I.; Zalibera, M.; Schaniel, D.; Telser, J.; Arion, B.V. Ruthenium-nitrosyl complexes as NO-releasing molecules, potential anticancer drugs, and photoswitches based on linkage isomerism. Dalton Trans. 2022, 51, 5367–5393. [Google Scholar] [CrossRef]
- da Silva Filho, P.M.; Paz, I.A.; Falcão do Nascimento, N.R.; Abreu, D.S.; de França Lopes, L.G.; Sousa, E.H.S.; Longhinotti, E. Nitroprusside-Expanding the Potential Use of an Old Drug Using Nanoparticles. Mol. Pharm. 2023, 20, 6–22. [Google Scholar] [CrossRef]
- Rose, M.J.; Mascharak, P.K. Photoactive ruthenium nitrosyls: Effects of light and potential application as NO donors. Coord. Chem. Rev. 2008, 252, 2093–2114. [Google Scholar] [CrossRef]
- Cvrtila, I.; Stilinović, V. New Tricks by Old Anions: Hydrogen Bonded Hexacyanoferrous Anionic Networks. Cryst. Growth Des. 2017, 17, 6793–6800. [Google Scholar] [CrossRef]
- Tanaka, R.; Matsushita, N. Crystal structure of bis(1-ethylpyridinium) dioxonium hexacyanidoferrate(II). Acta Cryst. 2017, E73, 219–222. [Google Scholar] [CrossRef]
- Tanaka, R.; Okazawa, A.; Kojima, N.; Matsushita, N. Ionic Crystal Containing Protons (H+) as Counter Cations: Preparation and Crystal Structure of a Salt of 4,4’-Bipiperidine-1,1’-diium and Hexacyanidoferrate(II). Chem. Lett. 2018, 47, 697–699. [Google Scholar] [CrossRef]
- Tanaka, R.; Okazawa, A.; Konaka, H.; Sasaki, A.; Kojima, N.; Matsushita, N. Unique Hydration/Dehydration-Induced Vapochromic Behavior of a Charge-Transfer Salt Comprising Viologen and Hexacyanidoferrate(II). Inorg. Chem. 2018, 57, 2209–2217. [Google Scholar] [CrossRef]
- Xydias, P.; Lymperopoulou, S.; Dokorou, V.; Manos, M.; Plakatouras, J.C. Supramolecular networks derived from hexacyanoferrates and nitrogen heterocyclic cations. Polyhedron 2019, 157, 341–357. [Google Scholar] [CrossRef]
- Córdoba, L.M.; Echeverría, G.A.; Piro, O.E.; Gómez, M.I. Ammonium, barium hexacyanoferrate(II) trihydrate: Synthesis, crystal structure, thermal decomposition and spectroscopic study. J. Therm. Anal.Calorim. 2015, 120, 1827–1834. [Google Scholar] [CrossRef]
- Reguera, E.; Fernández-Bertrán, J. Effect of the water of crystallization on the Mössbauer spectra of hexacyanoferrates (II and III). Hyperfine Interact. 1994, 88, 49–58. [Google Scholar] [CrossRef]
- Ferlay, S.; Hellwig, P.; Hosseini, M.W. Partially Reversible Thermal-Induced Oxidation During a Dehydration Process in an H-bonded Supramolecular System. ChemPhysChem 2018, 19, 3219–3225. [Google Scholar] [CrossRef]
- Foretić, B.; Picek, I.; Damjanović, V.; Cvijanović, D.; Milić, D. The structures and stabilities of biologically active 1-phenacyl- and 1-benzoylethyl-derivatives of the pyridinium cation. J. Mol. Struct. 2012, 1019, 196–205. [Google Scholar] [CrossRef]
- Oksana, T.; Leroux, M.; Mercier, N.; Allain, M.; Kassiba, A.H.; Swamy, S.K.K.; Dittmer, J. Bipyridinium-bis(carboxylate) Radical Based Materials: X-ray, EPR and Paramagnetic Solid-State NMR Investigations. Eur. J. Inorg. Chem. 2016, 2016, 1036–1043. [Google Scholar]
- Willans, J.M.; Wasylishen, R.E.; McDonald, R. Polymorphism of Potassium Ferrocyanide Trihydrate as Studied by Solid-State Multinuclear NMR Spectroscopy and X-ray Diffraction. Inorg. Chem. 2009, 48, 4342–4353. [Google Scholar] [CrossRef]
- Clemente-León, M.; Coronado, E.; Galán-Mascarós, J.R.; Gómez-García, C.J.; Woike, T.; Clemente-Juan, J.M. Bimetallic Cyanide-Bridged Complexes Based on the Photochromic Nitroprusside Anion and Paramagnetic Metal Complexes. Inorg. Chem. 2001, 40, 87–94. [Google Scholar] [CrossRef]
- Balmaseda, J.; Reguera, E.; Gomez, A.; Roque, J.; Vazquez, C.; Autie, M. On the Microporous Nature of Transition Metal Nitroprussides. J. Phys. Chem. B 2003, 107, 11360–11369. [Google Scholar] [CrossRef]
- Cano, A.; Lartundo-Rojas, L.; Shchukarevc, A.; Reguera, E. Contribution to the coordination chemistry of transition metal nitroprussides: A cryo-XPS study. New J. Chem. 2019, 43, 4835–4848. [Google Scholar] [CrossRef]
- Di Santo, A.; Osiry, H.; Reguera, E.; Alborés, P.; Carbonio, R.E.; Altabef, A.B.; Gil, D.M. New coordination polymers based on 2-methylimidazole and transition metal nitroprusside containing building blocks: Synthesis, structure and magnetic properties. New J. Chem. 2018, 42, 1347–1355. [Google Scholar] [CrossRef]
- Gil, D.M.; Osiry, H.; Rodriguez, A.; Lemus-Santana, A.A.; Carbonio, R.E.; Reguera, E. Layered Transition Metal Nitroprussides—Their Preparation, Crystal Structure, and Magnetic Properties. Eur. J. Inorg. Chem. 2016, 2016, 1690–1696. [Google Scholar] [CrossRef]
- Klencsar, Z.; Kuzmann, E.; Vertes, A. User-friendly software for Mössbauer spectrum analysis. J. Radioanal. Nucl. Chem. 1996, 201, 105–118. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.A. Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.G.; Shields, P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picek, I.; Matković-Čalogović, D.; Dražić, G.; Kapun, G.; Šket, P.; Popović, J.; Foretić, B. Supramolecular Solid Complexes between Bis-pyridinium-4-oxime and Distinctive Cyanoiron Platforms. Molecules 2024, 29, 1698. https://doi.org/10.3390/molecules29081698
Picek I, Matković-Čalogović D, Dražić G, Kapun G, Šket P, Popović J, Foretić B. Supramolecular Solid Complexes between Bis-pyridinium-4-oxime and Distinctive Cyanoiron Platforms. Molecules. 2024; 29(8):1698. https://doi.org/10.3390/molecules29081698
Chicago/Turabian StylePicek, Igor, Dubravka Matković-Čalogović, Goran Dražić, Gregor Kapun, Primož Šket, Jasminka Popović, and Blaženka Foretić. 2024. "Supramolecular Solid Complexes between Bis-pyridinium-4-oxime and Distinctive Cyanoiron Platforms" Molecules 29, no. 8: 1698. https://doi.org/10.3390/molecules29081698