
Study of CHF3/CH2F2 Adsorption Separation in TIFSIX-2-Cu-i

 and
and
Abstract
:1. Introduction
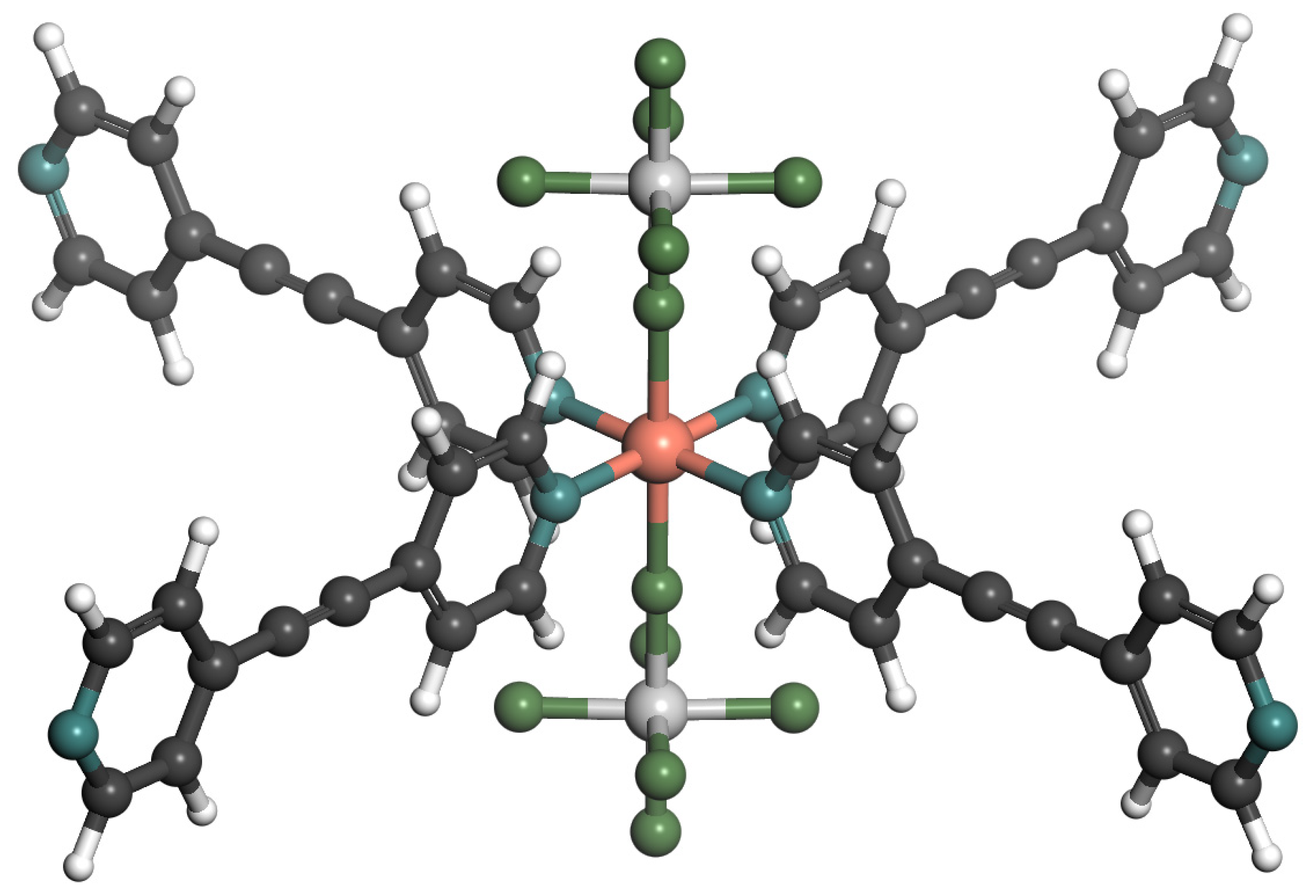
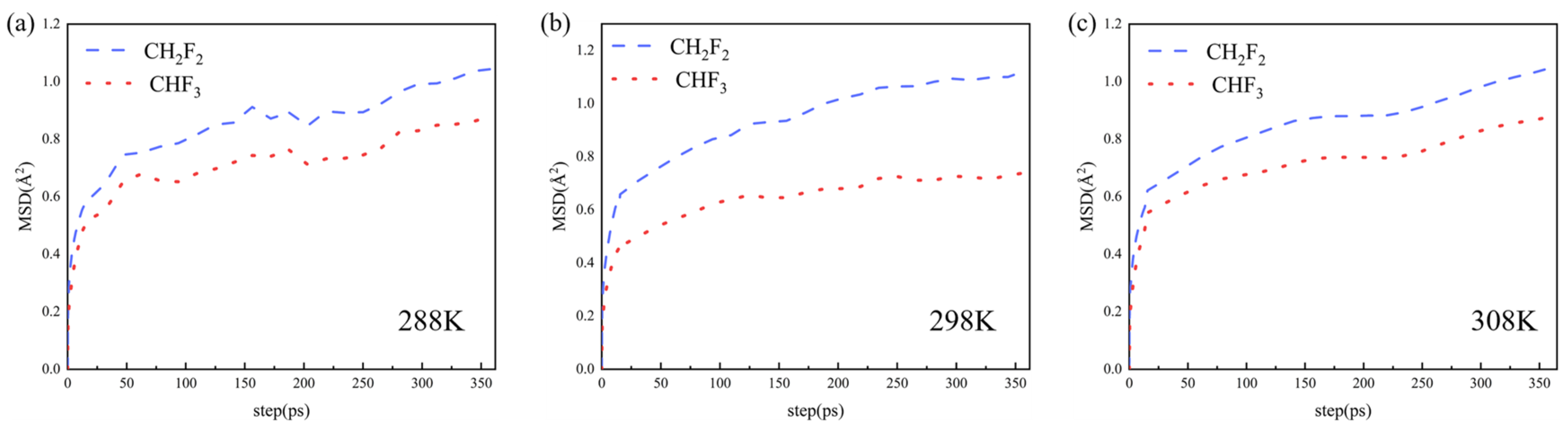
2. Results and Discussion
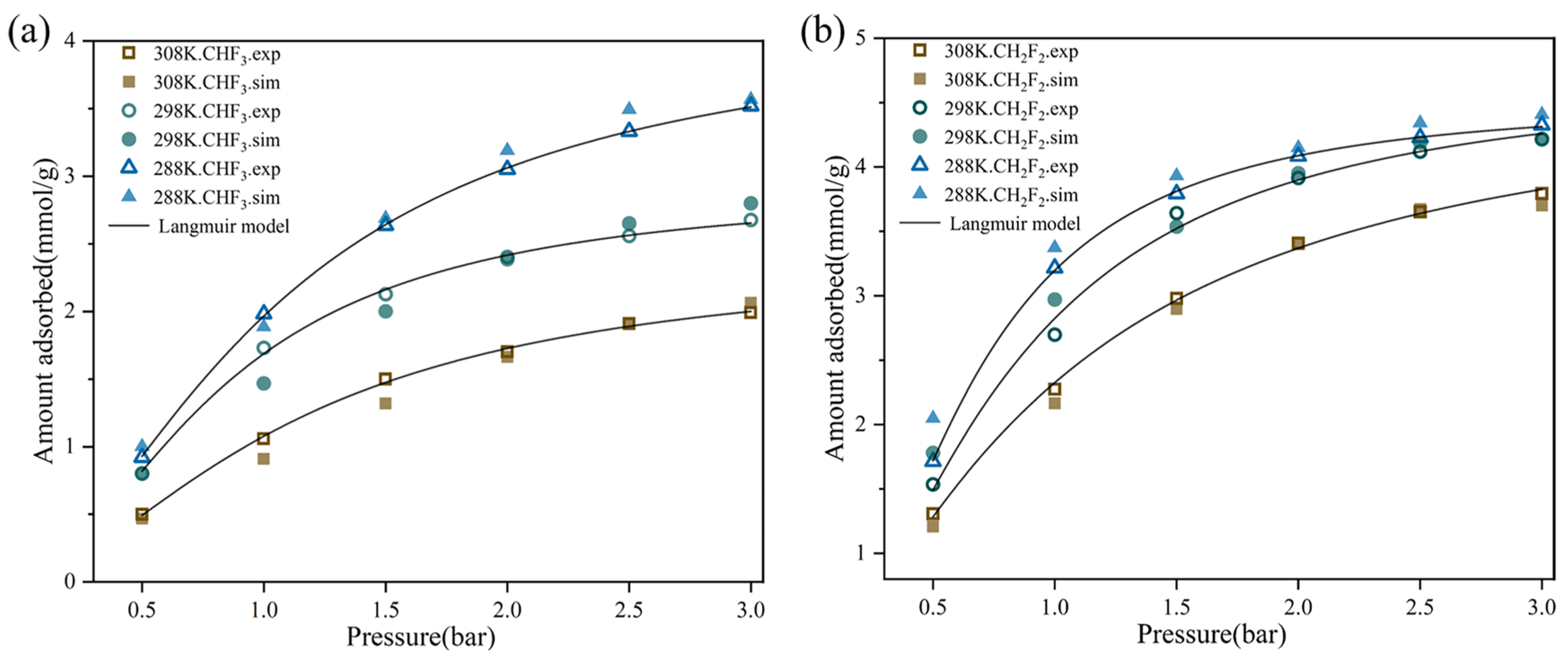
2.1. Adsorption Isotherm
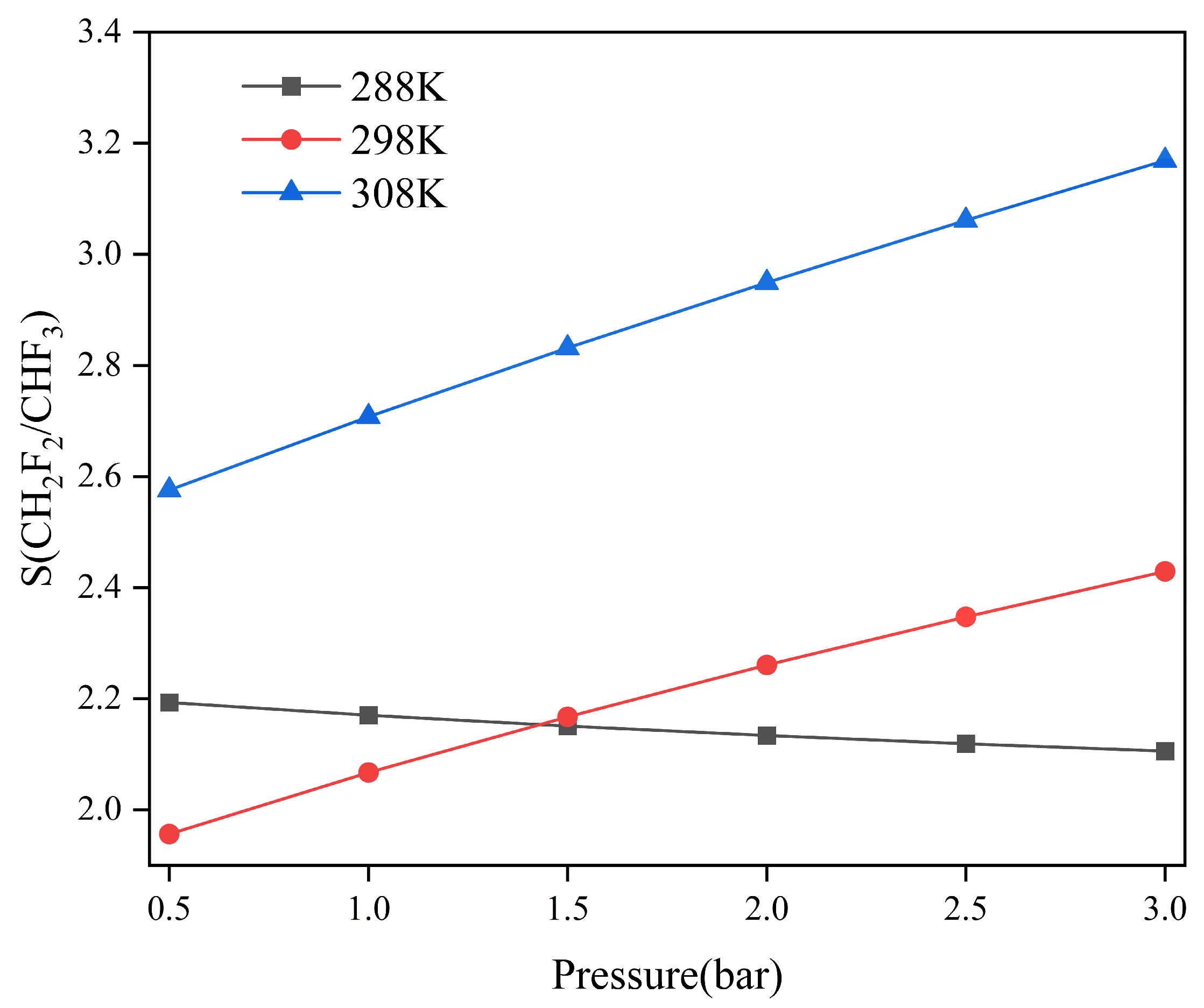
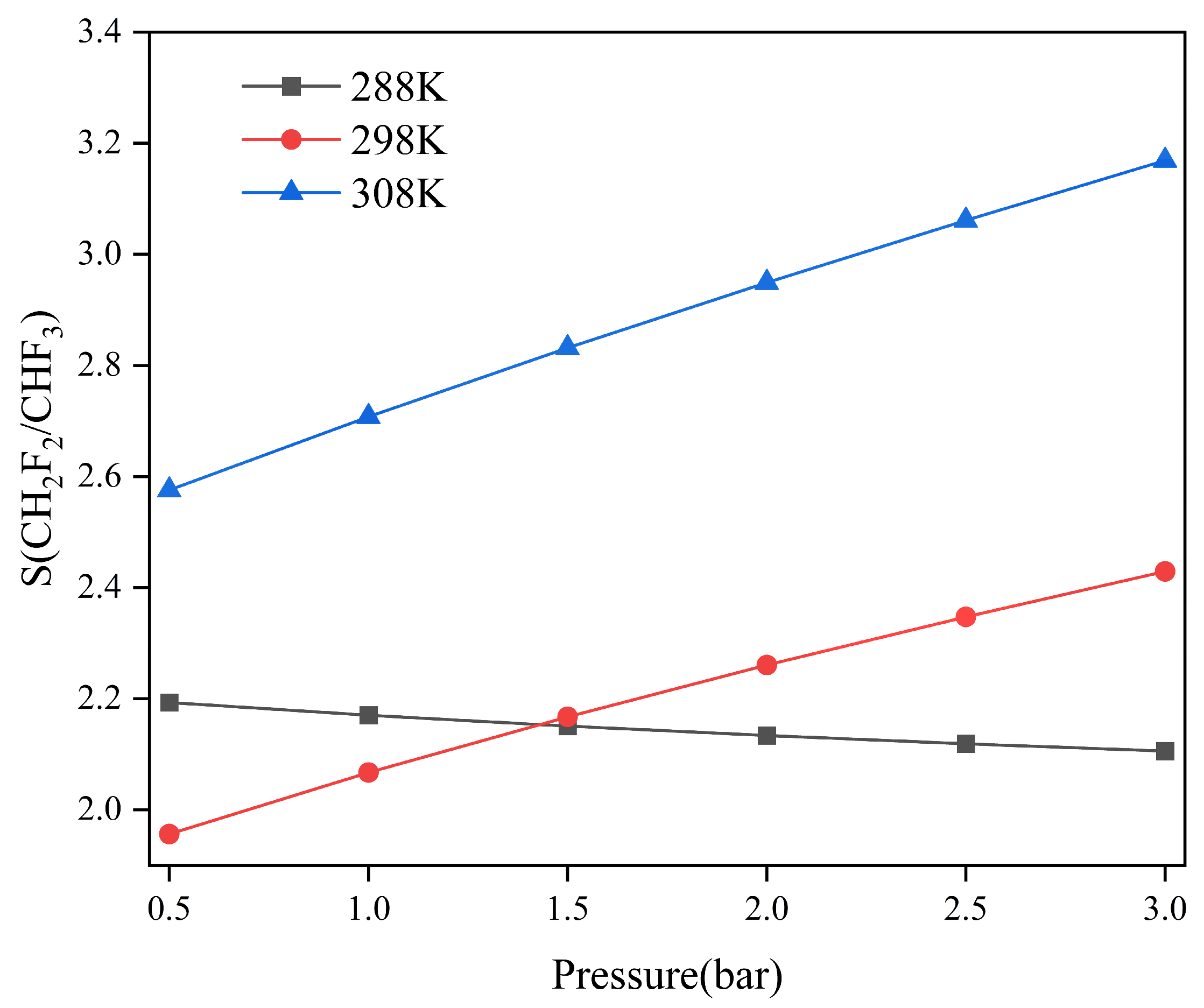
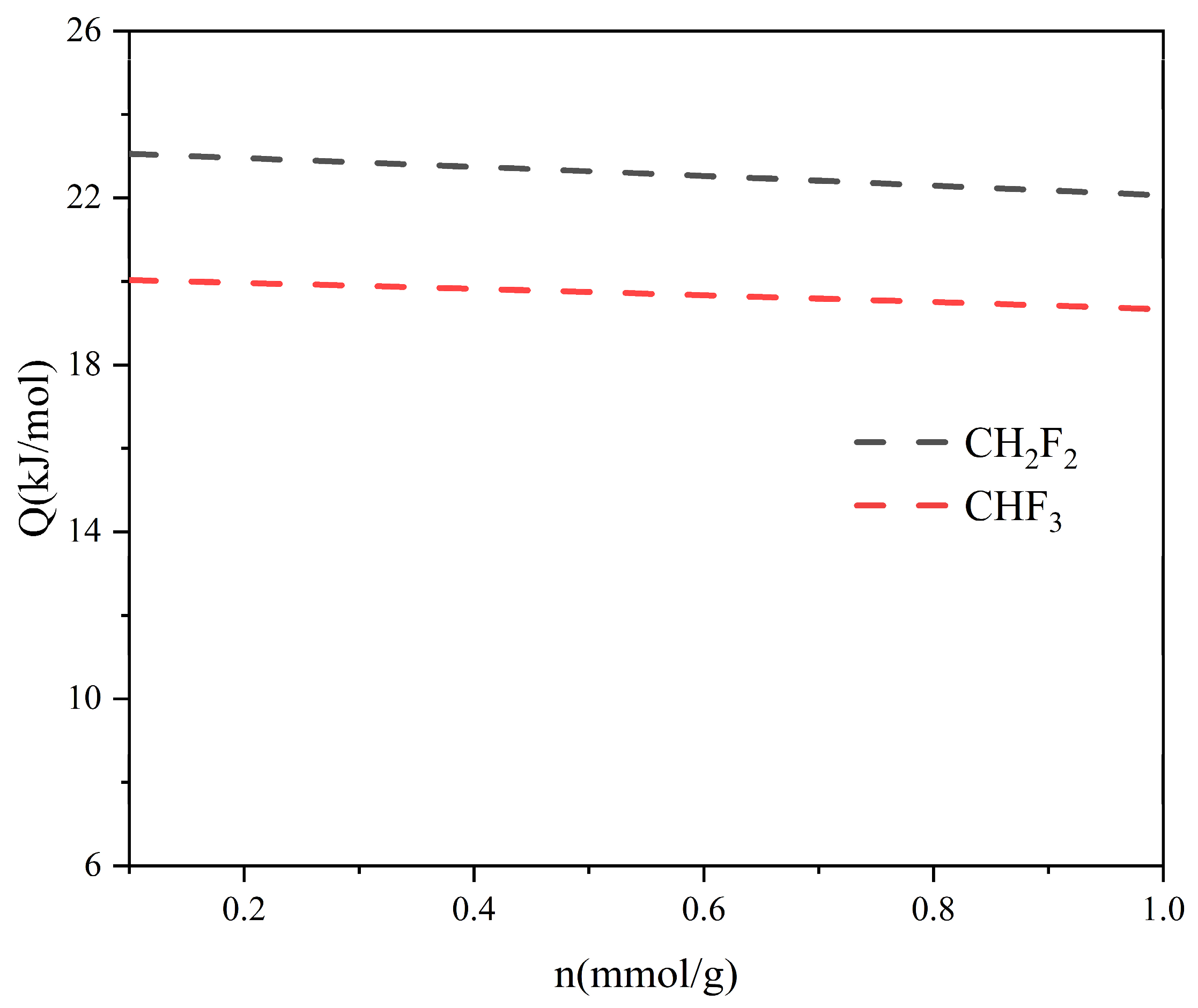
2.2. Adsorption Selectivity and Heat
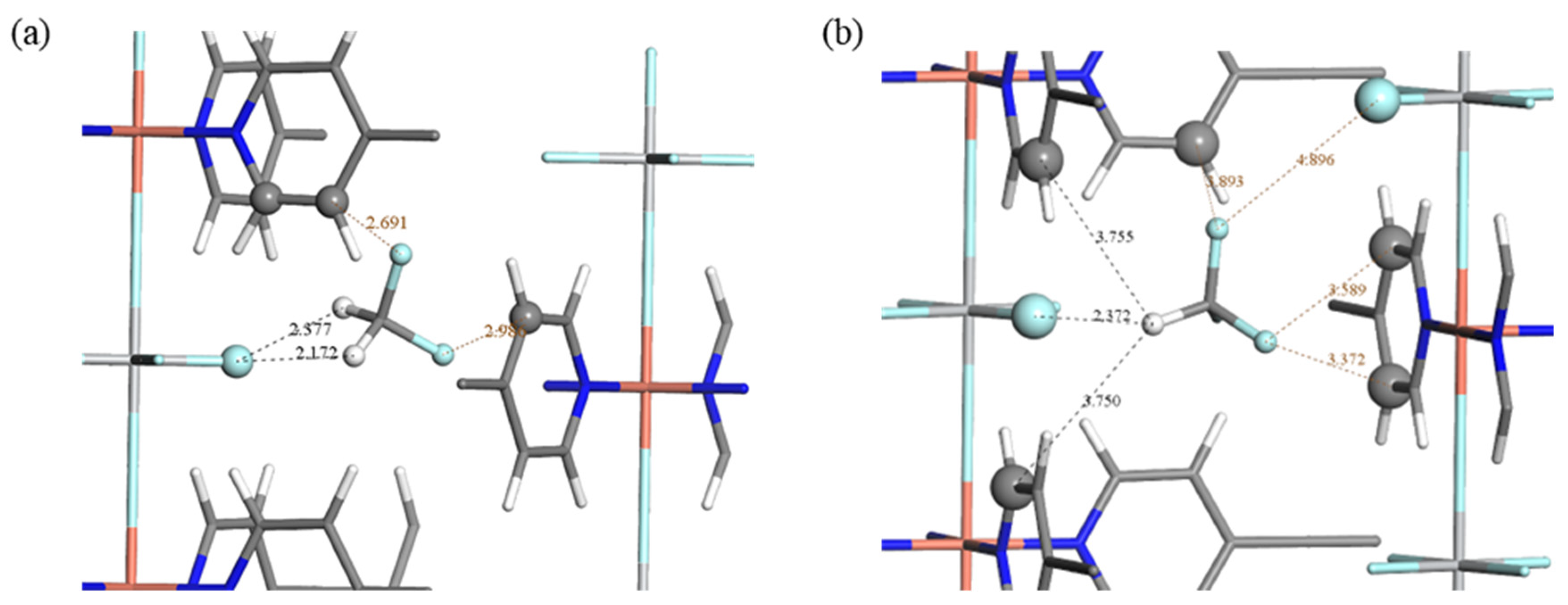
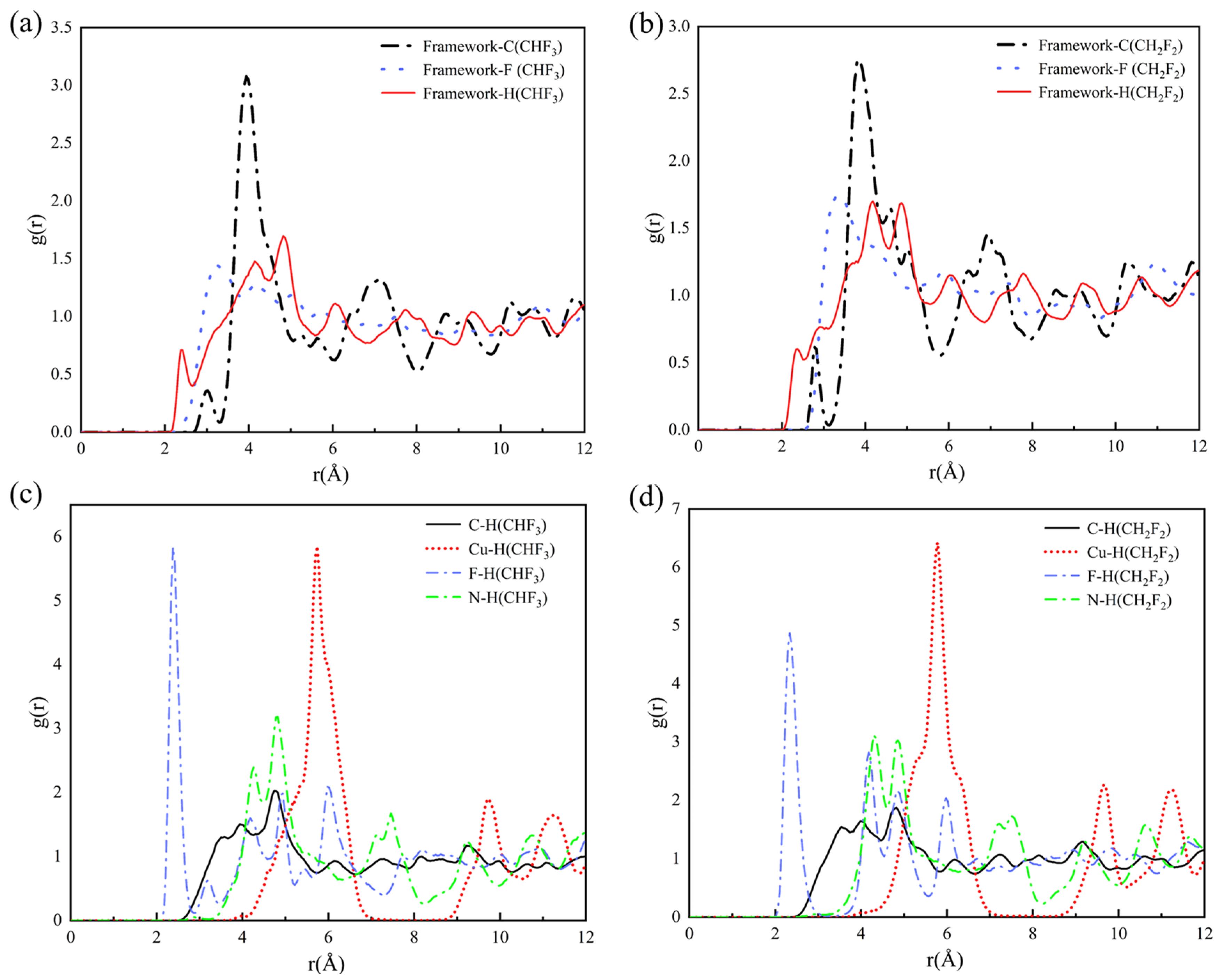
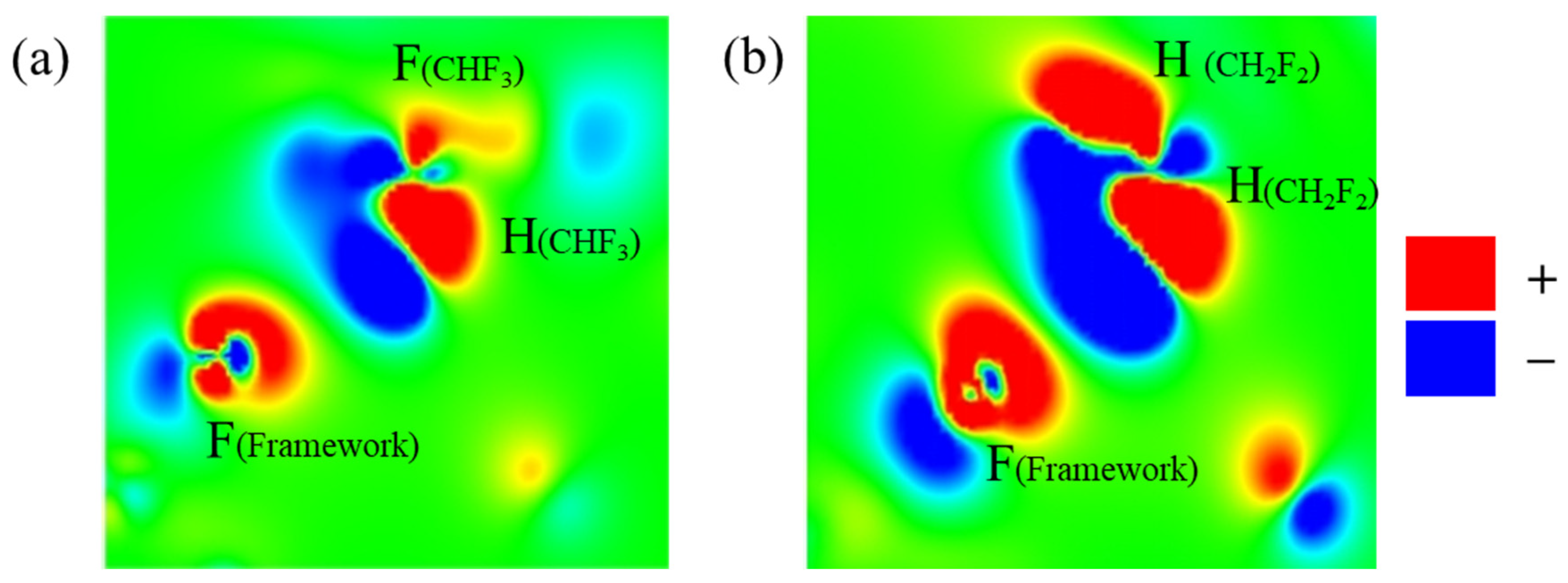
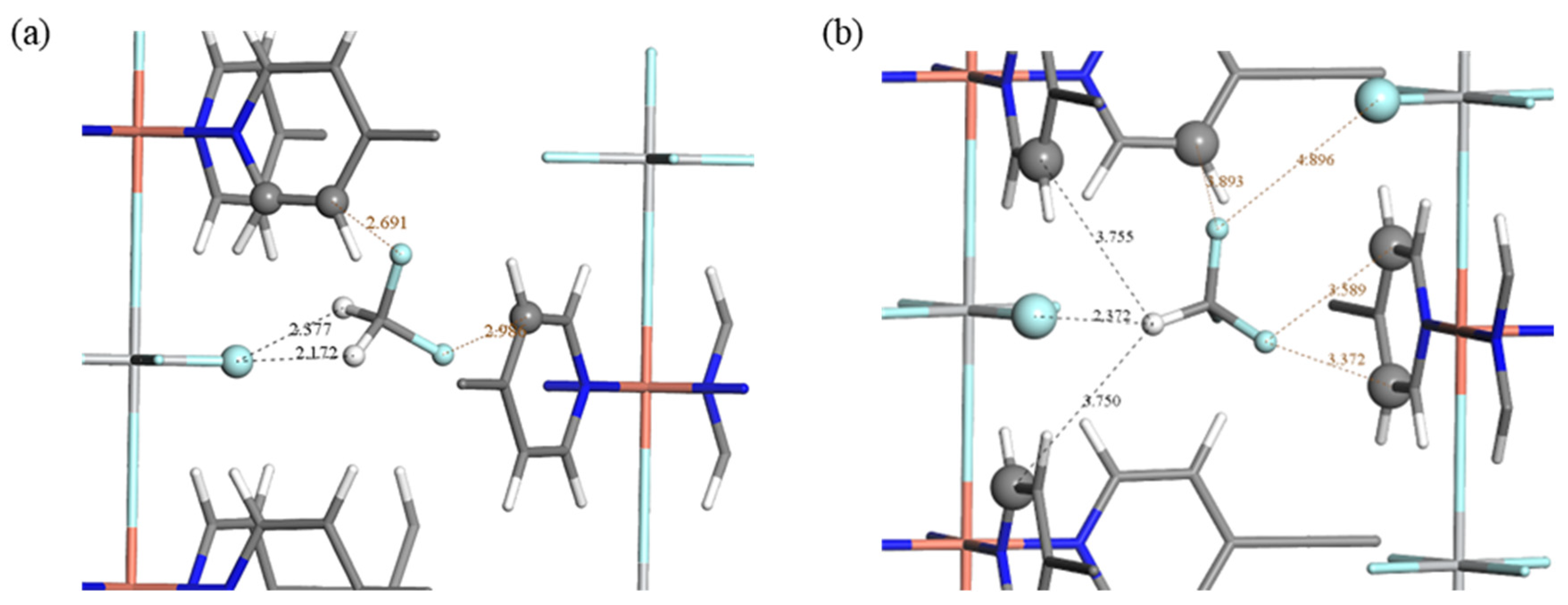
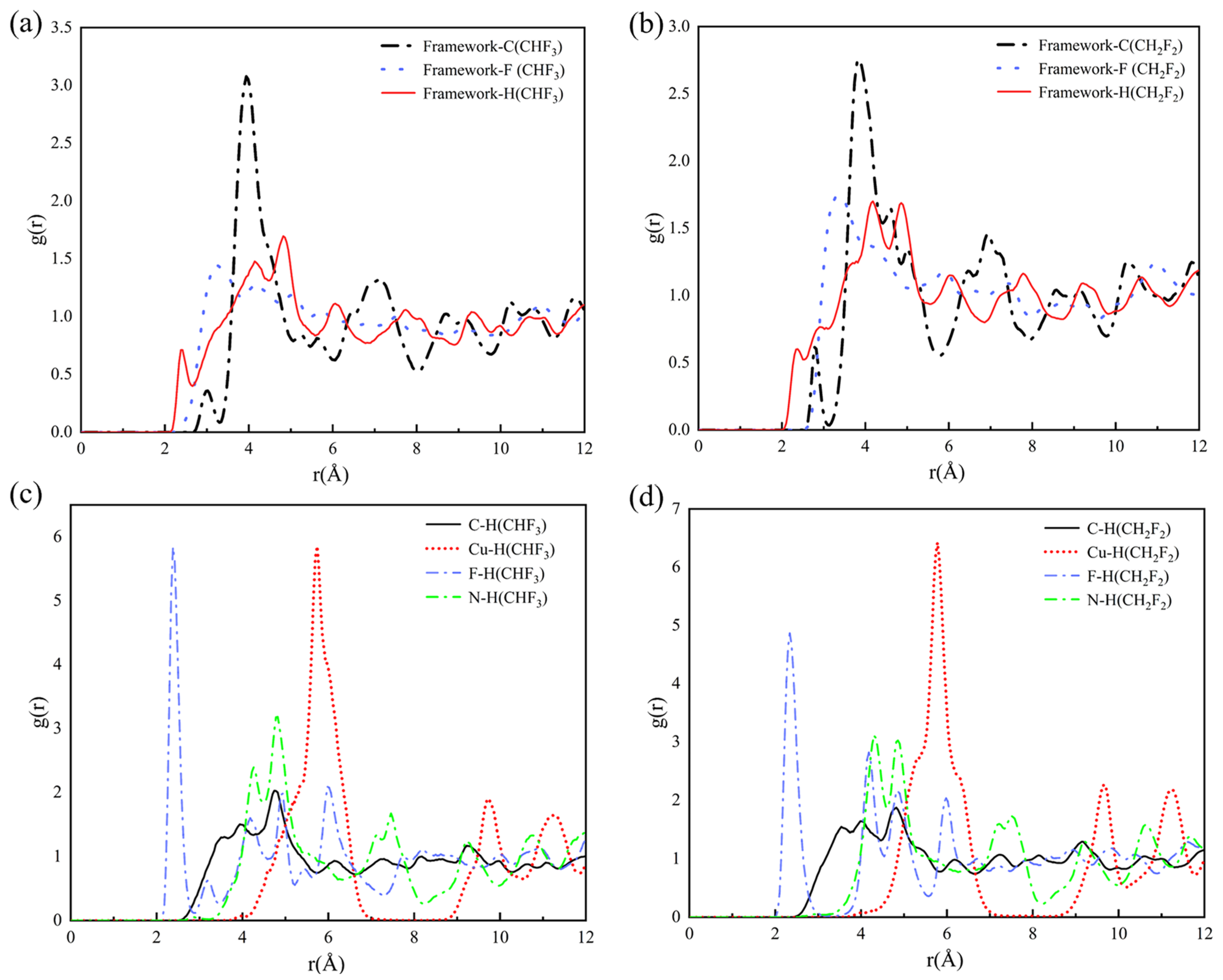
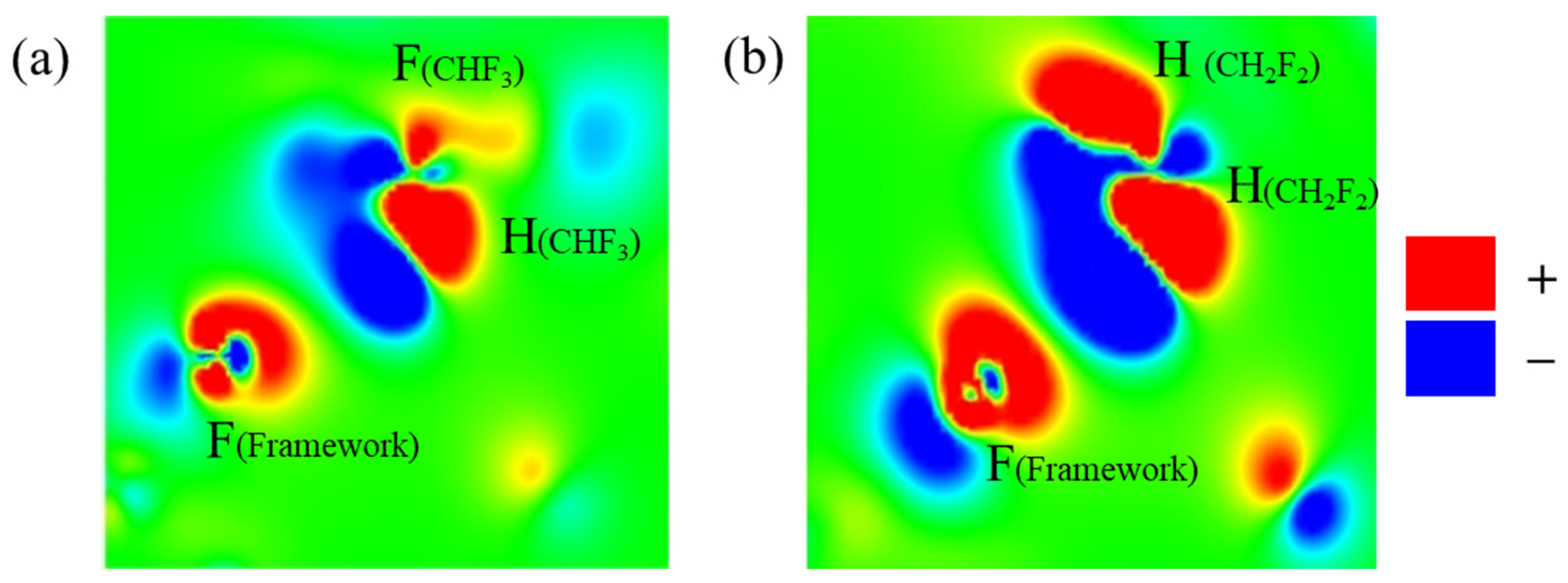
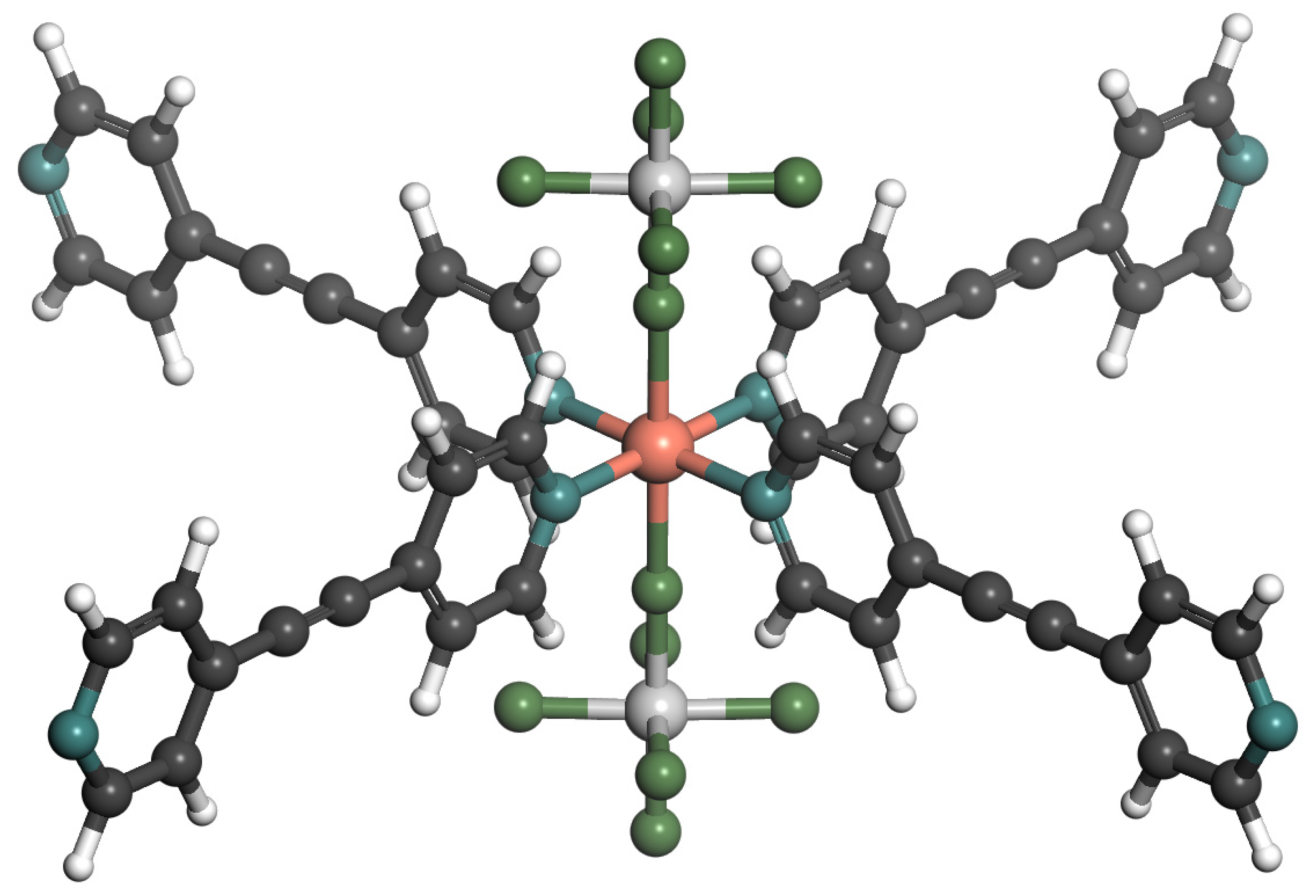
2.3. Adsorption Sites
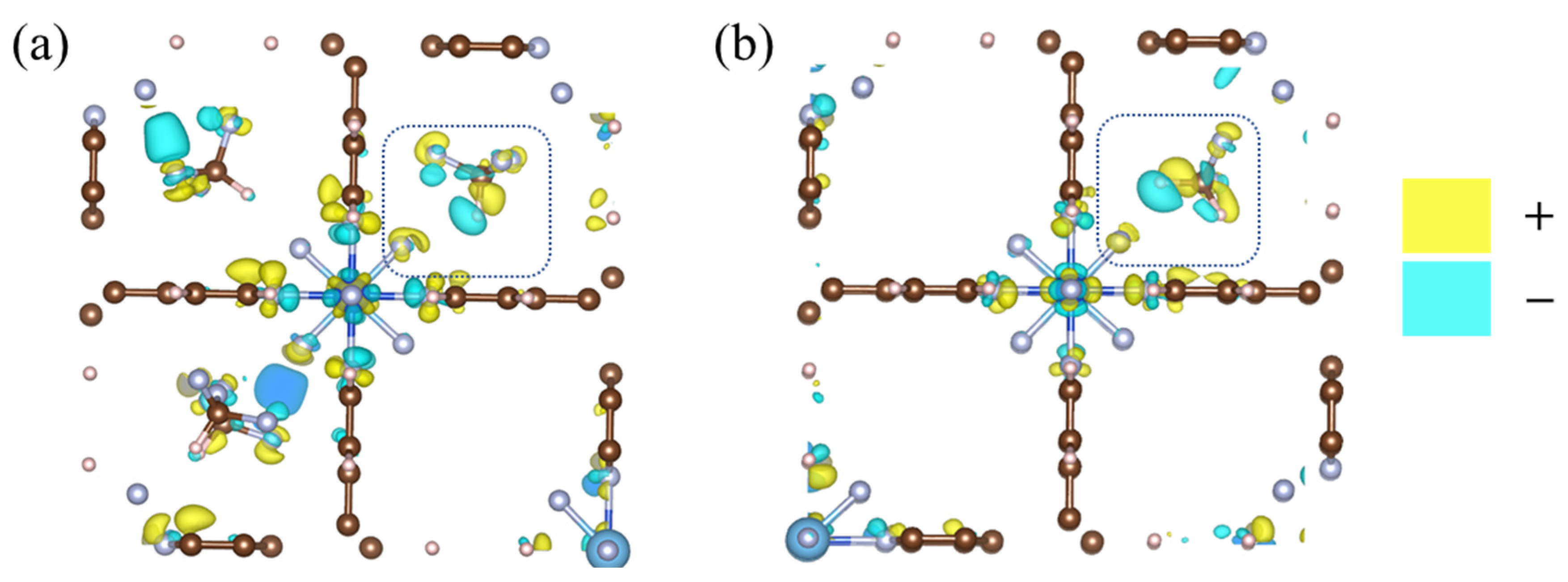
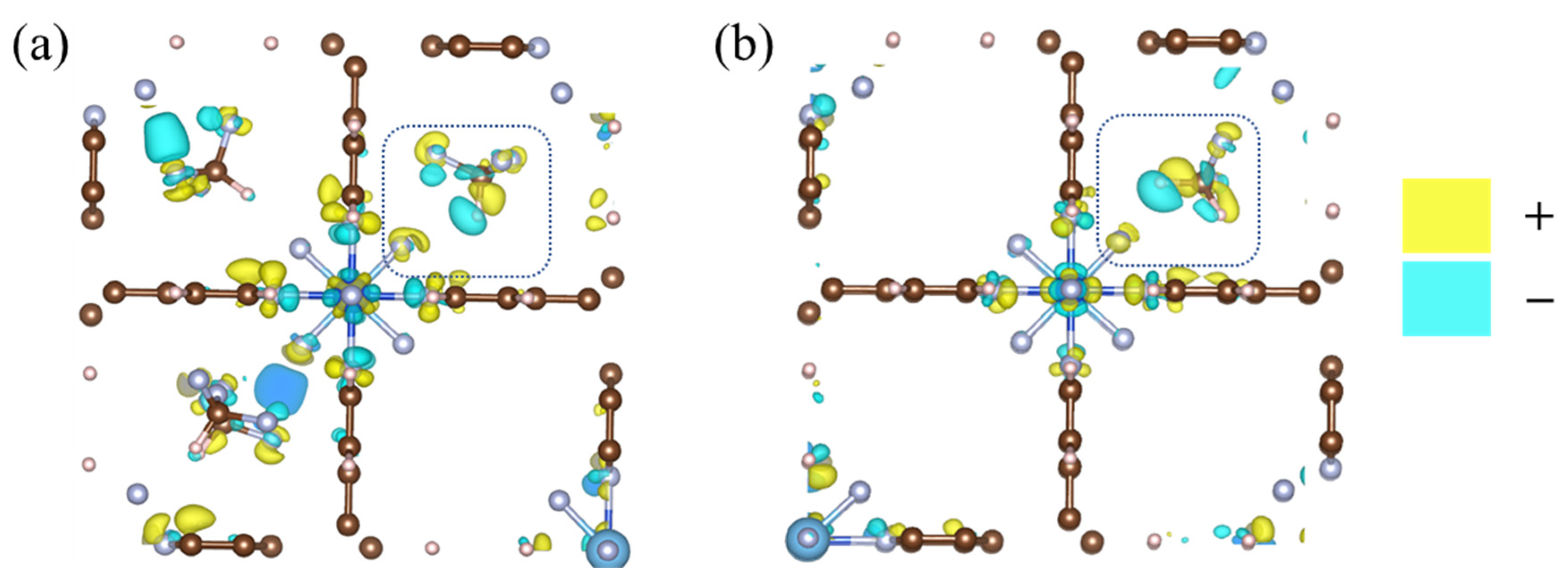
2.4. Redistribution of Charge Density
3. Experiment Section
3.1. Preparation of Materials
3.2. Synthetic Procedures
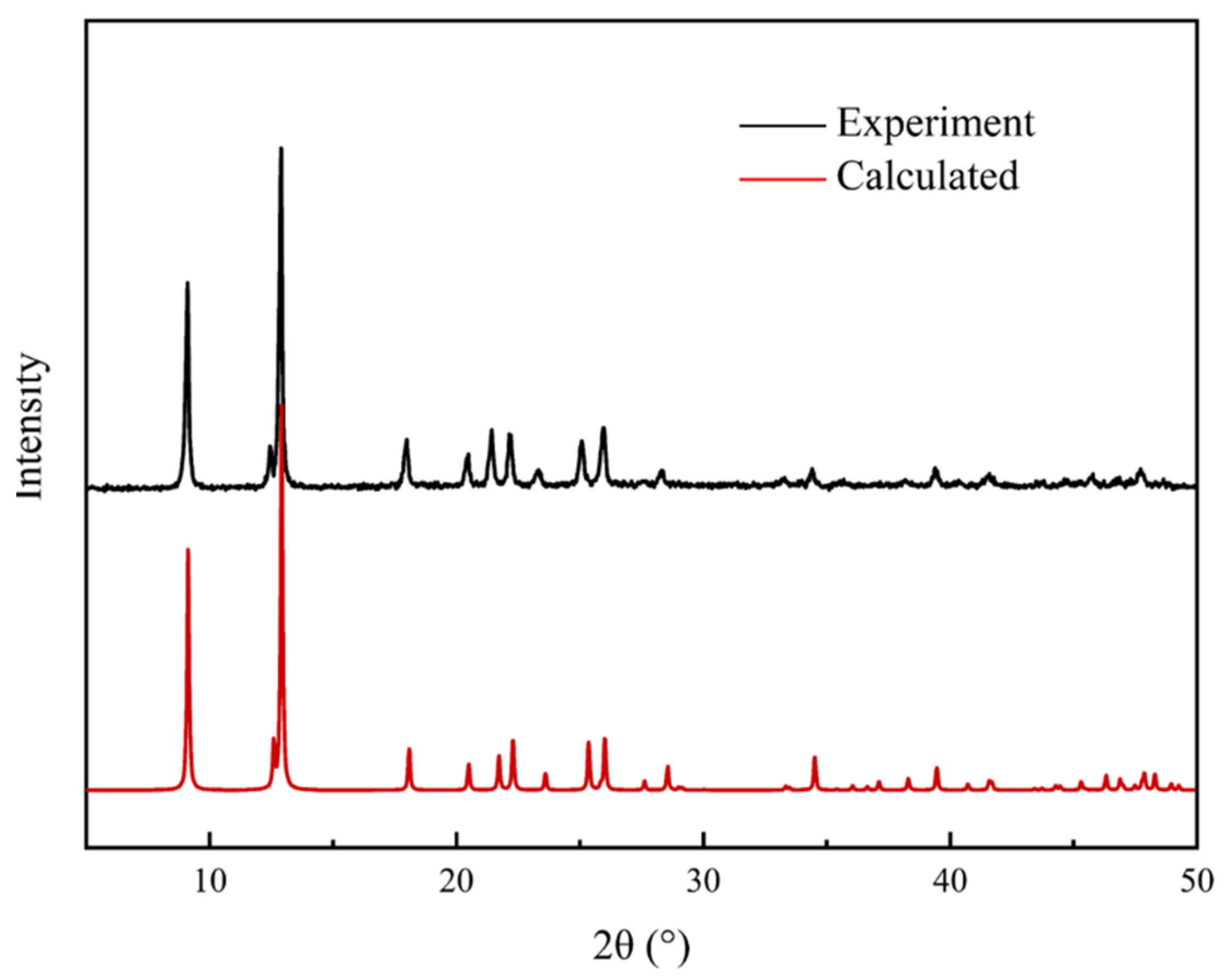
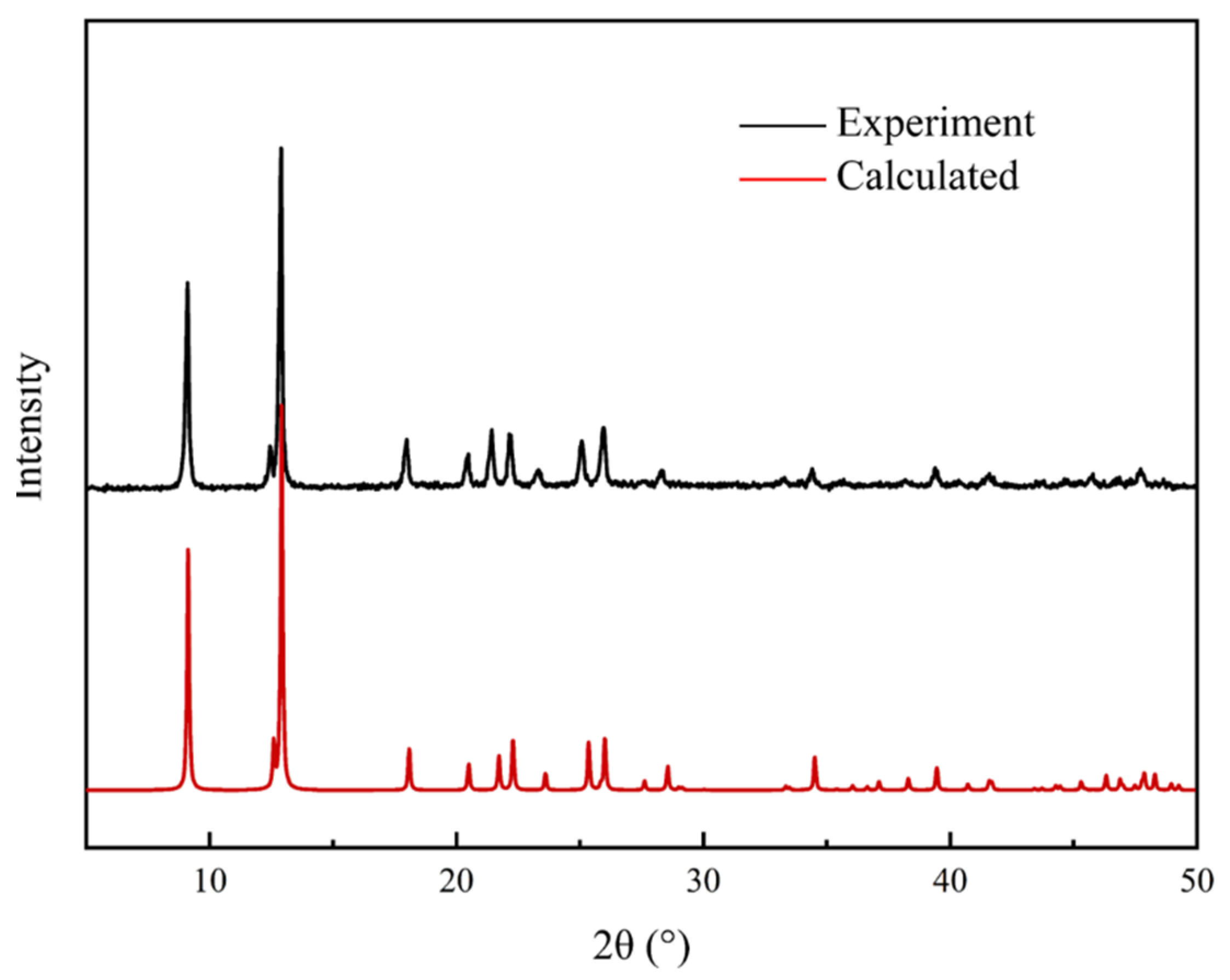
3.3. Characterization
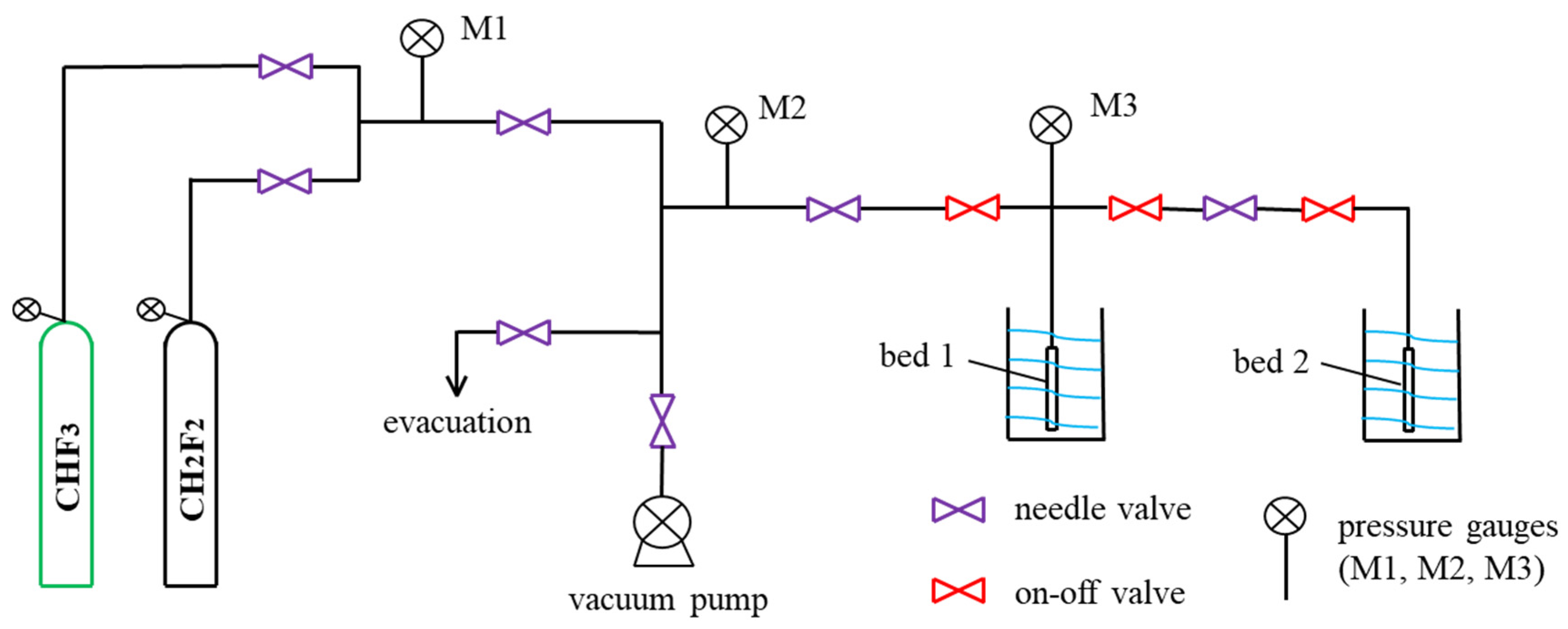
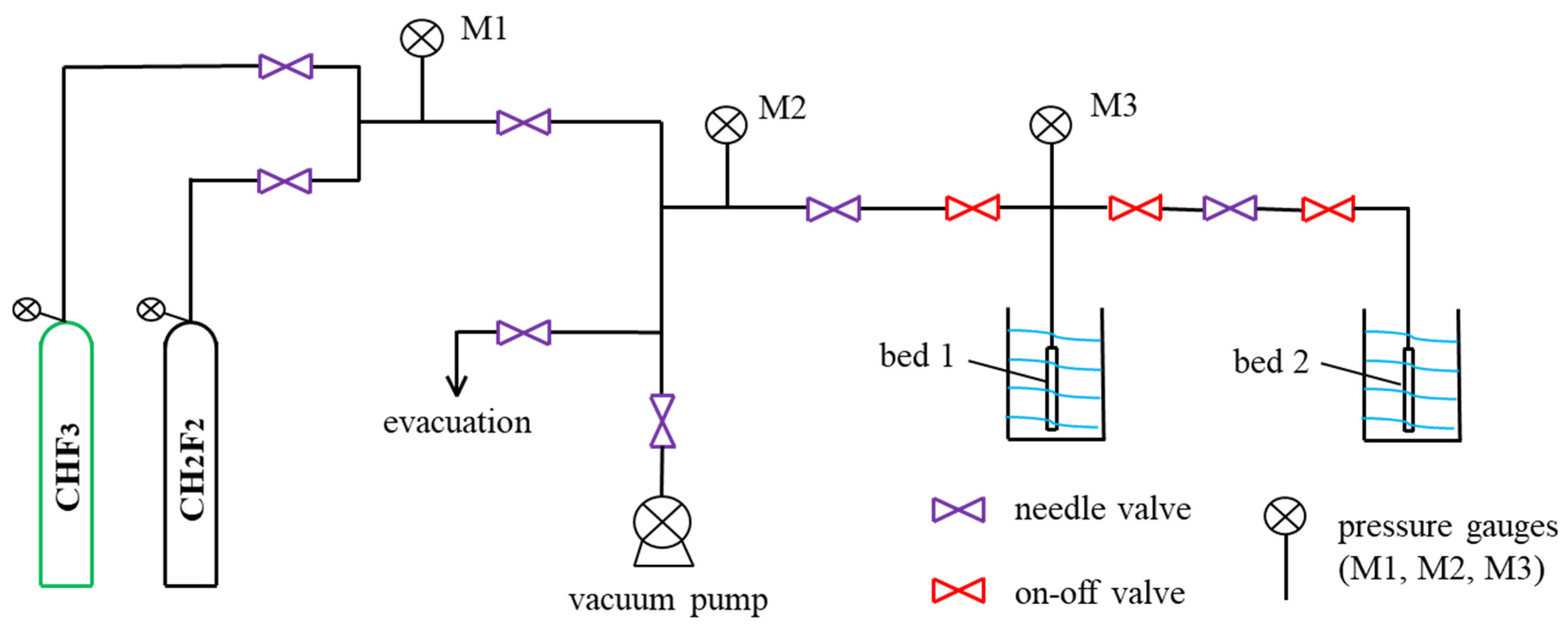
3.4. Single-Component Adsorption Measurements
4. Models and Methods
4.1. Models
4.2. Density Functional Theory Calculations
4.3. Grand Canonical Monte Carlo Simulations
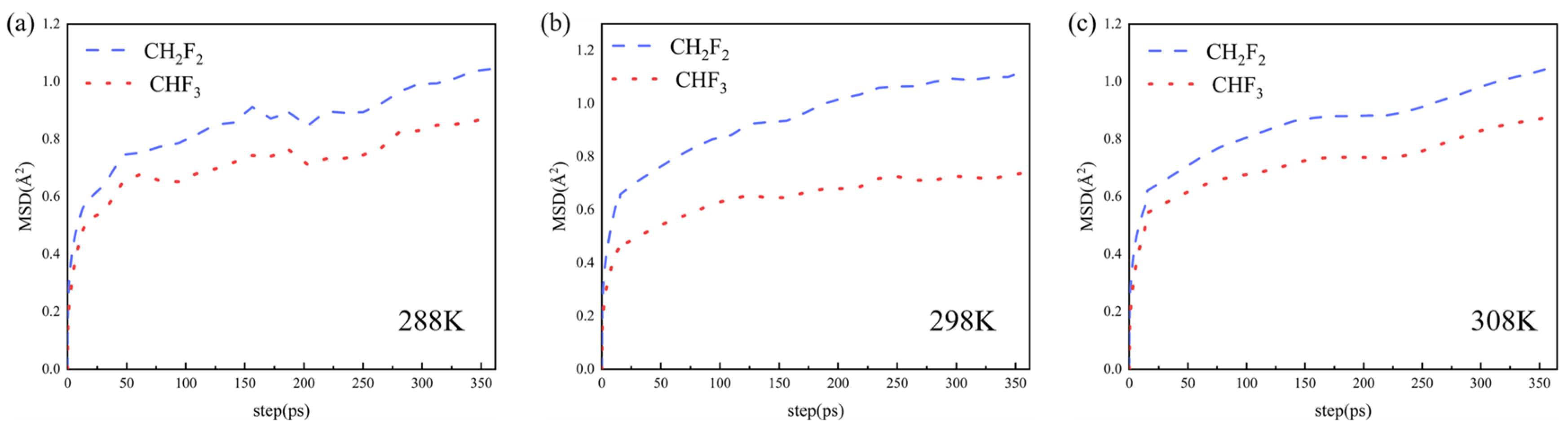
4.4. Molecular Dynamics Simulations Details
