
New Pyranone Derivatives and Sesquiterpenoid Isolated from the Endophytic Fungus Xylaria sp. Z184
 , and
, and
Abstract
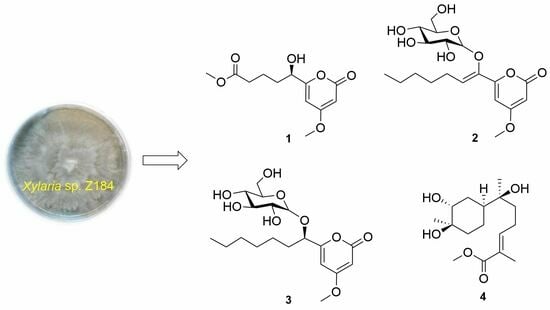
:
1. Introduction
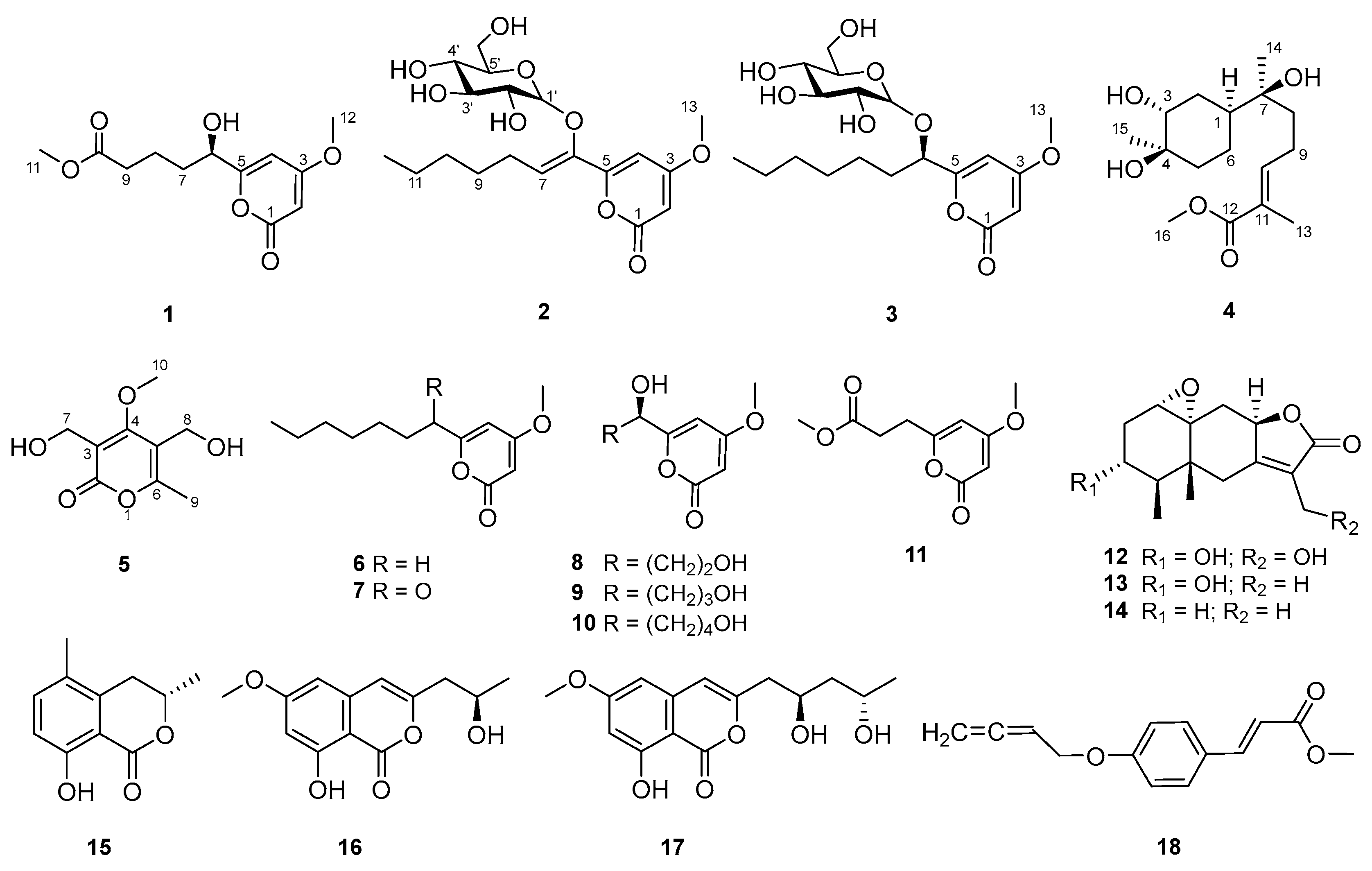
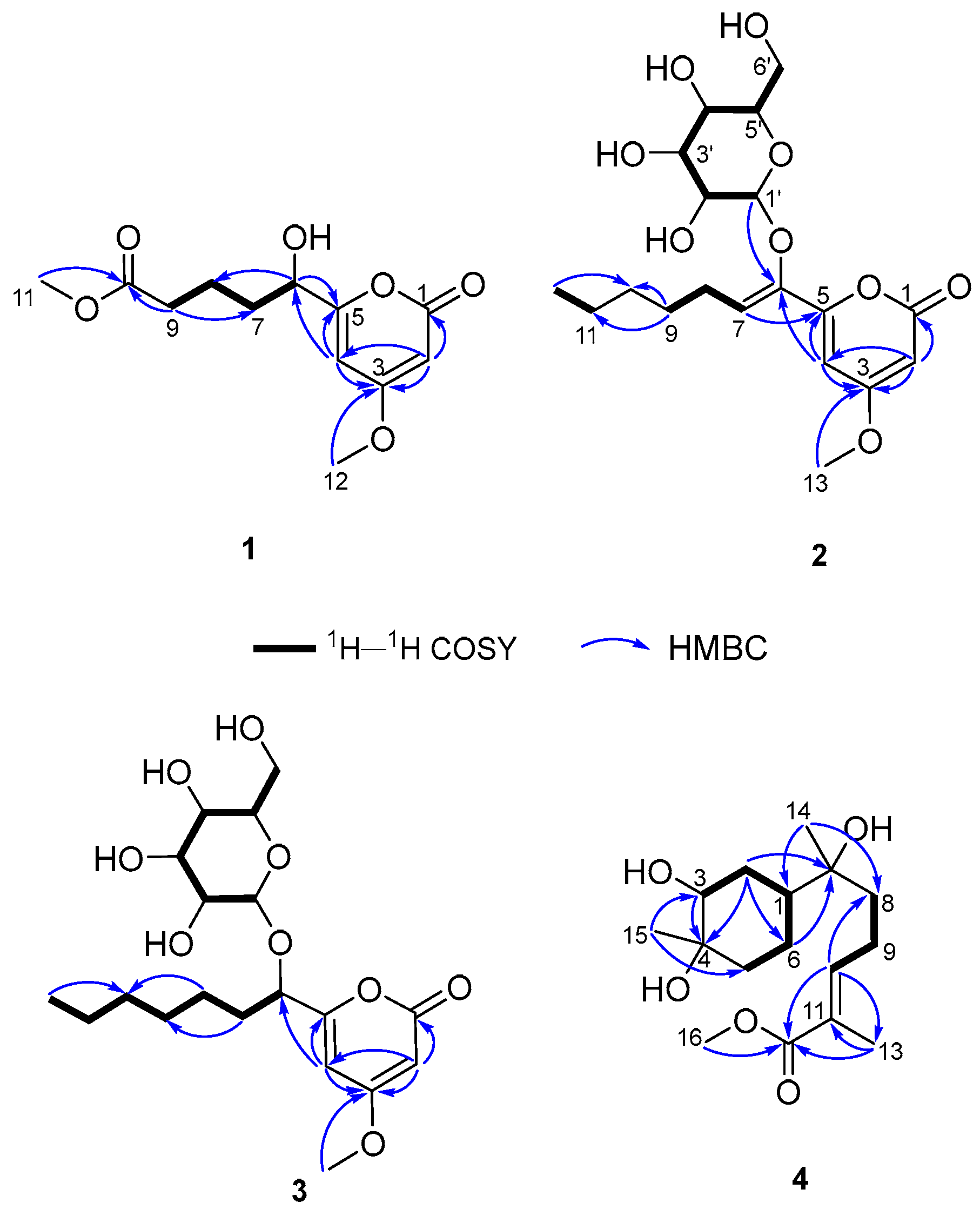
