3. Materials and Methods
1H and 13C NMR spectra were recorded with a Varian Mercury 300 (at 300 MHz, and 75 MHz, respectively) or a JEOL 400 (at 400 MHz and 101 MHz, respectively).
Unless otherwise stated, NMR spectra were recorded using residual solvent as the internal standard 1H NMR: TMS = 0.00; (CD3)2SO = 2.50; and 13C NMR: CDCl3 = 77.16; (CD3)2SO = 39.52. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), integration, multiplicity, and coupling constants (Hz). Data for 13C NMR spectra are reported in terms of chemical shift (δ ppm). Interpretation of spectra has been made also with the aid of gCOSY, gHSQC, and gHMBC experiments. The following abbreviations are used to indicate the multiplicity in NMR spectra: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet.
IR spectra were recorded directly on solid, oil, or foamy samples, with the ATR (attenuated total reflectance) technique, using a FT Perkin Elmer Spectrum 65 spectrophotometer. TLC analyses were carried out on silica gel plates, viewed at UV (ν = 254 nm) and developed with Hanessian stain (dipping into a solution of (NH4)4MoO4·4H2O (21 g) and Ce(SO4)2·4H2O (1 g) in H2SO4 (31 mL) and H2O (469 mL) and warming. Rf values were measured after an elution of 7–9 cm. Chiral HPLC analyses for the determination of enantiomeric excess were performed on a Daicel Chiral Pak AD 250 × 4.6 mm column, at 25–26 °C with a flow of about 0.8 mL/min (UV detection at ν = 220 nm). HPLC-MS analyses were performed on Synergi Hydro RP 150 × 3 mm column, at 30 °C with a flow of 0.5 mL/min (where not otherwise stated). For MS, the ESI+ ionization method was used. HPLC-UV analyses were carried out on a HP-1100 system (Agilent, Santa Clara, CA, USA) equipped with (a) a HYDRO RP column (150 × 3 mm, 4 μ) at 25 °C with flow = 0.5 mL/min and isocratic elution (CH3CN/H2O 50:50). Detection was conducted with UV at 220 nm; (b) a C6 PHENYLIC RP column (150 × 3 mm, 3 μ) at 25 °C with flow = 0.38 mL/min and gradient H2O/CH3CN, A = CH3CN—B = H2O, 0 min B = 70%, 20 min B = 0%. HRMS: samples, provided at 10 mM in DMSO, were diluted at 50 µM with acetonitrile/water 1:1, and analyzed on a UPLC Acquity system coupled to a Synapt G2 QToF mass spectrometer. MS signals were acquired from 50 to 1200 m/z ESI positive ionization mode. UPLC was carried out with H2O–CH3CN–HCO2H with an Acquity UPLC BEH C18, 1.7 µM, 2.1 × 50 mm column at 45 °C. Column chromatography was performed with the “flash” methodology using 220–400 mesh silica. Melting points were determined with an electrothermal apparatus (Büchi B-535). Petroleum ether (40–60 °C) is abbreviated as PE. All reactions employing dry solvents were carried out under a nitrogen atmosphere. After extractions, the aqueous phases were always re-extracted three times with the appropriate organic solvent, and the organic extracts were always dried over Na2SO4 and filtered before evaporation to dryness.
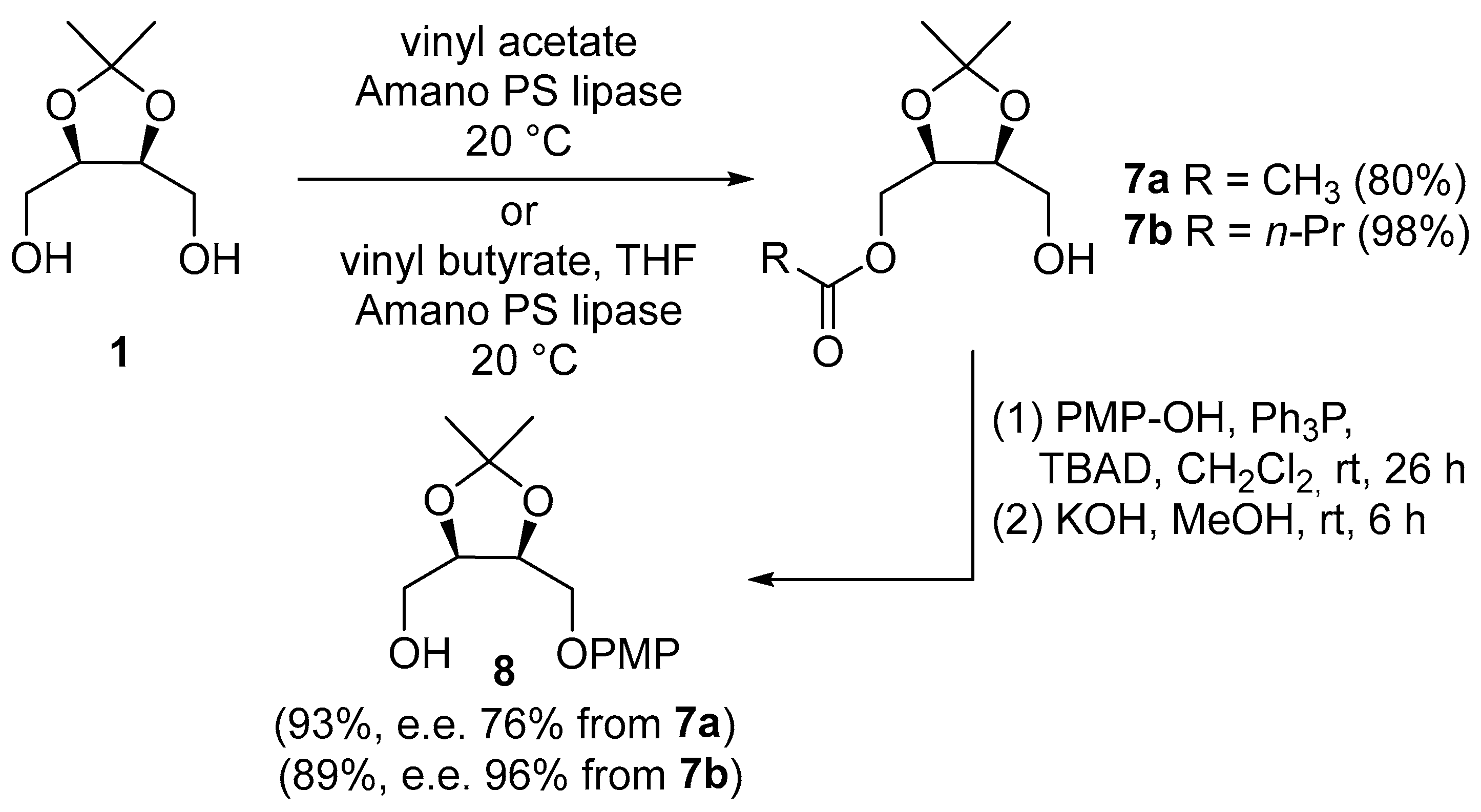
((4R,5S)-5-((4-methoxyphenoxy)methyl)-2,2-dimethyl-1,3-dioxolan-4-yl)methanol 8: from 7a: To a solution of 7a (110 mg, 0.50 mmol), triphenylphosphine (193 mg, 0.73 mmol) and p-methoxyphenol (182 mg, 1.50 mmol) in dry CH2Cl2 (5 mL) at 0 °C was added to tert-butyl azodicarboxylate (175 mg, 0.74 mmol). The mixture was stirred at room temperature for 37 h. Then, the reaction mixture was concentrated and filtered through a short column of silica gel with PE/CH2Cl2/Et2O (1:1:1). The residue was directly diluted with MeOH (2 mL) and treated with KOH (0.80 mL, 6 M in MeOH). The reaction mixture was stirred at room temperature for 6 h, then diluted with saturated NH4Cl aq, extracted with Et2O, dried (Na2SO4), and concentrated. The crude residue was eluted from a column of silica gel with PE/AcOEt 3:1 to give 8 (152 mg, 93%, e.e. 76%) as a colorless oil.
From 7b: To a solution of 7b (4.71 g, 21.57 mmol), triphenylphosphine (8.49 g, 32.35 mmol) and p-methoxyphenol (8.03 g, 64.70 mmol) in dry CH2Cl2 (215 mL) at 0 °C was added to tert-butyl azodicarboxylate (7.45 g, 32.36 mmol). The mixture was stirred at room temperature for 37 h. Then, the reaction mixture was concentrated and filtered through a short column of silica gel with PE/CH2Cl2/Et2O (1:1:1). The residue was directly diluted with MeOH (108 mL) and treated with KOH (32 mL, 1 M in MeOH). The reaction mixture was stirred at room temperature for 6 h, then diluted with saturated NH4Cl aq, extracted with Et2O, dried (Na2SO4), and concentrated. The crude residue was eluted from a column of silica gel with PE/AcOEt (from 2:1 to 1:1) to give 8 (5.16 g, 89%, e.e. 95%) as a colorless oil. The enantiomeric excess was determined using HPLC on a chiral stationary phase. Conditions: column Daicel Chiral Pak AD (250 × 4.6 mm); detector DAD (220 nm); flow 0.8 mL min−1. Isocratic elution with n-hexane/isopropanol 90: 10. Temperature: 25 °C. Rt 14.3 min. (4R,5S) and 17.0 min (4S,5R). Rf = 0.27 (PE/AcOEt 2:1); [α]D20 = +8.2 (c 1.0, CHCl3); m.p. 52.3–54.2 °C (CH2Cl2); IR (ATR): ν = 3519, 3058, 2988, 2938, 2887, 2836, 1509, 1460, 1374, 1335, 1289, 1216, 1182, 1164, 1111, 1089, 1050, 1035, 996, 907, 845, 830, 818, 803, 751, 715, 650 cm−1; 1H NMR (CDCl3, 400 MHz): δ = 6.91–6.78 (m, 4H, aromatic H), 4.55 (q, J = 6.3 Hz, 1H, CH-CH2OPMP), 4.40 (q, J = 6.3 Hz, 1H, CH-CH2OH), 4.08–4.01 (m, 2H, CH2OPMP), 3.88–3.74 (m, 2H, CH2OH), 3.77 (s, 3H, OCH3), 2.22 (q, J = 6.4 Hz, 1H, OH), 1.50 (s, 3H, CH3 of acetonide), 1.41 (s, 3H, CH3 of acetonide); 13C NMR (CDCl3, 101 MHz): δ = 154.5 (Cq Ar), 152.3 (Cq Ar), 115.7 (2 CH Ar), 114.8 (2 CH Ar), 109.0 (Cq acetonide), 77.4 (CH-CH2OH), 75.0 (CH-CH2OPMP), 67.2 (CH2OPMP), 61.1 (CH2OH), 55.8 (OCH3), 27.9 (CH3 acetonide), 25.3 (CH3 acetonide); HRMS (ESI+) m/z: [M + Na]+ Calcd for C14H20NaO5+: 291.1203; Found: 291.1106.