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sovacool, B.K.; Griffiths, S.; Kim, J.; Bazilian, M. Climate change and industrial F-gases: A critical and systematic review of developments, sociotechnical systems and policy options for reducing synthetic greenhouse gas emissions. Renew. Sustain. Energy Rev. 2021, 141, 110759. [Google Scholar] [CrossRef]
- Mota-Babiloni, A.; Navarro-Esbrí, J.; Barragán-Cervera, Á.; Molés, F.; Peris, B. Analysis based on EU Regulation No 517/2014 of new HFC/HFO mixtures as alternatives of high GWP refrigerants in refrigeration and HVAC systems. Int. J. Refrig. 2015, 52, 21–31. [Google Scholar] [CrossRef]
- Mota-Babiloni, A.; Makhnatch, P.; Khodabandeh, R. Recent investigations in HFCs substitution with lower GWP synthetic alternatives: Focus on energetic performance and environmental impact. Int. J. Refrig. 2017, 82, 288–301. [Google Scholar] [CrossRef]
- Velders, G.J.M.; Andersen, S.O.; Daniel, J.S.; Fahey, D.W.; McFarland, M. The importance of the Montreal Protocol in protecting climate. Proc. Natl. Acad. Sci. USA 2007, 104, 4814–4819. [Google Scholar] [CrossRef]
- Heath, E.A. Amendment to the Montreal Protocol on Substances that Deplete the Ozone Layer (Kigali Amendment). Int. Leg. Mater. 2017, 56, 193–205. [Google Scholar] [CrossRef]
- Benhadid-Dib, S.; Benzaoui, A. Refrigerants and their Environmental Impact Substitution of Hydro Chlorofluorocarbon HCFC and HFC Hydro Fluorocarbon. Search for an Adequate Refrigerant. Energy Procedia 2012, 18, 807–816. [Google Scholar] [CrossRef]
- Heredia-Aricapa, Y.; Belman-Flores, J.; Mota-Babiloni, A.; Serrano-Arellano, J.; García-Pabón, J.J. Overview of low GWP mixtures for the replacement of HFC refrigerants: R134a, R404A and R410A. Int. J. Refrig. 2020, 111, 113–123. [Google Scholar] [CrossRef]