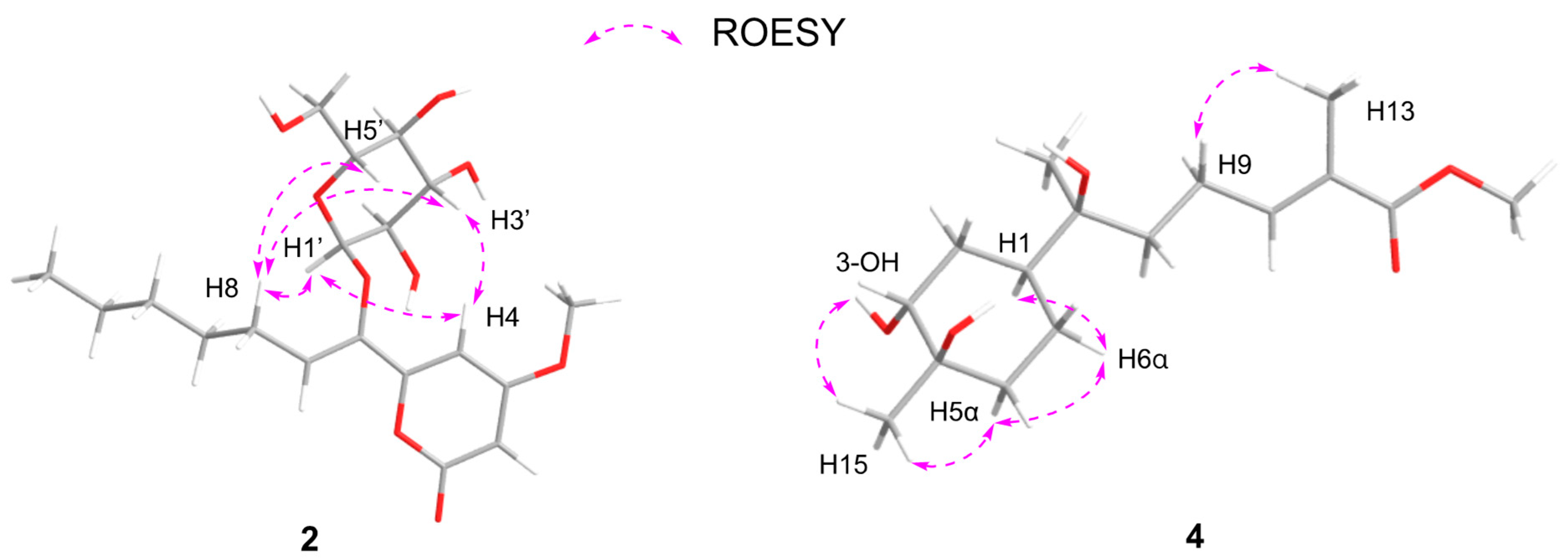