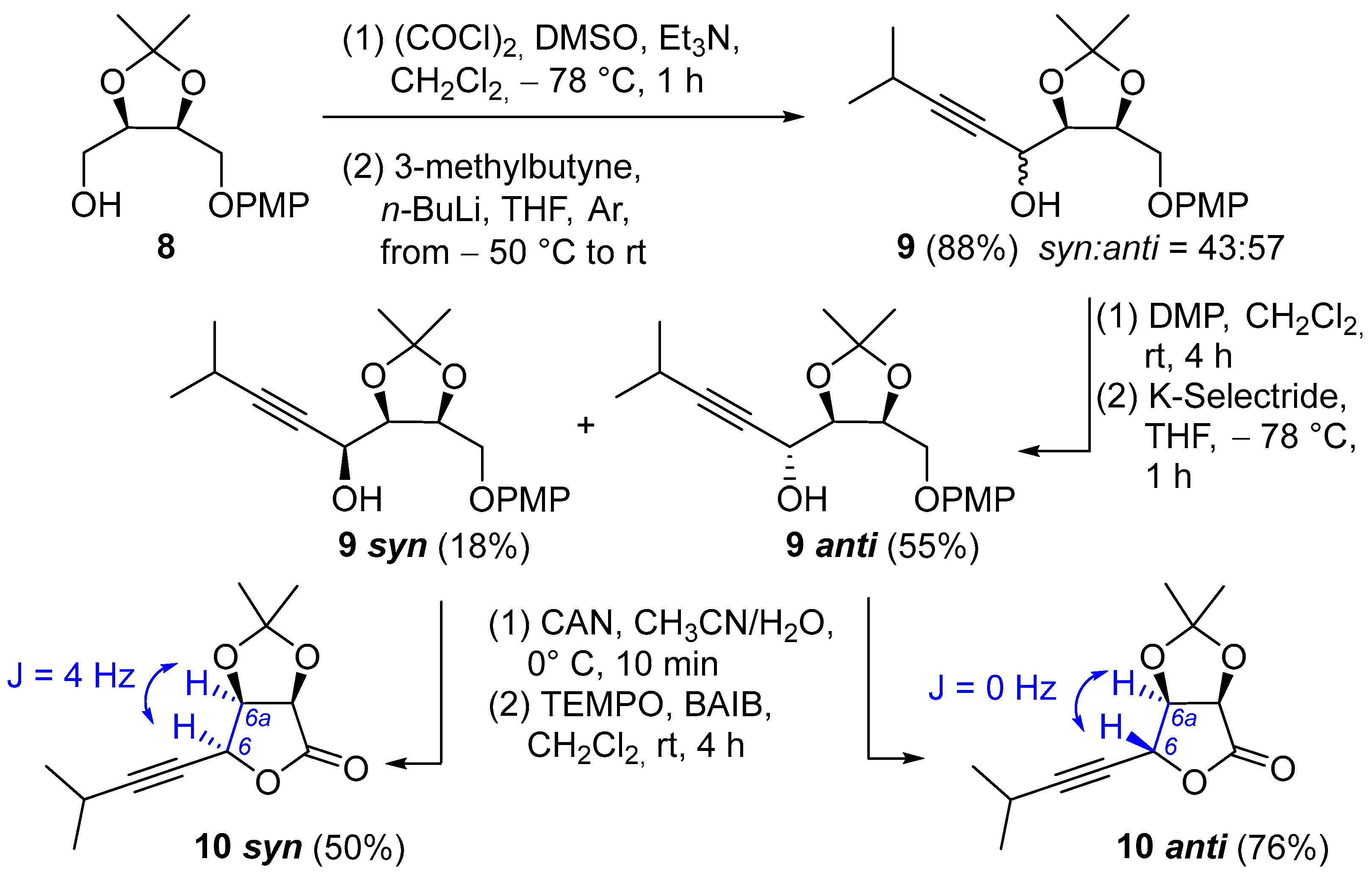
1-((4R,5S)-5-((4-methoxyphenoxy)methyl)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-methylpent-2-yn-1-ol (9): To a solution of DMSO (1.1 mL, 15.08 mmol), in dry CH2Cl2 (36 mL), at −70 °C, under a nitrogen atmosphere, a solution of oxalyl chloride in dry CH2Cl2 (1.43 M, 9.8 mL) was added. The solution was stirred for approximately 10 min, until effervescence ceased. A solution of 8 (1.50 g, 5.59 mmol) in dry CH2Cl2 (20 + 10 + 6 mL) was added dropwise, and the solution was stirred for 10 min at −70 °C. NEt3 (4.3 mL, 30.73 mmol) was then added, and the solution was stirred for 2 h at −70 °C. After this time, the reaction mixture was poured into a mixture of 5% aq (NH4)H2PO4 (90 mL) and 1 M HCl (10 mL) (final pH 4) and extracted with Et2O (100 + 30 mL). The organic layer was washed with brine (20 mL), dried (Na2SO4), and concentrated. The resulting crude aldehyde was rapidly solubilized in THF (20 mL) under Ar and used as such for the next reaction. To a solution of 2,2′-bipyridine (catalytic amount) in dry THF (30 mL) under Ar, at −50 °C, n-BuLi (11 mL, 1.6 M in hexane) was added, until a deep red color persisted. Then, 3-methyl-1-butyne (2 mL, 19.55 mmol) was added, and the mixture was stirred for 25 min. After this time, the temperature was kept at −70 °C, and the solution of aldehyde was slowly added to the mixture. The reaction was stirred for 1 h at −50 °C and overnight at room temperature. The reaction was diluted with saturated NH4Cl aq., extracted with AcOEt, dried (Na2SO4) and concentrated. The residue was eluted from a column of silica gel with PE/Et2O 3:1 to give 9 (1.64 g, 88%) as a 57:43 mixture of diastereoisomers (PHENYLIC RP column 150 × 3 mm, 3 μm, temp 25 °C, flow = 0.38 mL/min, mobile phase H2O/CH3CN, A = CH3CN—B = H2O, 0 min B = 90%, 30 min B = 0%. Rt (syn) = 12.0 min, Rt (anti) = 12.4 min).
Oxidation and diastereoselective reduction to give 9 anti: To a solution of 9 (969 mg, 2.90 mmol), in dry CH2Cl2 (22 mL), under a nitrogen atmosphere, Dess Martin periodinane (1.35 g, 3.19 mmol) was added at 0 °C, and the reaction was stirred at room temperature for 4.5 h. The mixture was quenched with NaHCO3 (5% w/v aqueous solution)/Na2S2O3 (0.4 M in water) (1:1), extracted with CH2Cl2, dried (Na2SO4), and concentrated to afford the corresponding ketone, which was used as such for the next reaction. To a solution of ketone in dry THF (29 mL) at −78 °C under a nitrogen atmosphere, K-Selectride (1 M in THF, 2.9 mL) was added. After stirring at room temperature for 5 h, the reaction was diluted with saturated NH4Cl (saturated aqueous solution), extracted with AcOEt, washed with brine, dried (Na2SO4), and concentrated. The residue was eluted from a column of silica gel with PE/AcOEt 3:1 to give first 9 anti (529 mg, 55%) as a white solid and 9 syn (168 mg, 18%) as a pale-yellow oil. The diastereoisomeric ratio (76:24) was determined on the crude after the reduction with HPLC (PHENYLIC RP column 150 × 3 mm, 3 μm, temp 25 °C, flow = 0.38 mL/min, mobile phase H2O/CH3CN, A = CH3CN—B = H2O, 0 min B = 90%, 30 min B = 0%. Rt (syn) = 12.0 min, Rt (anti) = 12.4 min).
9 anti: Rf = 0.73 (PE/AcOEt 6:4); [α] D25 = −13.0 (c 1.0, CHCl3); m.p. 85.8–88.3 °C (CHCl3); IR (ATR): ν = 3455, 3222, 2970, 2934, 2835, 1507, 1458, 1381, 1319, 1289, 1228, 1214, 1167, 1125, 1106, 1082, 1038, 884, 856, 824, 727, 639 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 6.93–6.86 (m, 2H, 2 CH Ar), 6.86–6.79 (m, 2H, 2 CH Ar), 4.65–4.55 (m, 2H, CH-4 and CHOH), 4.36 (dd, J = 10.2, 5.2 Hz, 1H, 1 H of CH2), 4.31 (dd, J = 6.5, 5.1 Hz, 1H, CH-5), 4.20 (dd, J = 10.1, 6.2 Hz, 1H, 1 H of CH2), 3.77 (s, 3H, OCH3), 2.70 (d, J = 5.7 Hz, 1H, OH), 2.57 (pd, J = 6.9, 1.8 Hz, 1H, CH of iPr), 1.55 (s, 3H, CH3 of acetonide), 1.42 (s, 3H, CH3 of acetonide), 1.14 (d, J = 6.9 Hz, 6H, 2 CH3 of iPr); 13C NMR (CDCl3, 75 MHz): δ = 154.1 (Cq Ar), 152.4 (Cq Ar), 115.6 (2 CH Ar), 114.5 (2 CH Ar), 109.1 (Cq acetonide), 92.6 (Cq alkyne), 79.3 (CH-5), 77.2 (Cq alkyne), 75.5 (CH-4), 67.3 (CH2), 61.7 (CHOH), 55.6 (OCH3), 27.3 (CH3 acetonide), 25.2 (CH3 acetonide), 22.6 (2 CH3 of iPr), 20.4 (CH of iPr); HRMS (ESI+) m/z: [M + Na]+ Calcd for C19H26NaO5+: 357.1672; Found: 357.1670.
9 syn: Rf = 0.65 (PE/AcOEt 6:4); [α] D20= −76.3 (c 1.0, CHCl3); IR (ATR): ν = 3455, 3222, 2970, 2934, 2835, 1507, 1458, 1381, 1319, 1289, 1228, 1214, 1167, 1125, 1106, 1082, 1038, 884, 856, 824, 727, 639 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 6.92–6.79 (m, 4H. 4 CH Ar), 4.57 (td, J = 6.5, 4.4 Hz, 1H, CH-5), 4.53–4.48 (m, 1H, CH-OH), 4.30 (dd, J = 10.1, 4.4 Hz, 1H, 1 H of CH2), 4.26 (dd, J = 7.3, 6.3 Hz, 1H, CH-4), 4.10 (dd, J = 10.0, 6.6 Hz, 1H, 1 H of CH2), 3.77 (s, 3H, OCH3), 2.55 (pd, J = 7.4, 1.9 Hz, 1H, CH of iPr), 2.53 (bs, 1H, OH), 1.54 (s, 3H, CH3 of acetonide), 1.44 (s, 3H, CH3 of acetonide), 1.13 (d, J = 6.9 Hz, 3H, CH3 of acetonide), 1.13 (d, J = 6.9 Hz, 3H, CH3 of acetonide); 13C NMR (CDCl3, 75 MHz): δ = 154.2 (Cq Ar), 152.7 (Cq Ar), 115.7 (2 CH Ar), 114.7 (2 CH Ar), 109.6 (Cq acetonide), 93.0 (Cq alkyne), 79.8 (CH-5), 76.8 (Cq alkyne), 75.6 (CH-4), 67.2 (CH2), 61.3 (CHOH), 55.8 (OCH3), 27.8 (CH3 acetonide), 25.4 (CH3 acetonide), 22.8 (2 CH3 iPr), 20.6 (CH iPr); HRMS (ESI+) m/z: [M + Na]+ Calcd for C19H26NaO5+: 357.1672; Found: 357.1670.