- Hu, J.; Liu, C.; Liu, L.; Li, Q. Thermal energy storage of R1234yf, R1234ze, R134a and R32/MOF-74 nanofluids: A molecular simulation study. Materials 2018, 11, 1164. [Google Scholar] [CrossRef]
- Hu, G.; Wang, Z.; Zhang, W.; He, H.; Zhang, Y.; Deng, X.; Li, W. MIL-161 Metal–Organic Framework for Efficient Au(III) Recovery from Secondary Resources: Performance, Mechanism, and DFT Calculations. Molecules 2023, 28, 5459. [Google Scholar] [CrossRef]
- Apriliyanto, Y.B.; Faginas-Lago, N.; Evangelisti, S.; Bartolomei, M.; Leininger, T.; Pirani, F.; Pacifici, L.; Lombardi, A. Multilayer Graphtriyne Membranes for Separation and Storage of CO2: Molecular Dynamics Simulations of Post-Combustion Model Mixtures. Molecules 2022, 27, 5958. [Google Scholar] [CrossRef]
- Tagliabue, M.; Farrusseng, D.; Valencia, S.; Aguado, S.; Ravon, U.; Rizzo, C.; Corma, A.; Mirodatos, C. Natural gas treating by selective adsorption: Material science and chemical engineering interplay. Chem. Eng. J. 2009, 155, 553–566. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, Y.; Li, Y.; Min, X.; Zhang, J.; Sun, T. Methane separation and capture from nitrogen rich gases by selective adsorption in microporous materials: A review. Sep. Purif. Technol. 2022, 283, 120206. [Google Scholar] [CrossRef]
- Yancey, A.D.; Terian, S.J.; Shaw, B.J.; Bish, T.M.; Corbin, D.R.; Shiflett, M.B. A review of fluorocarbon sorption on porous materials. Microporous Mesoporous Mater. 2022, 331, 111654. [Google Scholar] [CrossRef]