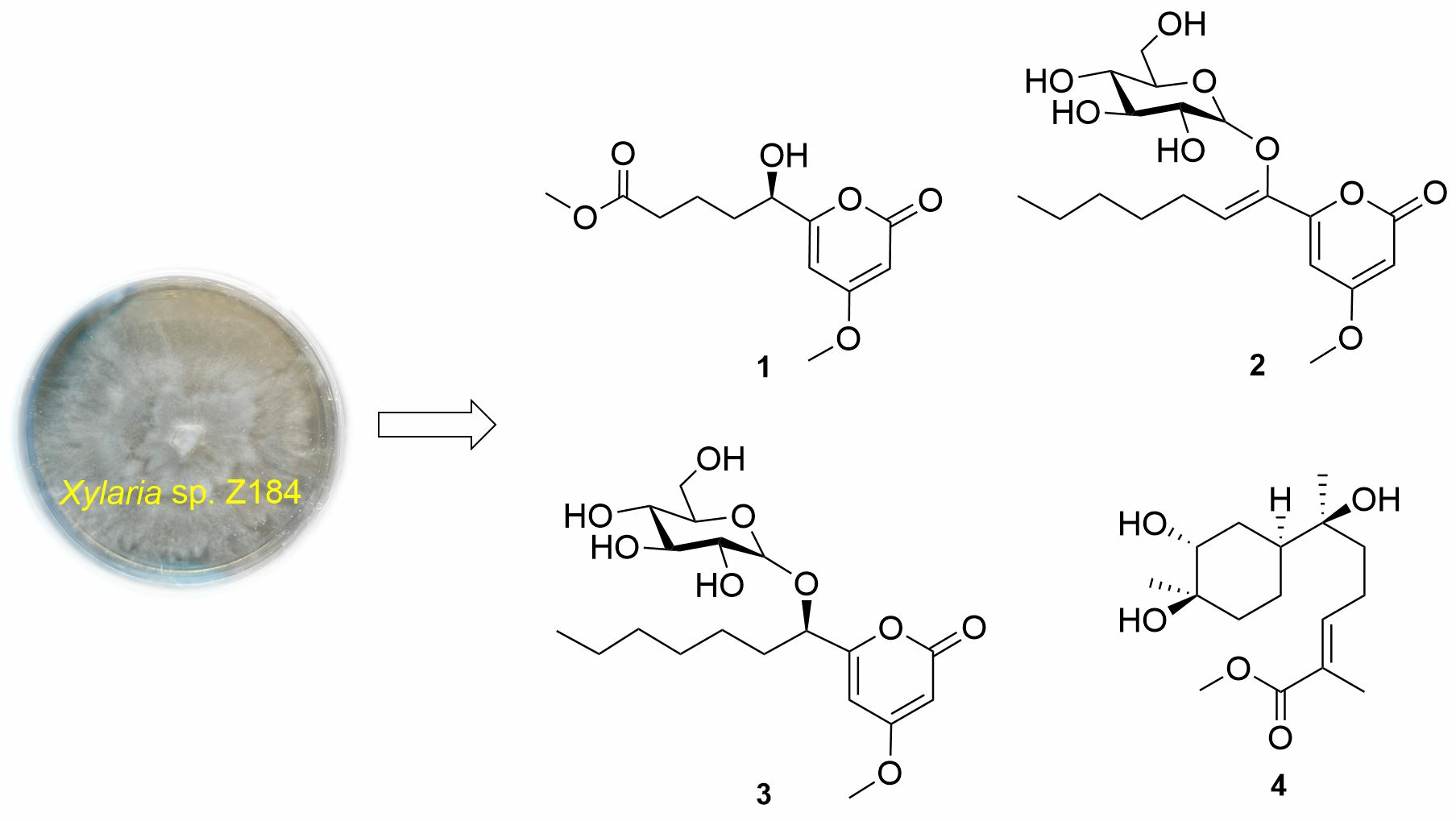
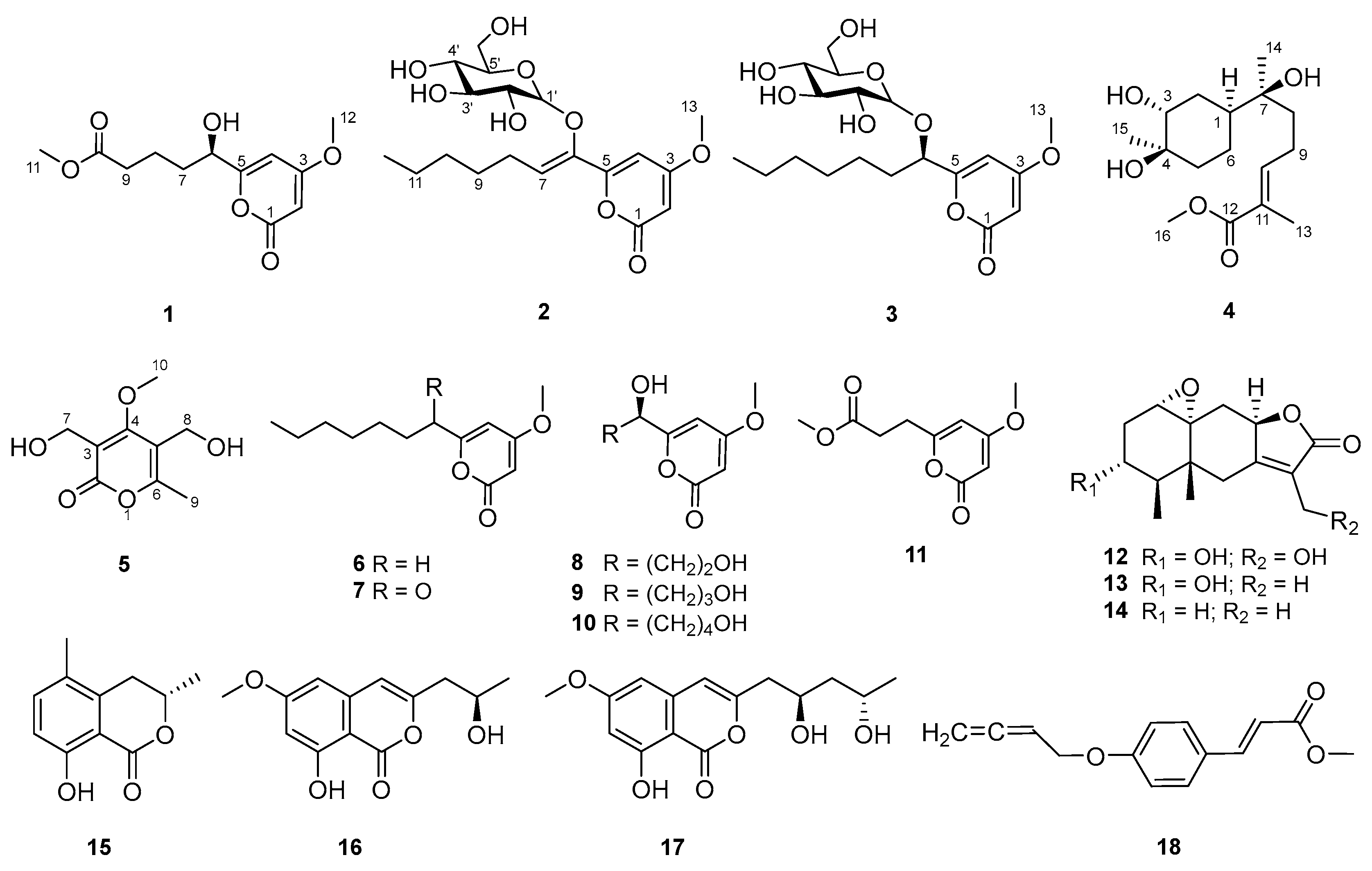