(3aR,6R,6aR)-2,2-dimethyl-6-(3-methylbut-1-yn-1-yl)dihydrofuro[3,4-d][1,3]dioxol-4(3aH)-one (10 anti): To a solution of 9 anti (30 mg, 0.09 mmol), in CH3CN (1.5 mL) and H2O (450 µL), at 0 °C, CAN (123 mg, 0.22 mmol) was added. After 10 min, the reaction mixture was diluted with saturated aqueous NaHCO3 and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and concentrated to afford the crude diol, which was directly used for the next reaction. To a solution of crude diol in CH2Cl2 (1 mL), under a nitrogen atmosphere, TEMPO (5 mg, 0.03 mmol) and BAIB (155 mg, 0.48 mmol) were added. After stirring for 4 h at room temperature, the reaction mixture was diluted with CH2Cl2, washed with Na2S2O3 (0.4 M in water), dried (Na2SO4), and concentrated. The residue was purified with chromatography using PE/AcOEt 8:1 to create 10 anti (15 mg, 74%) as a pale-yellow oil. Rf = 0.86 (PE/AcOEt = 3:2); [α] D20 = +50.4 (c 2.2, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 5.14 (dt, J = 2.0, 0.5 Hz, 1H, propargylic CH), 4.86 (d, J = 5.2 Hz, 1H, CH-4), 4.75 (d, J = 5.3 Hz, 1H, CH-3), 2.59 (heptd, J = 6.9, 2.0 Hz, 1H, CH of iPr), 1.47 (s, 3H, CH3 of acetonide), 1.39 (s, 3H, CH3 of acetonide), 1.17 (d, J = 6.9 Hz, 6H, 2 CH3 of CH(CH3)2); 13C NMR (CDCl3, 75 MHz): δ = 173.7 (Cq of lactone), 114.6 (Cq of acetonide), 96.8 (Cq alkyne), 80.9 (CH-3), 75.0 (CH-4), 73.0 (Cq alkyne), 71.8 (propargylic CH), 26.9 (CH3 acetonide), 26.1 (CH3 acetonide), 22.5 (2 CH3 of iPr), 20.6 (CH of iPr); HRMS (ESI+) m/z: [M + Na]+ Calcd for C12H16NaO4+: 247.0941; Found: 247.0940.
(3aR,6S,6aR)-2,2-dimethyl-6-(3-methylbut-1-yn-1-yl)dihydrofuro[3,4-d][1,3]dioxol-4(3aH)-one (10 syn): Compound 10 syn was obtained as a white foam (18 mg, 50%), starting from 9 syn, using the same procedure as above described for 10 anti. Rf = 0.70 (PE/AcOEt = 3:2); [α] D20 = +87.9 (c 0.8, CHCl3); 1H NMR (CDCl3, 300 MHz): δ 5.17 (dd, J = 3.6, 1.8 Hz, 1H, propargylic CH), 4.81 (dd, J = 5.6, 3.6 Hz, 1H, CH-4), 4.78 (d, J = 5.6 Hz, 1H, CH-3), 2.67 (heptd, J = 6.9, 1.9 Hz, 1H, CH of iPr), 1.51 (s, 3H, CH3 of acetonide), 1.44 (s, 3H, CH3 of acetonide), 1.21 (dd, J = 6.9, 1.0 Hz, 6H, 2 CH3 of iPr); 13C NMR (CDCl3, 75 MHz): δ = 173.1 (Cq lactone), 114.7 (Cq acetonide), 97.4 (Cq alkyne), 76.9 (CH-4), 75.7 (CH-3), 71.1 (propargylic CH), 70.4 (Cq alkyne), 26.9 (CH3 acetonide), 26.3 (CH3 acetonide), 22.6 (2 CH3 of iPr), 20.8 (CH of iPr); HRMS (ESI+) m/z: [M + Na]+ Calcd for C12H16NaO4+: 247.0941; Found: 247.0940.
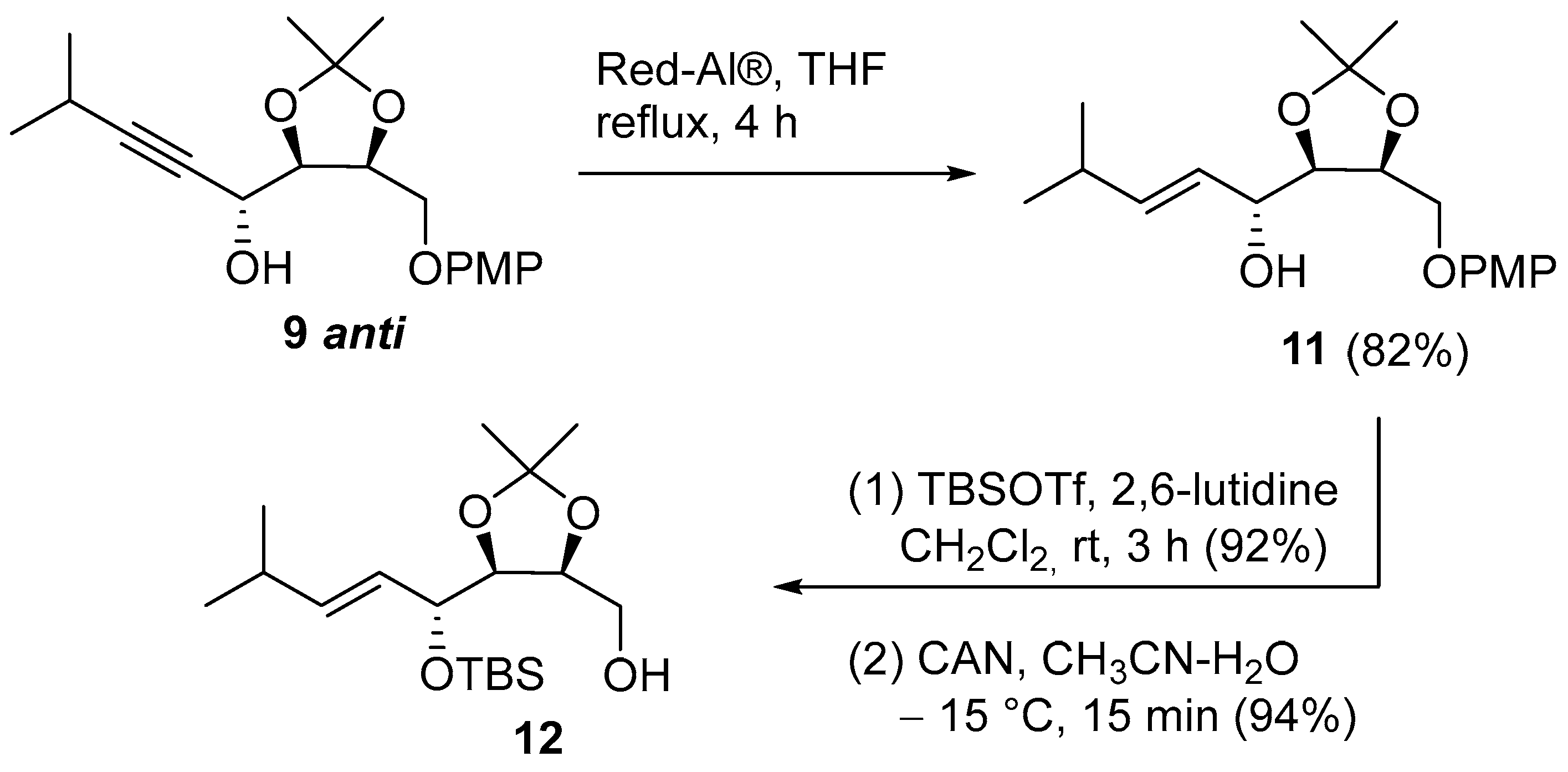
(R,E)-1-((4R,5S)-5-((4-methoxyphenoxy)methyl)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-methylpent-2-en-1-ol (11): To a solution of 9 anti (556 mg, 1.66 mmol), in dry THF (17 mL), under an argon atmosphere, Red-Al® (3.5 M in toluene, 1.2 mL) was added dropwise at 0 °C and the reaction was stirred under reflux for 4 h. Then, it was cooled to 0 °C and carefully quenched with 1:1 Rochelle salt (30% aqueous solution) and saturated NH4Cl aqueous solution. The mixture was stirred for 1 h and then extracted with AcOEt. The organic phase was washed with brine, dried (Na2SO4), and concentrated. The crude residue was purified using silica gel column chromatography (PE/Et2O 3:1) to afford 11 (451 mg, 82%) as a white solid. Rf = 0.55 (PE/AcOEt 4:1); [α]D25 = +4.9 (c 1.2, CHCl3). m.p. 44.6–47.1 °C; IR (ATR): ν = 3487, 2990, 2957, 2939, 2883, 2867, 2837, 1858, 1670, 1624, 1591, 1506, 1458, 1441, 1412, 1379, 1367, 1329, 1302, 1290, 1250, 1220, 1183, 1167, 1137, 1113, 1081, 1039, 1013, 971, 958, 936, 923, 906, 861, 822, 799, 778, 721, 669, 659, 642, 605 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 6.93–6.79 (m, 4H, 4 CH Ar), 5.79 (ddd, J = 15.6, 6.5, 1.2 Hz, 1H, iPr-CH=), 5.57 (ddd, J = 15.6, 5.7, 1.3 Hz, 1H, iPr-CH=CH), 4.55 (dt, J = 6.7, 5.6 Hz, 1H, CH-CH2OPMP), 4.38–4.25 (m, 1H, CHOH), 4.19 (dd, J = 9.8, 6.8 Hz, 1H, 1 H of CH2), 4.13 (dd, J = 7.8, 5.7 Hz, 1H, CH-CHOH), 4.02 (dd, J = 9.7, 5.5 Hz, 1H, 1 H of CH2), 3.77 (s, 3H, OCH3), 2.72 (d, J = 3.6 Hz, 1H, OH), 2.33 (hept, J = 6.9 Hz, 1H, CH of iPr), 1.47 (s, 3H, CH3 of acetonide), 1.39 (s, 3H, CH3 of acetonide), 1.01 (dd, J = 6.8, 1.1 Hz, 6H, 2 CH3 of iPr); 13C NMR (CDCl3, 75 MHz): δ = 154.6 (Cq Ar), 152.2 (Cq Ar), 140.6 (iPr-CH=), 125.9 (iPr-CH=CH), 115.8 (2 CH Ar), 114.8 (2 CH Ar), 109.0 (Cq acetonide), 80.2 (CH-CHOH), 75.7 (CH-CH2), 70.3 (CHOH), 67.8 (CH2), 55.9 (OCH3), 31.0 (CH of iPr), 28.1 (CH3 acetonide), 25.5 (CH3 acetonide), 22.4 (CH3 of iPr), 22.3 (CH3 of iPr); HRMS (ESI+) m/z: [M + Na]+ Calcd for C19H28NaO5+: 359.1829; Found: 359.1816.