- Peng, J.; Fu, C.; Zhong, J.; Ye, B.; Xiao, J.; Duan, C.; Lv, D. Efficient Selective Capture of Carbon Dioxide from Nitrogen and Methane Using a Metal-Organic Framework-Based Nanotrap. Molecules 2023, 28, 7908. [Google Scholar] [CrossRef]
- Li, Y.; Bai, Y.; Wang, Z.; Gong, Q.; Li, M.; Bo, Y.; Xu, H.; Jiang, G.; Chi, K. Exquisitely Constructing a Robust MOF with Dual Pore Sizes for Efficient CO2 Capture. Molecules 2023, 28, 6276. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, Y.; Wu, J.; Lv, D.; Yan, J.; Wang, X.; Chen, X.; Liu, Z.; Peng, J. A Microporous Zn(bdc)(ted)0.5 with Super High Ethane Uptake for Efficient Selective Adsorption and Separation of Light Hydrocarbons. Molecules 2023, 28, 6000. [Google Scholar] [CrossRef] [PubMed]
- Ghazvini, M.F.; Vahedi, M.; Nobar, S.N.; Sabouri, F. Investigation of the MOF adsorbents and the gas adsorptive separation mechanisms. J. Environ. Chem. Eng. 2021, 9, 104790. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, P.-F.; Yang, G.-P.; Hou, L.; Zhang, W.-Y.; Han, Y.-F.; Liu, P.; Wang, Y.-Y. Supramolecular control of MOF pore properties for the tailored guest adsorption/separation applications. Coord. Chem. Rev. 2021, 434, 213709. [Google Scholar] [CrossRef]
- Wanigarathna, D.K.; Gao, J.; Liu, B. Metal organic frameworks for adsorption-based separation of fluorocompounds: A review. Mater. Adv. 2020, 1, 310–320. [Google Scholar] [CrossRef]