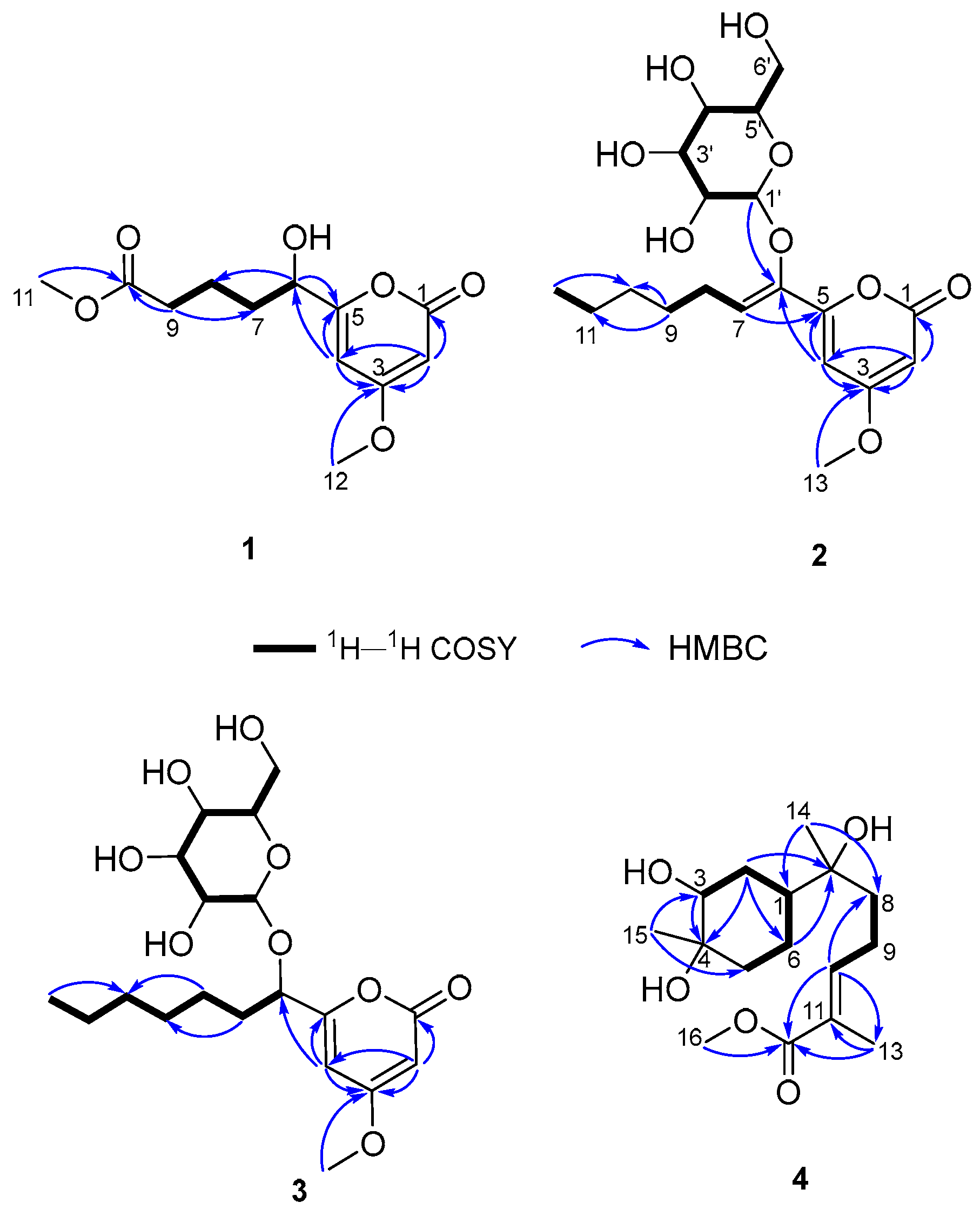