((4S,5S)-5-((R,E)-1-((tert-butyldimethylsilyl)oxy)-4-methylpent-2-en-1-yl)-2,2-dimethyl-1,3-dioxolan-4-yl)methanol (12): A solution of 11 (381 mg, 1.13 mmol), in dry CH2Cl2 (6 mL), under a nitrogen atmosphere was treated with 2,6-lutidine (527 μL, 4.53 mmol) and TBS-OTf (624 μL, 2.72 mmol) at 0 °C. After stirring at room temperature for 3 h, the reaction was diluted with saturated aqueous NH4Cl and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and concentrated and the crude residue was filtered through a short column of silica gel (PE/Et2O 8:1) and the free alcohol obtained (470 mg, 92%) is directly subjected to the next reaction. To a solution of free alcohol (75 mg, 0.17 mmol) in CH3CN (3 mL), a solution of CAN in deionized water (1 mL, 0.4 M) was added dropwise at −15 °C. After stirring for 15 min at −15 °C, the mixture was diluted with NaHCO3 (5% w/v aqueous solution)/Na2S2O3 (0.4 M in water) (1:1) and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and concentrated, and the crude residue was purified with silica gel column chromatography (PE/CH2Cl2/Et2O 4:1:0.5) to afford 12 (54 mg, 94%) as a pale-yellow oil. Rf = 0.18 (PE/CH2Cl2/Et2O 4:1:0.5); [α]D25= −17.8 (c 1.2, CHCl3); 1H NMR (CDCl3, 300 MHz): δ = 5.66 (dd, J = 15.3, 6.8 Hz, 1H, iPr-CH=), 5.39 (ddd, J = 15.4, 7.2, 1.3 Hz, 1H, iPr-CH=CH), 4.44 (dd, J = 7.1, 5.5 Hz, 1H, CH-OTBS), 4.19 (q, J = 5.8 Hz, 1H, CH-CH2OH), 4.01 (t, J = 5.6 Hz, 1H, CH-CHOTBS), 3.75 (ddd, J = 11.8, 7.7, 5.8 Hz, 1H, 1 H of CH2OH), 3.67 (ddd, J = 11.9, 6.2, 5.7 Hz, 1H, 1 H of CH2OH), 2.95 (dd, J = 7.6, 6.2 Hz, 1H, OH), 2.33 (hept, J = 6.7 Hz, 1H, CH of iPr), 1.45 (s, 3H, CH3 acetonide), 1.34 (s, 3H, CH3 acetonide), 1.00 (dd, J = 6.8, 1.8 Hz, 6H, 2 CH3 of iPr), 0.90 (s, 9H, 3 CH3 of TBS), 0.12 (s, 3H, CH3 of TBS), 0.08 (s, 3H, CH3 of TBS); 13C NMR (CDCl3, 75 MHz): δ = 141.4 (iPr-CH=CH), 126.2 (iPr-CH=CH), 108.1 (Cq of acetonide), 79.8 (CH-CHOTBS), 77.8 (CH-CH2OH), 72.9 (CHOTBS), 61.7 (CH2OH), 30.9 (CH of iPr), 27.9 (CH3 acetonide), 26.0 (3 CH3 of TBS), 25.8 (CH3 acetonide), 22.2 (CH3 of iPr), 22.0 (CH3 of iPr), 18.3 (Cq of TBS), −3.7 (CH3 of TBS), −4.4 (CH3 of TBS).; HRMS (ESI+) m/z: [M+Na]+ Calcd for C18H36NaO4Si+: 367.2275; Found: 367.2322.
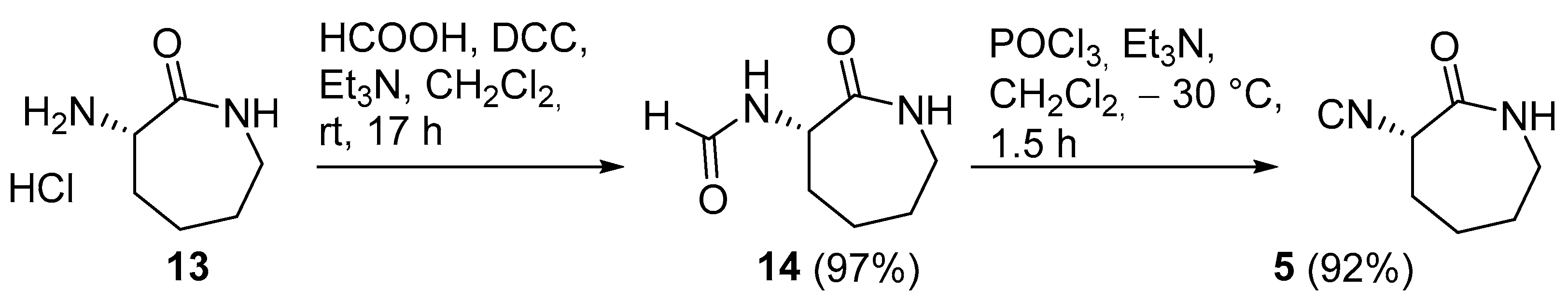
(S)-N-(2-oxoazepan-3-yl)formamide (14): To a solution of L -(−)-α-amino-ε-caprolactam hydrochloride 13 (502 mg, 3.05 mmol) in dry CH2Cl2 (15 mL), Et3N (593 µL, 4.25 mmol), formic acid (183 μL, 4.86 mmol), and DCC (877 mg, 4.25 mmol) were added at 0 °C, and the reaction was stirred at room temperature for 19 h. The mixture was filtered through a pad of celite, washing it with CH2Cl2, and the solvent was removed under reduced pressure. The residue was purified using silica gel column chromatography (from AcOEt + 2% MeOH to AcOEt + 10% MeOH) to afford 14 as a white amorphous solid (460 mg, 97%). The optical purity of formamide was checked using chiral HPLC analysis on Daicel Chiralpak AD 250 × 4.6 mm column, after standardization with a racemic sample. Flow 1.0 mL/min; isocratic elution with n-hexane/iPrOH 90:10; temp.: 25 °C; UV detection at 220 nm. Rt 23.4 min (D) and 27.6 min (L). Rf = 0.42 (CH2Cl2/MeOH 9:1); [α]D20 = +80.04 (c 1.01, CHCl3); IR (ATR): ν = 3268, 3089, 2972, 2912, 2866, 2850, 1695, 1628, 1517, 1482, 1437, 1381, 1370, 1361, 1335, 1316, 1292, 1278, 1222, 1212, 1122, 1092, 1057, 1043, 978, 946, 910, 851, 835, 804, 759 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 8.20 (s, 1H, CHO), 7.14 (bs, 1H, NH), 6.50 (bs, 1H, NH), 4.61 (dd, J = 11.2, 6.1 Hz, 1H, CH), 3.52–3.12 (m, 2H, CH2C=O), 2.35–1.96 (m, 2H, 2 H of CH2), 1.96–1.72 (m, 2H, 2 H of CH2), 1.62–1.34 (m, 2H, 2 H of CH2); 13C NMR (CDCl3, 75 MHz): δ = 175.2 (C=O caprolactame), 160.3 (C=O formamide), 51.2 (CH), 42.2 (CH2), 31.6 (CH2), 28.9 (CH2), 28.0 (CH2); HRMS (ESI+) m/z: [M + Na]+ Calcd for C7H12N2NaO2+: 179.0791: Found: 179.0802.
(S)-3-isocyanoazepan-2-one (5): To a solution of
14 (122 mg, 0.78 mmol), in dry CH
2Cl
2 (4 mL), Et
3N (512 μL, 3.67 mmol) and POCl
3 (179 μL, 1.17 mmol) were added dropwise at −30 °C. After stirring for 90 min at −30 °C, the reaction was diluted with saturated NaHCO
3 aq, extracted with AcOEt, dried (Na
2SO
4), and concentrated. The crude residue was purified using silica gel column chromatography (PE/AcOEt 1:5) to afford
5 as a white amorphous solid (98 mg, 91%). The optical purity of isocyanide was not confirmed in this crude product due to the presence of unresolved peaks in the chromatogram. Therefore, the optical purity was checked on model compounds
16 derived from a Passerini reaction of
5 and
7a (See
Supporting Information). R
f = 0.63 (CH
2Cl
2/MeOH 9:1); [α]
D25= −11.2 (
c 1.0, CHCl
3); IR (ATR): ν = 3328, 3223, 3099, 2992, 2948, 2925, 2858, 2148, 1670, 1478, 1466, 1436, 1428, 1359, 1331, 1323, 1291, 1274, 1248, 1111, 1092, 1075, 1038, 1015, 964, 944, 885, 823, 789, 776, 687 cm
−1:
1H NMR (CDCl
3, 300 MHz):
δ = 7.50 (bs, 1H, NH), 4.49 (dd,
J = 9.6, 2.2 Hz, 2H, CH), 3.50–3.32 (m, 1H, 1 H of
CH2NH), 3.13 (dddd,
J = 15.5, 10.1, 5.7, 1.3 Hz, 1H, 1 H of
CH2NH), 2.08 (dtt,
J = 17.4, 11.0, 3.4 Hz, 3H, 3 H of CH
2), 1.90–1.67 (m, 2H, 2 H of CH
2), 1.66–1.49 (m, 1H, 1 H of CH
2);
13C NMR (CDCl
3, 75 MHz):
δ = 170.1 (C=O), 159.5 (NC), 57.8 (CH), 41.7 (CH
2), 31.3 (CH
2), 28.5 (CH
2), 26.8 (CH
2); HRMS (ESI+)
m/
z: [M + Na]
+ Calcd for C
7H
10N
2NaO
2+: 161.0685; Found: 161.0694.