- Jiang, M.; Cui, X.; Yang, L.; Yang, Q.; Zhang, Z.; Yang, Y.; Xing, H. A thermostable anion-pillared metal-organic framework for C2H2/C2H4 and C2H2/CO2 separations. Chem. Eng. J. 2018, 352, 803–810. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, L.; Tan, B.; Hu, J.; Wang, Q.; Cui, X.; Xing, H. Remarkable separation of C5 olefins in anion-pillared hybrid porous materials. Nano Res. 2020, 14, 541–545. [Google Scholar] [CrossRef]
- Li, B.; Chen, B. Fine-Tuning Porous Metal-Organic Frameworks for Gas Separations at Will. Chem 2016, 1, 669–671. [Google Scholar] [CrossRef]
- Bajpai, A.; O’Nolan, D.; Madden, D.G.; Chen, K.-J.; Pham, T.; Kumar, A.; Lusi, M.; Perry, J.J.; Space, B.; Zaworotko, M.J. The effect of centred versus offset interpenetration on C2H2 sorption in hybrid ultramicroporous materials. Chem. Commun. 2017, 53, 11592–11595. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, X.; Fu, Q.; Qin, H.; Zhang, H.; Li, H.; Wang, S.; Dong, Z.; Zhang, Z.; Wang, M. Study on the Adsorption Separation of CHF3/N2 by an SIFSIX-3-Ni Metal–Organic Framework. Ind. Eng. Chem. Res. 2023, 62, 13627–13636. [Google Scholar] [CrossRef]
- Kołaczkiewicz, J.; Bauer, E. Clausius-Clapeyron equation analysis of two-dimensional vaporization. Surf. Sci. 1985, 155, 700–714. [Google Scholar] [CrossRef]
- Chen, K.-J.; Scott, H.S.; Madden, D.G.; Pham, T.; Kumar, A.; Bajpai, A.; Lusi, M.; Forrest, K.A.; Space, B.; Perry, J.J.; et al. Benchmark C2H2/CO2 and CO2/C2H2 Separation by Two Closely Related Hybrid Ultramicroporous Materials. Chem 2016, 1, 753–765. [Google Scholar] [CrossRef]
- Bokii, G. Kristallokhimiya, Izd; Nauka: Moskva, Russia, 1971. [Google Scholar]
- Zhou, L.; Zhou, Y.; Li, M.; Chen, P.; Wang, Y. Experimental and Modeling Study of the Adsorption of Supercritical Methane on a High Surface Activated Carbon. Langmuir 2000, 16, 5955–5959. [Google Scholar] [CrossRef]
- Nugent, P.; Rhodus, V.; Pham, T.; Tudor, B.; Forrest, K.; Wojtas, L.; Space, B.; Zaworotko, M. Enhancement of CO2 selectivity in a pillared pcu MOM platform through pillar substitution. Chem. Commun. 2013, 49, 1606–1608. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 atomic population analysis: Part 2. Computed results for a wide range of periodic and nonperiodic materials. RSC Adv. 2016, 6, 45727–45747. [Google Scholar] [CrossRef]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 atomic population analysis: Part 4. Efficient parallel computation of net atomic charges, atomic spin moments, bond orders, and more. RSC Adv. 2018, 8, 2678–2707. [Google Scholar] [CrossRef] [PubMed]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Dubbeldam, D.; Calero, S.; Ellis, D.E.; Snurr, R.Q. RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 2016, 42, 81–101. [Google Scholar] [CrossRef]
- Schnabel, T.; Vrabec, J.; Hasse, H. Unlike Lennard–Jones parameters for vapor–liquid equilibria. J. Mol. Liq. 2007, 135, 170–178. [Google Scholar] [CrossRef]
- Fu, Q.; Tanaka, H.; Miyahara, M.T.; Qin, Y.; Shen, Y.; Zhang, D. CHF3–CHClF2 Binary Competitive Adsorption Equilibria in Graphitic Slit Pores: Monte Carlo Simulations and Breakthrough Curve Experiments. Ind. Eng. Chem. Res. 2018, 57, 6440–6450. [Google Scholar] [CrossRef]