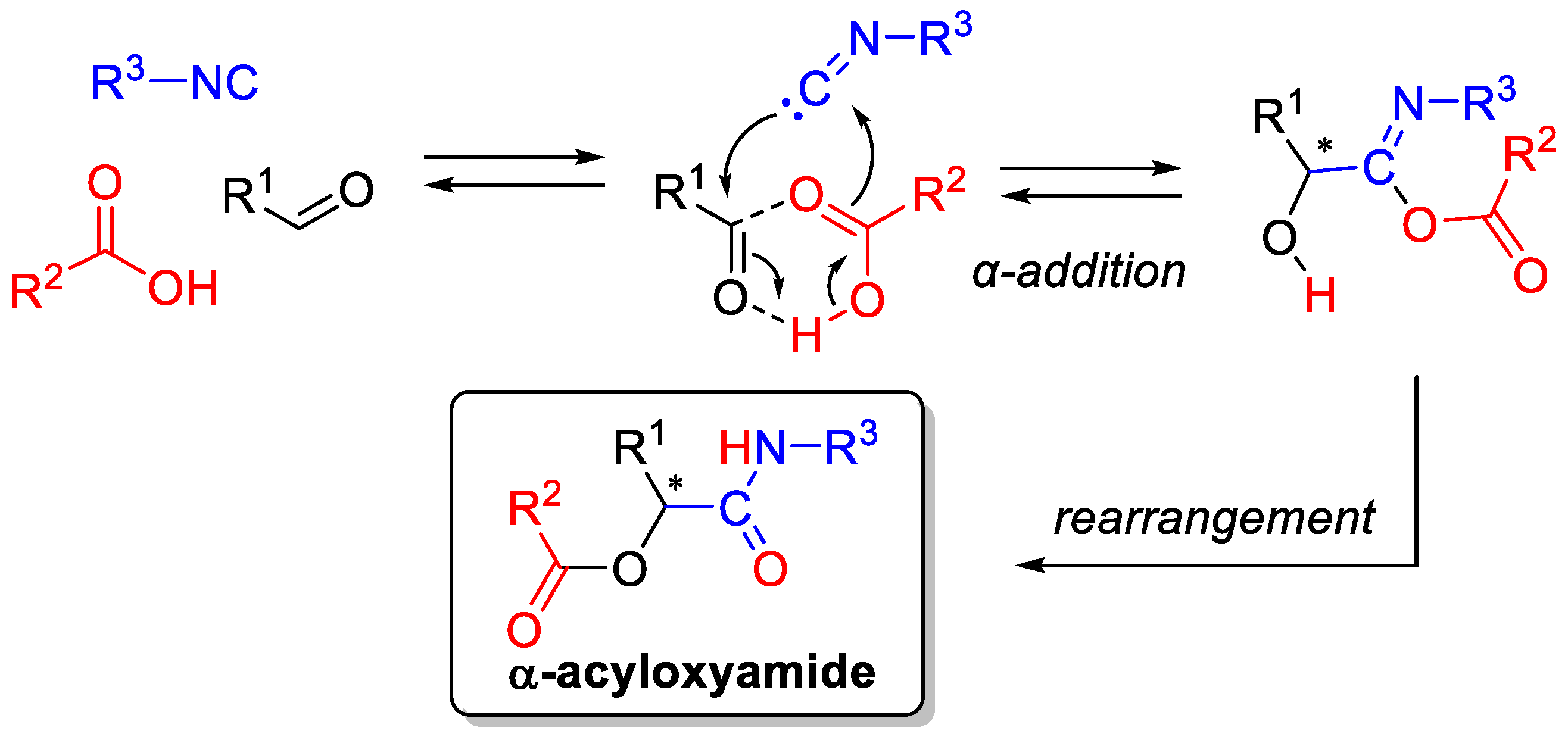
((4R,5R)-5-(1-acetoxy-2-oxo-2-(((S)-2-oxoazepan-3-yl)amino)ethyl)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl acetate (16): To a solution of DMSO (44 µL, 0.61 mmol), in dry CH2Cl2 (3 mL), at −70 °C, under a nitrogen atmosphere, a solution of oxalyl chloride in dry CH2Cl2 (2 M, 0.26 mL) was added. The solution was stirred for approximately 10 min, until effervescence ceased. A solution of 7a (50 mg, 0.24 mmol) in dry CH2Cl2 (1 + 1 + 0.5 mL) was added dropwise, and the solution was stirred for 10 min at −70 °C. NEt3 (160 µL, 1.15 mmol) was then added, and the solution was stirred for 2 h at −50 °C. After this time, the reaction mixture was poured into a mixture of 5% aq (NH4)H2PO4 (5 mL) and 1 M HCl (0.1 mL) (final pH 4) and extracted with Et2O (20 + 10 mL). The organic layer was washed with brine (5 mL), dried (Na2SO4), and concentrated. The resulting crude aldehyde 15 was rapidly solubilized in CH2Cl2 (1 mL) under N2 and isocyanide 5 (37 mg, 0.27 mmol) and acetic acid (15 μL, 0.27 mmol) were added. After stirring for 7 h at room temperature, the solvent was removed and the residue was filtered on silica gel (PE/Acetone 3:2) to give 16 (68 mg, 69%) as a 20:80 (syn/anti) mixture of diastereoisomers (Colonna Hydro RP (2) 150 × 3 mm, 4 micron; flow = 0.5 mL/min; Vinj 5 µL; Temp: 26 °C Term. ON, VWD = 210 nm; MS: FullScan 100–800 m/z Positive, tic volt: 750V, Gradient A = H2O+0.1%FA C = MeOH+0.1% FA, 0 min A = 80%, 30 min A = 0%. Rt (anti) = 12.9 min, Rt (syn) = 13.3 min). 16 anti and 16 syn can be separated performing column chromatography on silica gel with PE/Acetone (from 1:1 to 3:2).
16 anti: amorphous solid; Rf = 0.14 (PE/Acetone 2:1); [α]D25 = +26.47 (c 0.85, CHCl3): 1H NMR (CDCl3, 300 MHz): δ = 7.57 (bd, J = 5.8 Hz, 1H, NHCH), 6.25 (bt, J = 6.4 Hz, 1H, NHCH2), 5.21 (d, J = 7.6 Hz, 1H, CHOAc), 4.57 (dd, J = 7.6, 5.8 Hz, 1H, CH-CHOAc), 4.54–4.43 (m, 2H, CH-CH2OAc and CHNH), 4.41 (dd, J = 11.4, 3.8 Hz, 2H, 1 H of CH2OAc), 4.11 (dd, J = 11.4, 6.8 Hz, 1H, 1 H of CH2OAc), 3.36–3.19 (m, 2H, CH2NH), 2.17 (s, 3H, OAc), 2.08 (s, 3H, OAc), 2.20–1.95 (m, 2H, 2 H of CH2), 1.90–1.74 (m, 2H, 2 H of CH2), 1.51 (s, 3H, CH3 acetonide), 1.56–1.30 (m, 2H, 2 H of CH2), 1.38 (s, 3H, CH3 acetonide); 13C NMR (CDCl3, 75 MHz): δ = 175.0 (C=O), 170.8 (C=O), 169.5 (C=O), 166.3 (C=O), 109.7 (Cq acetonide), 75.3 (CHNH), 75.3 (CH-CHOAc), 71.4 (CH-OAc), 62.4 (CH2OAc), 52.6 (CH-CH2OAc), 42.3 (CH2NH), 31.1 (CH2), 29.0 (CH2), 28.0 (CH2), 27.7 (CH3 acetonide), 25.3 (CH3 acetonide), 21.0 (CH3 of Ac), 20.8 (CH3 of Ac); HRMS (ESI+) m/z: [M + Na]+ Calcd for C18H28N2NaO8+: 423.1738; Found: 423.1736.
16 syn: colorless oil; Rf = 0.15 (PE/Acetone 2:1); 1H NMR (CDCl3, 300 MHz): δ = 7.46 (bd, J = 6.3 Hz, 1H, CHNH), 6.02 (bt, J = 6.4 Hz, 1H, CH2NH), 5.27 (d, J = 3.5 Hz, 1H, CHOAc), 4.62 (dd, J = 6.5, 3.5 Hz, 1H, CH-CHOAc), 4.53–4.39 (m, 2H, CHNH and CH-CH2OAc), 4.23 (dd, J = 11.4, 5.2 Hz, 1H, 1 H of CH2OAc), 4.15 (dd, J = 11.4, 6.9 Hz, 1H, 1 H of CH2OAc), 3.31–3.20 (m, 2H, CH2NH), 2.23 (s, 3H, OAc), 2.16–1.96 (m, 2H, CH2), 2.08 (s, 3H, OAc), 1.91–1.78 (m, 2H, CH2), 1.52 (s, 3H, CH3 acetonide), 1.48–1.37 (m, 2H, CH2), 1.35 (s, 3H, CH3 acetonide); 13C NMR (CDCl3, 75 MHz): δ = 174.9 (C=O), 170.8 (C=O), 169.7 (C=O), 166.8 (C=O), 109.7 (Cq acetonide), 75.8 (CH-CHOAc), 74.8 (CH-CH2OAc), 71.8 (CHOAc), 62.8 (CH2OAc), 52.4 (CHNH), 42.3 (CH2NH), 31.3 (CH2), 29.0 (CH2), 28.0 (CH2), 27.2 (OAc), 25.4 (OAc), 21.0 (CH3 acetonide), 20.9 (CH3 acetonide); HRMS (ESI+) m/z: [M + Na]+ Calcd for C18H28N2NaO8+: 423.1738; Found: 423.1736.