- Yang, T.; Siepmann, J.I.; Wu, J. Phase Equilibria of Difluoromethane (R32), 1,1,1,2-Tetrafluoroethane (R134a), and trans-1,3,3,3-Tetrafluoro-1-propene (R1234ze(E)) Probed by Experimental Measurements and Monte Carlo Simulations. Ind. Eng. Chem. Res. 2020, 60, 739–752. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Becker, T.M.; Dubbeldam, D.; Lin, L.-C.; Vlugt, T.J.H. Investigating polarization effects of CO2 adsorption in MgMOF-74. J. Comput. Sci. 2016, 15, 86–94. [Google Scholar] [CrossRef]
- Becker, T.M.; Heinen, J.; Dubbeldam, D.; Lin, L.-C.; Vlugt, T.J.H. Polarizable Force Fields for CO2 and CH4 Adsorption in M-MOF-74. J. Phys. Chem. C 2017, 121, 4659–4673. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Dielectric polarizabilities of ions in oxides and fluorides. J. Appl. Phys. 1993, 73, 348–366. [Google Scholar] [CrossRef]
- Van Duijnen, P.T.; Swart, M. Molecular and atomic polarizabilities: Thole’s model revisited. J. Phys. Chem. A 1998, 102, 2399–2407. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | qm (mmol/g) | b (bar−1) | R-Squared | |
|---|---|---|---|---|
| 288 | 6.22417 | 0.85526 | 0.9582 | |
| CH2F2 | 298 | 7.06367 | 0.57236 | 0.97048 |
| 308 | 6.97756 | 0.44233 | 0.9712 | |
| 288 | 4.92555 | 0.51897 | 0.976526 | |
| CHF3 | 298 | 4.22734 | 0.47127 | 0.96737 |
| 308 | 3.8638 | 0.38144 | 0.9911 |
| SBET (m2/g) | Sample Density (cm3/g) | Total Pore Volume (cm3/g) | t-Plot Micropore Volume (cm3/g) | Mesopore Volume (cm3/g) | Average Pore Size (Å) |
|---|---|---|---|---|---|
| 372 | 1.211 | 0.211 | 0.131 | 0.079 | 3.988 |