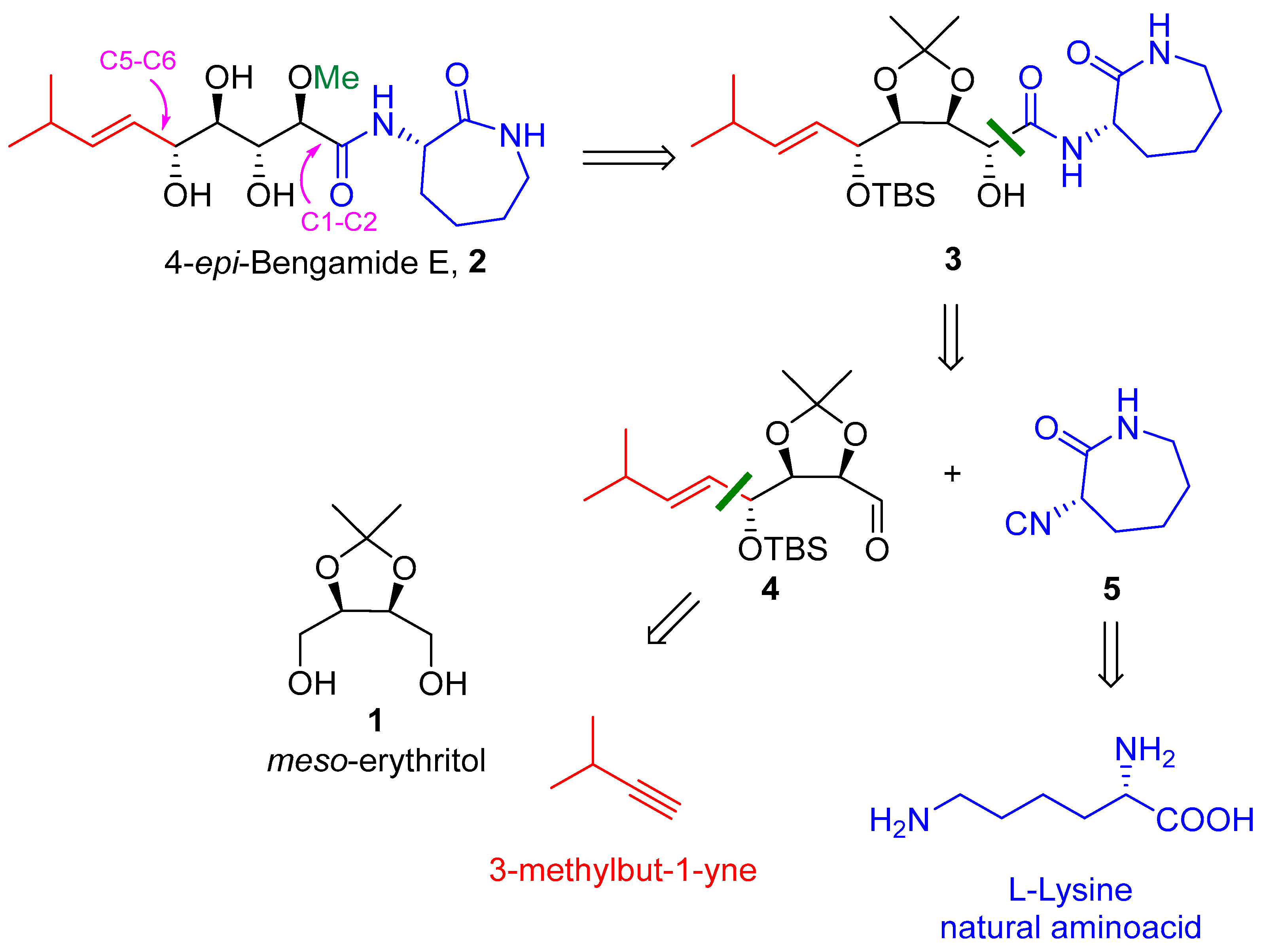
2-((4S,5S)-5-((R,E)-1-((tert-butyldimethylsilyl)oxy)-4-methylpent-2-en-1-yl)-2,2-dimethyl-1,3-dioxolan-4-yl)-2-hydroxy-N-((S)-2-oxoazepan-3-yl)acetamide (3): To a solution of DMSO (37 µL, 0.52 mmol), in dry CH2Cl2 (3 mL), at −70 °C, under a nitrogen atmosphere, a solution of oxalyl chloride in dry CH2Cl2 (1.43 M, 0.33 mL) was added. The solution was stirred for approximately 10 min, until effervescence ceased. A solution of 12 (67 mg, 0.19 mmol) in dry CH2Cl2 (1 + 0.5 mL) was added dropwise, and the solution was stirred for 10 min at −70 °C. NEt3 (160 µL, 1.15 mmol) was then added, and the solution was stirred for 2 h at −50 °C. After this time, the reaction mixture was poured into a mixture of 5% aq (NH4)H2PO4 (5 mL) and 1 M HCl (0.1 mL) (final pH 4) and extracted with Et2O (20 + 10 mL). The organic layer was washed with brine (5 mL), dried (Na2SO4), and concentrated. The resulting crude aldehyde 4 was rapidly solubilized in iPr2O (500 µL) under N2, and isocyanide 5 (53 mg, 0.38 mmol) and acetic acid (22 μL, 0.38 mmol) were added. After stirring for 48 h at room temperature, the solvent was removed and the residue was filtered on silica gel (PE/AcOEt 3:4) to give a mixture of products 18 and 3 (73 mg), which was treated with MeOH/H2O/Et3N (5:1:1) and stirred at room temperature for 48 h. Then, the solvent was removed. The diastereomeric ratio was determined as 80:20 (anti:syn) using reverse-phase HPLC on the crude mixture (C6 PHENYLIC RP column (150 × 3 mm, 3 μ) at 30 °C with flow = 0.34 mL/min and gradient H2O/MeOH, A = MeOH + 0.1% FA—B = H2O + 0.1% FA, 0 min B = 30%, 20 min B = 20%. Detection was carried out with UV at 210 nm, Rt (anti) = 13.2 min, Rt (syn) = 15.6 min). The crude residue was purified with column chromatography of silica gel (PE/Et2O 1:20) to give 3 anti (55 mg, 58%) and 3 syn (14 mg, 16%). 3 anti: pale-yellow oil, Rf = 0.35 (Et2O/PE 20:1); [α]D20 = +16.9 (c 1.0, CHCl3); IR (ATR): ν = 3462, 3346, 2956, 2931, 2858, 1685, 1598, 1525, 1463, 1375, 1254, 1215, 1168, 1069, 1045, 974, 834, 800, 777, 667 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 7.79 (bd, J = 6.5 Hz, 1H, NHCH), 6.20 (bs, 1H, NHCH2), 5.63 (dd, J = 15.5, 6.6 Hz, 1H, iPr-CH=CH), 5.47 (dd, J = 15.5, 7.8 Hz, 1H, iPr-CH=CH), 4.64–4.53 (m, 3H, OH, NHCH and CH-OTBS), 4.30–4.19 (m, 2H, CHOH and CH-CHOH), 4.06 (t, J = 4.5 Hz, 1H, CH-CHOTBS), 3.38–3.15 (m, 2H, CH2NH), 2.31 (h, J = 6.6 Hz, 1H, CH of iPr), 2.21–2.06 (m, 1H, 1 H of CH2), 2.07–1.94 (m, 1H, 1 H of CH2), 1.93–1.70 (m, 2H, 2 H of CH2), 1.62–1.47 (m, 1H, 1 H of CH2), 1.52 (s, 3H, CH3 acetonide), 1.47–1.36 (m, 1H, 1 H of CH2), 1.33 (s, 3H, CH3 acetonide), 1.005 (d, J = 6.7 Hz, 3H, CH3 of iPr), 1.00 (d, J = 6.8 Hz, 3H, CH3 of iPr), 0.90 (s, 9H, 3 CH3 of TBS), 0.14 (s, 3H, CH3 of TBS), 0.11 (s, 3H, CH3 of TBS); 13C NMR (CDCl3, 75 MHz): δ = 175.5 (C=O), 170.6 (C=O), 142.0 (iPr-CH=CH), 125.9 (iPr-CH=CH), 108.5 (Cq acetonide), 80.8 (CH-CHOTBS), 77.9 (CHOH), 73.6 (CHOTBS), 70.2 (CH-CHOH), 52.2 (CHNH), 42.1 (CH2NH), 31.4 (CH2), 30.8 (CH of iPr), 29.0 (CH2), 28.0 (CH2), 27.7 (CH3 acetonide), 25.9 (3 CH3 of TBS), 25.6 (CH3 acetonide), 22.1 (CH3 of iPr), 21.9 (CH3 of iPr), 18.3 (Cq of TBS), −3.9 (CH3 of TBS), −4.3 (CH3 of TBS); HRMS (ESI+) m/z: [M + Na]+ Calcd for C25H46N2NaO6Si+: 521.3017; Found: 521.3018. 3 syn: pale-yellow foam Rf = 0.25 ((Et2O/PE 20:1); [α]D20 = −33.7 (c 1.2, CHCl3); IR (ATR): ν = 3462, 3346, 2956, 2931, 2858, 1685, 1598, 1525, 1463, 1375, 1254, 1215, 1168, 1069, 1045, 974, 834, 800, 777, 667 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 7.98 (d, J = 7.0 Hz, 1H, NHCH), 6.16 (bs, 1H, NHCH2), 5.72 (dd, J = 15.6, 6.9 Hz, 1H, iPr-CH=CH), 5.38 (dd, J = 15.5, 6.1 Hz, 1H, iPr-CH=CH), 4.68 (t, J = 4.9 Hz, 1H, CH-OTBS), 4.64–4.56 (m, 2H, NHCH and CH-CHOH), 4.54 (d, J = 2.6 Hz, 1H, OH), 4.29 (d, J = 2.2 Hz, 1H, CHOH), 4.17 (dd, J = 6.5, 4.4 Hz, 1H, CH-CHOTBS), 3.40–3.13 (m, 2H, CH2NH), 2.31 (h, J = 6.9 Hz, 1H, CH of iPr), 2.13–1.92 (m, 2H, 2 H of CH2), 1.92–1.64 (m, 2H, 2 H of CH2), 1.61–1.51 (m, 1H, 1 H of CH2), 1.49 (s, 3H, CH3 acetonide), 1.32 (s, 3H, CH3 acetonide), 0.99 (d, J = 6.7 Hz, 6H, 2 CH3 of iPr), 0.92 (s, 9H, 3 CH3 of TBS), 0.14 (s, 3H, CH3 of iPr), 0.11 (s, 3H, CH3 of iPr); 13C NMR (CDCl3, 75 MHz): δ = 175.6 (C=O), 170.8 (C=O), 141.2 (iPr-CH=CH), 124.6 (iPr-CH=CH), 108.3 (Cq acetonide), 79.2 (CH-CHOTBS), 77.8 (CH-CHOH), 72.5 (CHOTBS), 71.5 (CHOH), 51.9 (CHNH), 42.3 (CH2NH), 31.9 (CH2), 31.0 (CH of iPr), 29.2 (CH2), 28.2 (CH2), 26.4 (CH3 acetonide), 26.0 (3 CH3 of iPr), 25.5 (CH3 acetonide), 22.3 (CH3 of iPr), 22.2 (CH3 of iPr), 18.6 (Cq TBS), −4.2 (CH3 of TBS), −4.7 (CH3 of TBS); HRMS (ESI+) m/z: [M + Na]+ Calcd for C25H46N2NaO6Si+: 521.3017; Found: 521.3018.