| Atom Types | q (|e|) | σ (Å) | ε (K) | α (Å3) |
|---|---|---|---|---|
| C_CHF3 | 0.719 | 3.52 | 55 | 1.2886 |
| H_CHF3 | 0.016 | 2.6 | 25.2 | 0.4138 |
| F_CHF3 | −0.245 | 2.92 | 25 | 0.44475 |
| C_CH2F2 | 0.385 | 3.46 | 42 | 1.2886 |
| H_CH2F2 | 0.049 | 2.2 | 29 | 0.4138 |
| F_CH2F2 | −0.2415 | 2.95 | 37 | 0.44475 |
| Atom Types | σ (Å) | ε (K) | α (Å3) |
|---|---|---|---|
| N | 3.25 | 85.5479 | 0.97157 |
| C | 3.5 | 40.258 | 1.2886 |
| H | 2.42 | 15.097 | 0.4138 |
| F | 3.0932 | 36.4834 | 0.44475 |
| Cu | 3.114 | 2.51 | 2.1963 |
| Ti | 2.8286 | 8.55473 | 3.2428 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Zhou, L.; Qin, H.; Dong, Z.; Li, H.; Liu, B.; Wang, Z.; Zhang, L.; Fu, Q.; Chen, X. Study of CHF3/CH2F2 Adsorption Separation in TIFSIX-2-Cu-i. Molecules 2024, 29, 1721. https://doi.org/10.3390/molecules29081721
Wang S, Zhou L, Qin H, Dong Z, Li H, Liu B, Wang Z, Zhang L, Fu Q, Chen X. Study of CHF3/CH2F2 Adsorption Separation in TIFSIX-2-Cu-i. Molecules. 2024; 29(8):1721. https://doi.org/10.3390/molecules29081721
Chicago/Turabian StyleWang, Shoudong, Lei Zhou, Hongyun Qin, Zixu Dong, Haoyuan Li, Bo Liu, Zhilu Wang, Lina Zhang, Qiang Fu, and Xia Chen. 2024. "Study of CHF3/CH2F2 Adsorption Separation in TIFSIX-2-Cu-i" Molecules 29, no. 8: 1721. https://doi.org/10.3390/molecules29081721
APA StyleWang, S., Zhou, L., Qin, H., Dong, Z., Li, H., Liu, B., Wang, Z., Zhang, L., Fu, Q., & Chen, X. (2024). Study of CHF3/CH2F2 Adsorption Separation in TIFSIX-2-Cu-i. Molecules, 29(8), 1721. https://doi.org/10.3390/molecules29081721