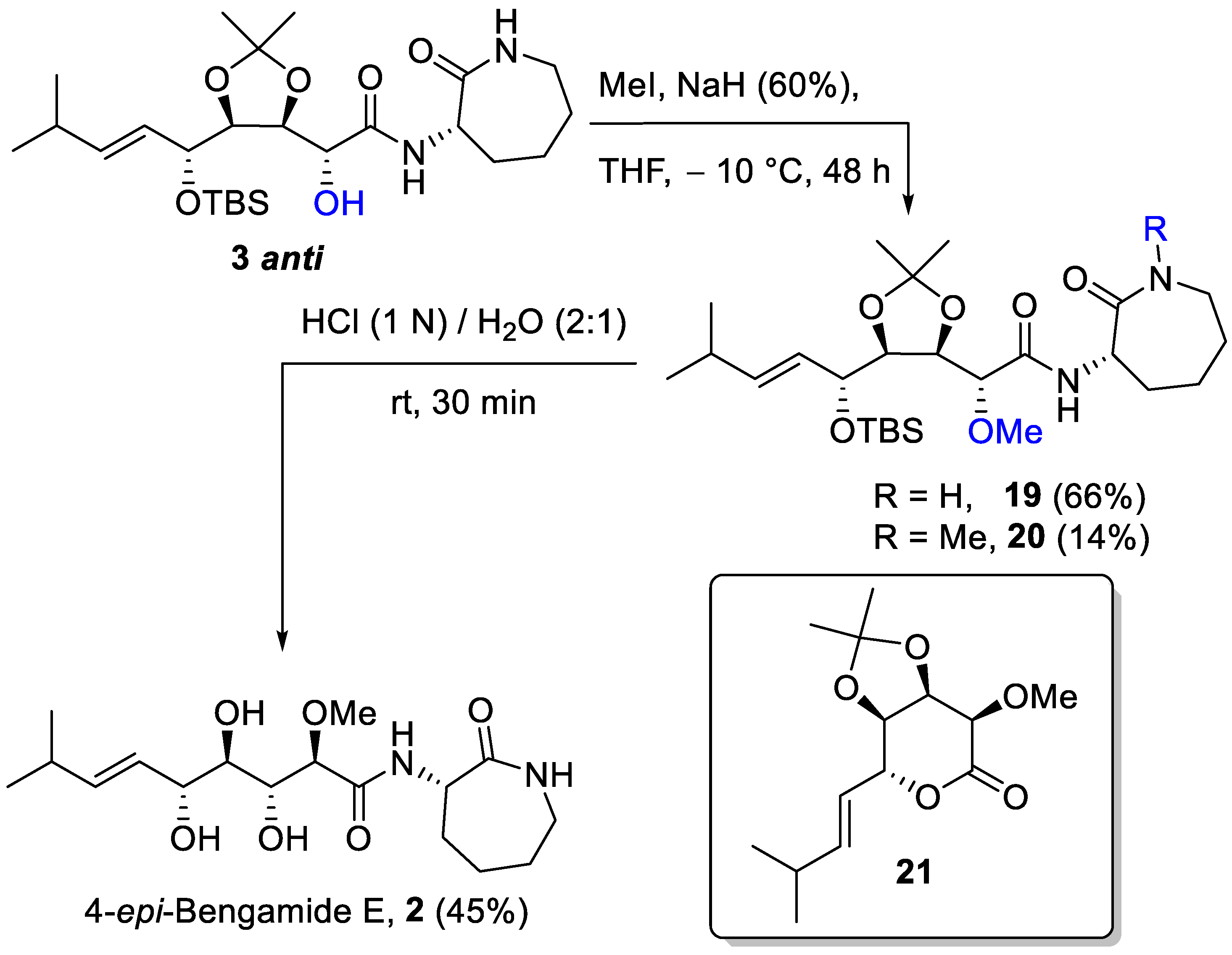
(R)-2-((4R,5S)-5-((R,E)-1-((tert-butyldimethylsilyl)oxy)-4-methylpent-2-en-1-yl)-2,2-dimethyl-1,3-dioxolan-4-yl)-2-methoxy-N-((S)-2-oxoazepan-3-yl)acetamide (19): A solution of 3 anti (64 mg, 0.128 mmol) in dry THF (1 mL) under a N2 atmosphere was cooled at −10 °C. NaH (60% in silicon oil, 8 mg, 0.199 mmol) was added and the mixture was stirred for 30 min. Then, MeI (17 µL, 0.265 mmol) was added and the reaction was stirred at −10 °C for 48 h. The reaction mixture was diluted with saturated NH4Cl solution, extracted with CH2Cl2, dried (Na2SO4), and concentrated. The crude residue was purified using silica gel column chromatography (PE/Et2O 1:20) to give 20 (14 mg, 14%) and 19 (59 mg, 66%) both as a colorless oil. 19: Rf = 0.31 (Et2O + 2% AcOEt); [α]D20 = +6.2 (c 1.7, CHCl3); IR (ATR): ν = 3383, 3292, 2955, 2930, 2858, 1714, 1662, 1504, 1474, 1435, 1362, 1334, 1250, 1217, 1169, 1103, 1073, 1047, 1017, 972, 941, 899, 875, 834, 808, 776, 754, 666 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 7.53 (d, J = 6.2 Hz, 1H, NHCH), 6.02 (t, J = 6.6 Hz, 1H, NHCH2), 5.66 (dd, J = 15.6, 5.8 Hz, 1H, iPr-CH=CH), 5.56 (dd, J = 15.6, 6.7 Hz, 1H, iPr-CH=CH), 4.75–4.52 (m, 2H, CHOTBS and NHCH), 4.22 (dd, J = 7.1, 6.2 Hz, 1H, CH-CHOCH3), 4.09 (dd, J = 6.1, 4.6 Hz, 1H, CH-CHOTBS), 4.05 (d, J = 7.0 Hz, 1H, CHOCH3), 3.34 (s, 3H, OCH3), 3.32–3.20 (m, 2H, CH2NH), 2.32 (h, J = 6.1 Hz, 1H, CH of iPr), 2.22–2.08 (m, 1H, 1 H of CH2), 2.04–1.94 (m, 1H, 1 H of CH2), 1.91–1.78 (m, 2H, 2 H of CH2), 1.60–1.40 (m, 2H, 2 H of CH2), 1.41 (s, 3H, CH3 acetonide), 1.29 (s, 3H, CH3 acetonide), 1.02 (d, J = 6.7 Hz, 3H, CH3 of iPr), 1.00 (d, J = 6.7 Hz, 3H, CH3 of iPr), 0.90 (s, 9H, 3 CH3 of TBS), 0.10 (s, 3H, CH3 of TBS), 0.07 (s, 3H, CH3 of TBS); 13C NMR (CDCl3, 75 MHz): δ = 175.3 (C=O), 169.7 (C=O), 140.9 (iPr-CH=CH), 127.3 (iPr-CH=CH), 108.5 (Cq acetonide), 81.0 (CH-CHOTBS), 80.9 (CHOCH3), 77.6 (CH-CHOCH3), 73.3 (CHOTBS), 57.4 (OCH3), 52.0 (CHNH), 42.2 (NHCH2), 31.5 (CH2), 30.9 (CH of iPr), 29.1 (CH2), 28.0 (CH2), 27.2 (CH3 acetonide), 26.1 (3 CH3 of TBS), 25.3 (CH3 acetonide), 22.4 (CH3 of iPr), 22.0 (CH3 of iPr), 18.5 (Cq of TBS), −3.5 (CH3 of TBS), −4.3 (CH3 of TBS); HRMS (ESI+) m/z: [M + Na]+ Calcd for C25H46N2NaO6Si+: 535.3174; Found: 535.3177.
(R)-2-((4R,5S)-5-((R,E)-1-((tert-butyldimethylsilyl)oxy)-4-methylpent-2-en-1-yl)-2,2-dimethyl-1,3-dioxolan-4-yl)-2-methoxy-N-((S)-1-methyl-2-oxoazepan-3-yl)acetamide (20): colorless oil; Rf = 0.38 (Et2O + 2% AcOEt); [α]D20 = +1.6 (c 0.6, CHCl3); IR (ATR): ν = 3388, 2955, 2930, 2858, 2246, 1648, 1495, 1461, 1403, 1381, 1370, 1339, 1251, 1214, 1157, 1138, 1102, 1075, 1047, 1016, 973, 910, 879, 834, 808, 777, 729, 646 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 7.61 (bd, J = 6.0 Hz, 1H, NHCH), 5.78–5.46 (m, 2H, CH=CH), 4.70 (dd, J = 9.5, 6.3 Hz, 1H, NHCH), 4.56 (dd, J = 6.4, 4.3 Hz, 1H, CHOTBS), 4.20 (dd, J = 7.4, 6.2 Hz, 1H, CH-CHOMe), 4.08 (dd, J = 6.0, 4.2 Hz, 1H, CH-CHOTBS), 4.02 (d, J = 7.5 Hz, 1H, CHOMe), 3.61 (dd, J = 15.3, 11.6 Hz, 1H, 1 H of CH2-N), 3.33 (s, 3H, OMe), 3.18 (dd, J = 15.0, 4.5 Hz, 1H, 1 H of CH2-N), 3.04 (s, 3H, NMe), 2.31 (h, J = 6.0 Hz, 1H, CH of iPr), 2.21–2.00 (m, 1H, 1 H of CH2), 2.00–1.72 (m, 3H, 3 H of CH2), 1.55–1.31 (m, 2H, 2 H of CH2), 1.42 (s, 3H, CH3 acetonide), 1.29 (s, 3H, CH3 acetonide), 1.01 (d, J = 6.7 Hz, 3H, CH3 of iPr), 1.00 (d, J = 6.7 Hz, 3H, CH3 of iPr), 0.90 (s, 9H, 3 CH3 of TBS), 0.10 (s, 3H, CH3 of TBS), 0.07 (s, 3H, CH3 of TBS); 13C NMR (CDCl3, 75 MHz): δ = 172.8 (C=O), 169.5 (C=O), 140.7 (CH=), 127.3 (C=H), 108.5 (Cq acetonide), 81.2 (CH-CHOTBS), 80.8 (CHOMe), 77.5 (CH-CHOMe), 73.3 (CHOTBS), 57.3 (OMe), 51.9 (NHCH), 50.5 (CH2N), 36.0 (NMe), 31.7 (CH2), 30.9 (CH of iPr), 27.8 (CH2), 27.3 (CH3 acetonide), 26.8 (CH2), 26.2 (3 CH3 of TBS), 25.3 (CH3 acetonide), 22.4 (CH3 of iPr), 22.1 (CH3 of iPr), 18.5 (Cq of TBS), −3.6 (CH3 of TBS), −4.3 (CH3 of TBS); HRMS (ESI+) m/z: [M + Na]+ Calcd for C27H50N2NaO6Si+: 549.3330; found: 549.3299.
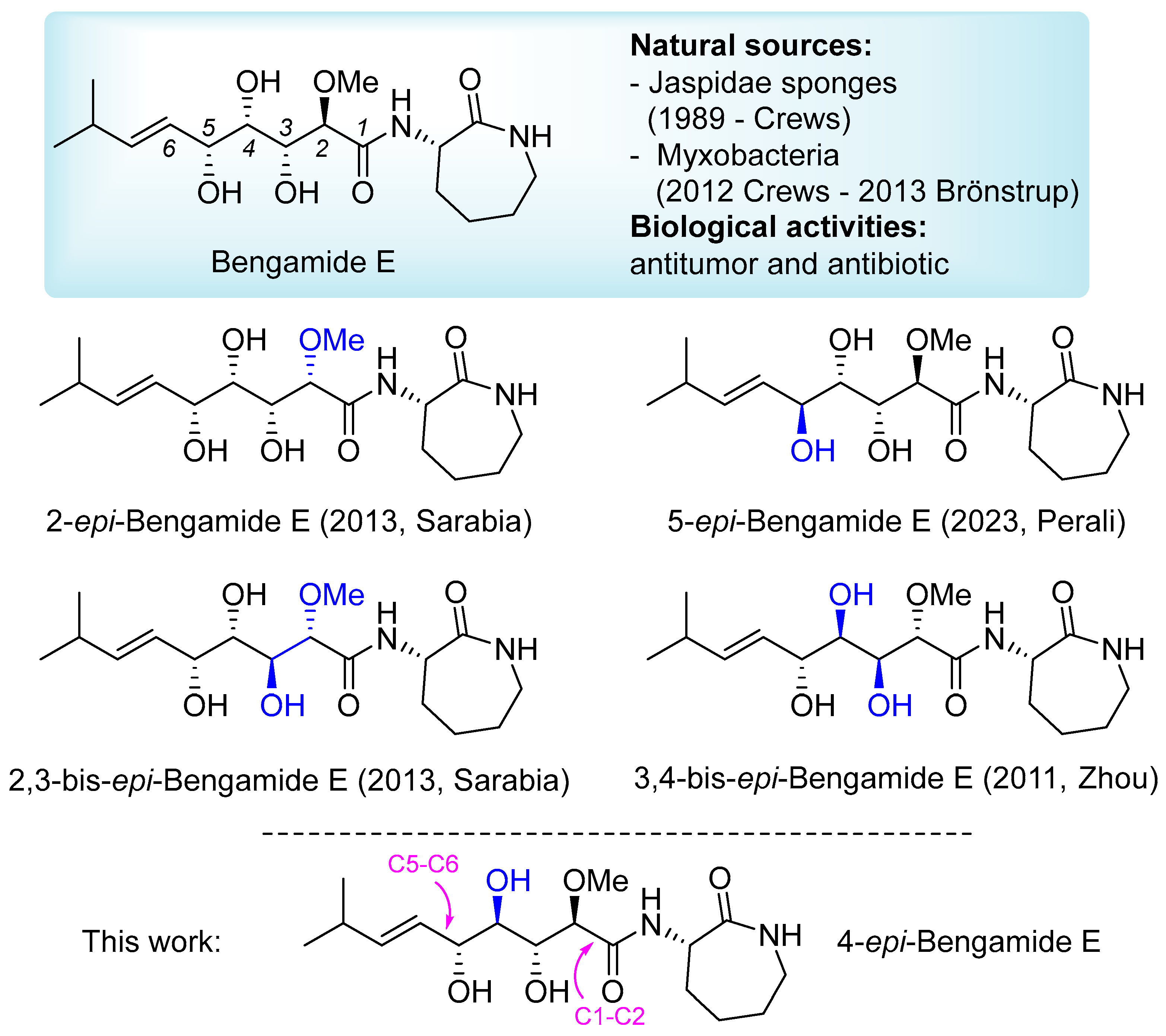
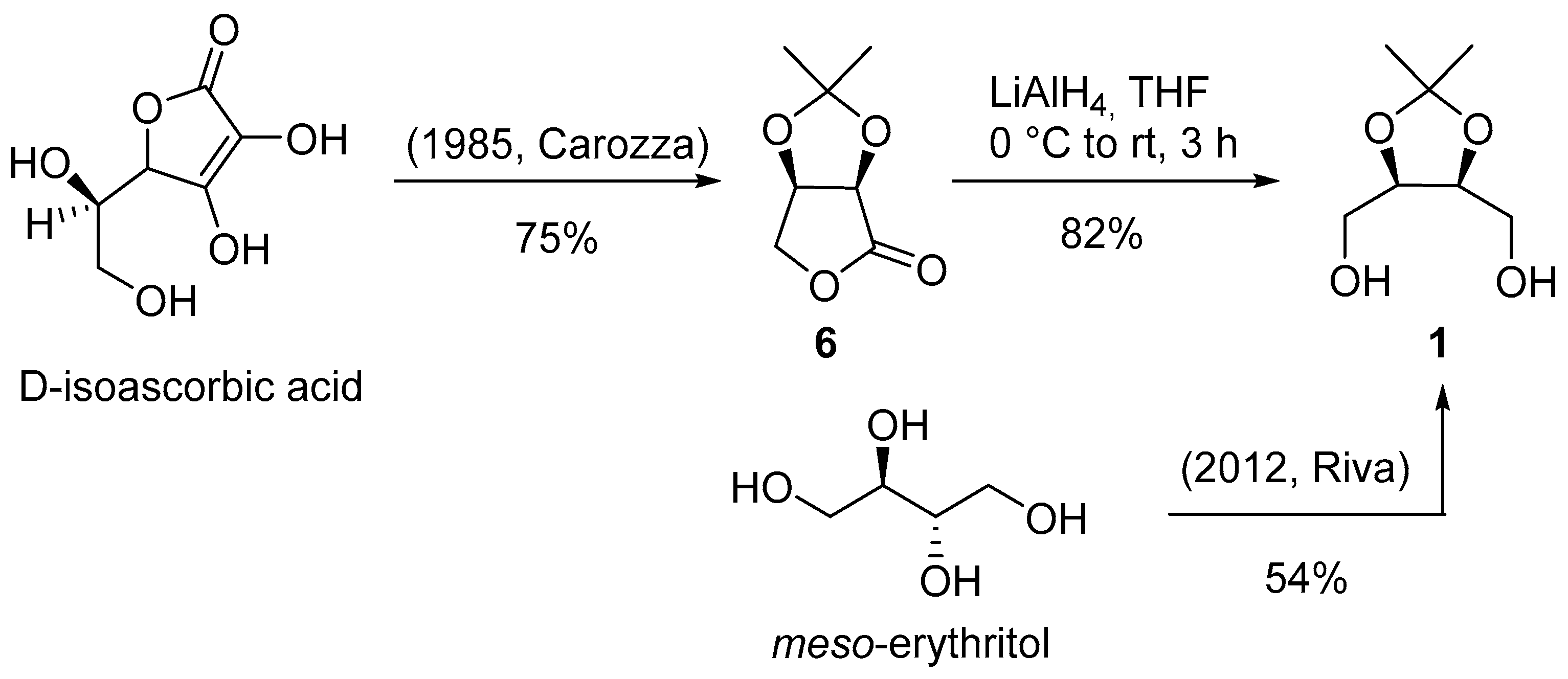
(2R,3R,4R,5R,E)-3,4,5-trihydroxy-2-methoxy-8-methyl-N-((S)-2-oxoazepan-3-yl)non-6-enamide (2): Pale-yellow foam; Rf = 0.42 (AcOEt + 2% MeOH); [α]D20 = +34.5 (c 0.3, CHCl3); IR (ATR): ν = 3334, 2956, 2930, 2868, 1639, 1519, 1483, 1437, 1361, 1334, 1291, 1261, 1066, 973, 943, 893, 800, 720 cm−1; 1H NMR (CDCl3, 300 MHz): δ = 7.63 (bd, J = 7.0 Hz, 1H, NHCH), 6.22 (bt, J = 6.1 Hz, 1H, NHCH2), 5.78 (ddd, J = 15.6, 6.4, 0.9 Hz, 1H, iPr-CH=CH), 5.55 (ddd, J = 15.6, 7.2, 1.3 Hz, 1H, iPr-CH=CH), 4.59 (dd, J = 10.3, 7.0 Hz, 1H, NHCH), 4.26 (bt, J = 5.3 Hz, 1H, CH-5), 4.07 (d, J = 3.3 Hz, 1H, CH-2), 4.07–3.95 (m, 1H, CH-3), 3.77 (d, J = 4.7 Hz, 1H, OH), 3.62 (dt, J = 8.2, 4.4 Hz, 1H, CH-4), 3.49 (s, 3H, OMe), 3.49–3.47 (m, 1H, OH), 3.39–3.18 (m, 2H, NHCH2), 2.77 (bs, 1H, OH), 2.32 (h, J = 6.3 Hz, 1H, CH of iPr), 2.11–2.00 (m, 2H, 2 H of CH2), 1.94–1.71 (m, 2H, 2 H of CH2), 1.61–1.36 (m, 2H, 2 H of CH2), 1.01 (d, J = 6.8 Hz, 6H, 2 CH3 of iPr); 13C NMR (CDCl3, 75 MHz): δ = 175.2 (C=O), 170.7 (C=O), 141.8 (iPr-CH=CH), 125.3 (iPr-CH=CH), 82.6 (CH-2), 74.7 (CH-5), 74.0 (CH-4), 73.0 (CH-3), 58.9 (OMe), 52.5 (NHCH), 42.2 (NHCH2), 31.0 (CH of iPr), 30.8 (CH2), 28.9 (CH2), 28.1 (CH2), 22.5 (CH3 of iPr), 22.3 (CH3 of iPr); HRMS (ESI+) m/z: [M + Na]+ Calcd for C17H30N2NaO6+: 381.1996; Found: 381.1993.
(3aR,4R,7R,7aR)-7-methoxy-2,2-dimethyl-4-((E)-3-methylbut-1-en-1-yl)tetrahydro-6H-[1,3]dioxolo[4,5-c]pyran-6-one (21): Colorless oil; Rf = 0.69 (AcOEt + 10% MeOH); 1H NMR (CDCl3, 300 MHz): δ = 5.82 (ddd, J = 15.8, 6.8, 2.1 Hz, 1H, iPr-CH=CH), 5.45 (ddd, J = 15.9, 4.1, 1.3 Hz, 1H, iPr-CH=CH), 5.07–4.98 (m, 1H, CH-5), 4.82 (dd, J = 7.6, 3.5 Hz, 1H, CH-3), 4.56 (dd, J = 7.6, 1.1 Hz, 1H, CH-4), 4.07 (d, J = 3.5 Hz, 1H, CH-2), 3.63 (s, 3H, OMe), 2.37 (sest, J = 6.8 Hz, 1H, CH of iPr), 1.50 (s, 3H, CH3 acetonide), 1.36 (s, 3H, CH3 acetonide), 1.02 (d, J = 6.7 Hz, 6H, 2 CH3 of iPr); 13C NMR (CDCl3, 75 MHz): δ = 168.6 (C=O), 142.7 (iPr-CH=CH), 121.3 (iPr-CH=CH), 111.0 (Cq acetonide), 79.7 (CH-5), 76.2 (CH-2), 75.7 (CH-4), 74.8 (CH-3), 59.9 (OMe), 31.3 (CH of iPr), 26.2 (CH3 acetonide), 24.4 (CH3 acetonide), 22.1 (CH3 of iPr), 22.0 (CH3 of iPr); HRMS (ESI+) m/z: [M + Na]+ Calcd for C14H22NaO5+: 293.1359; Found: 293.1349.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