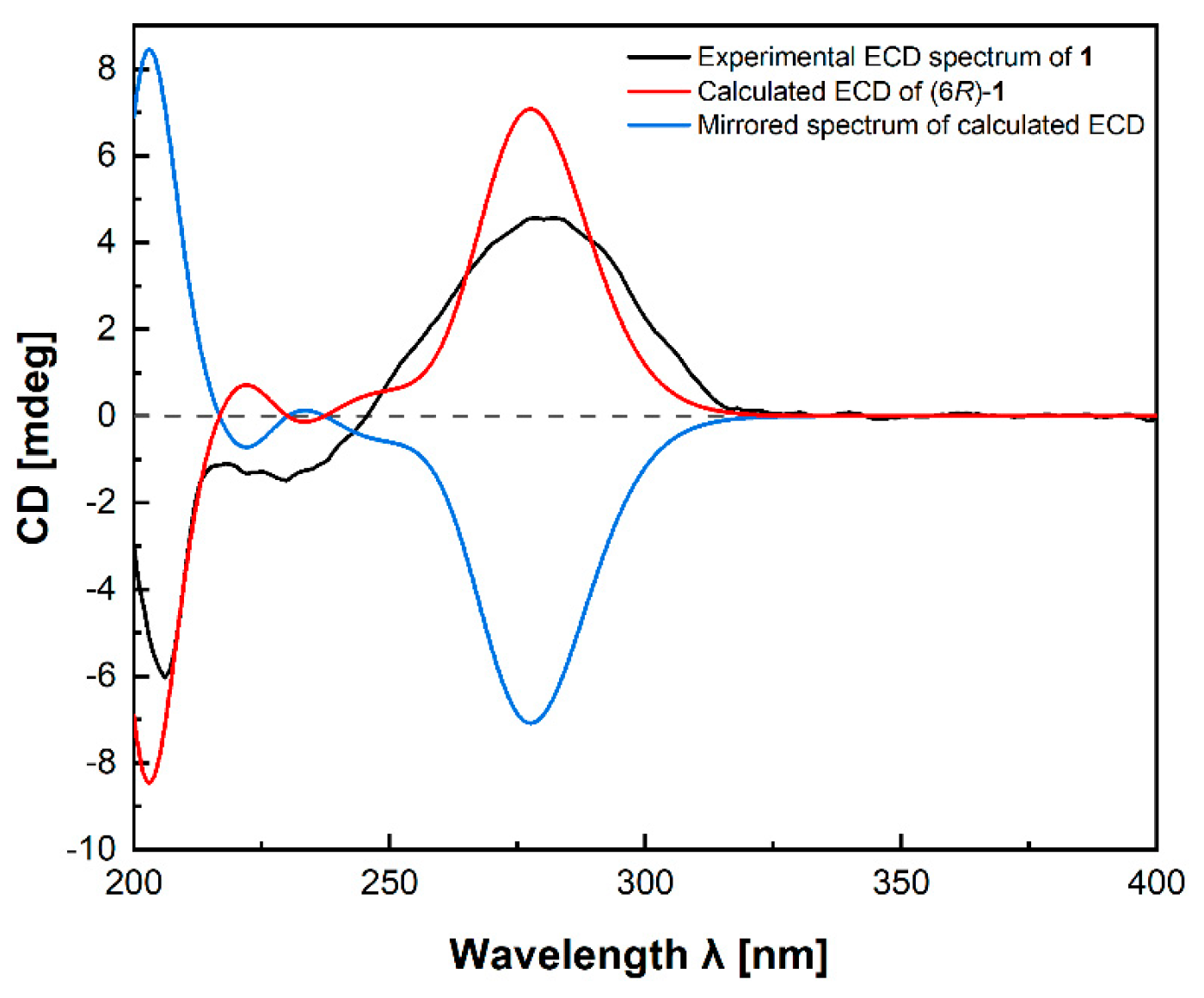
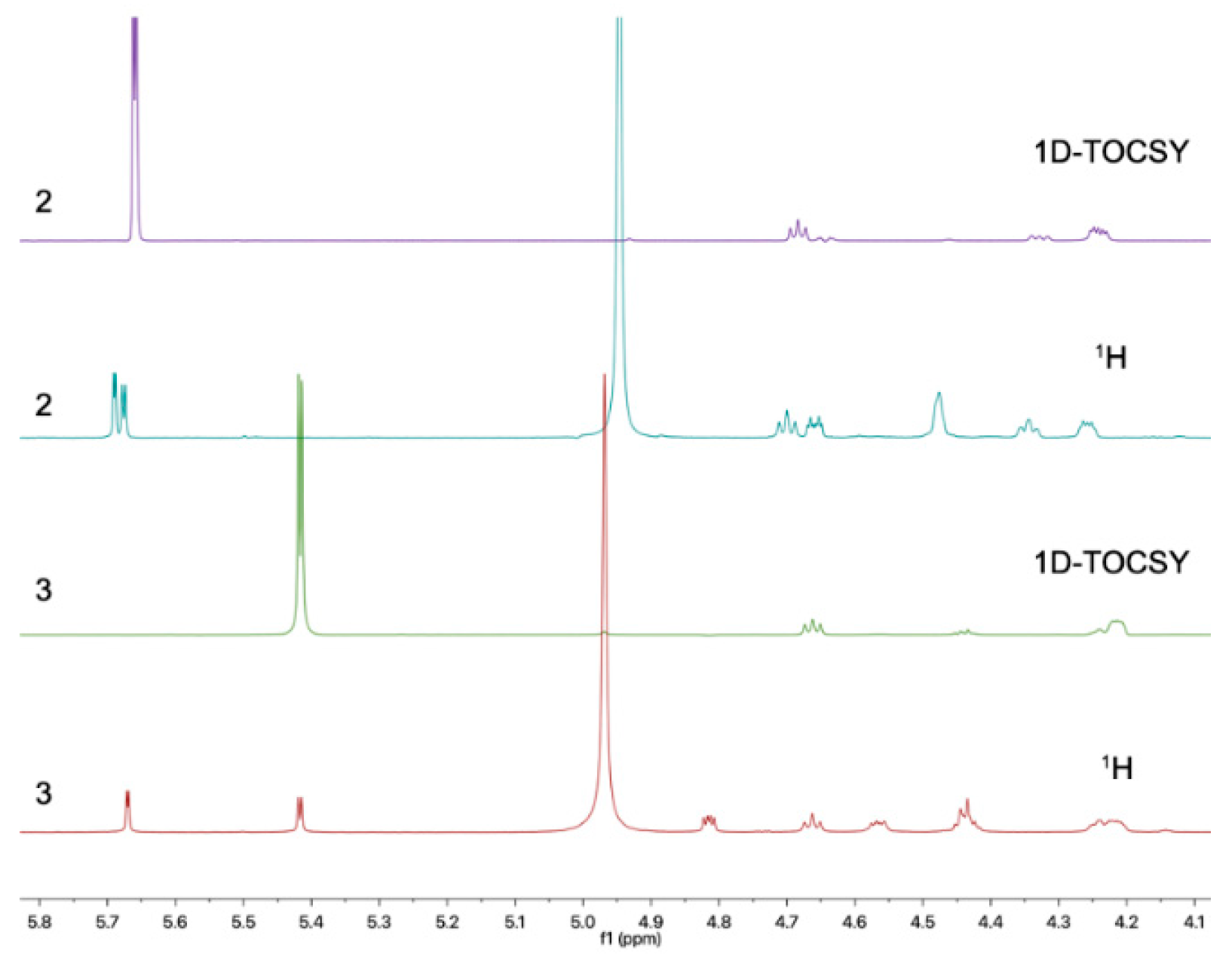
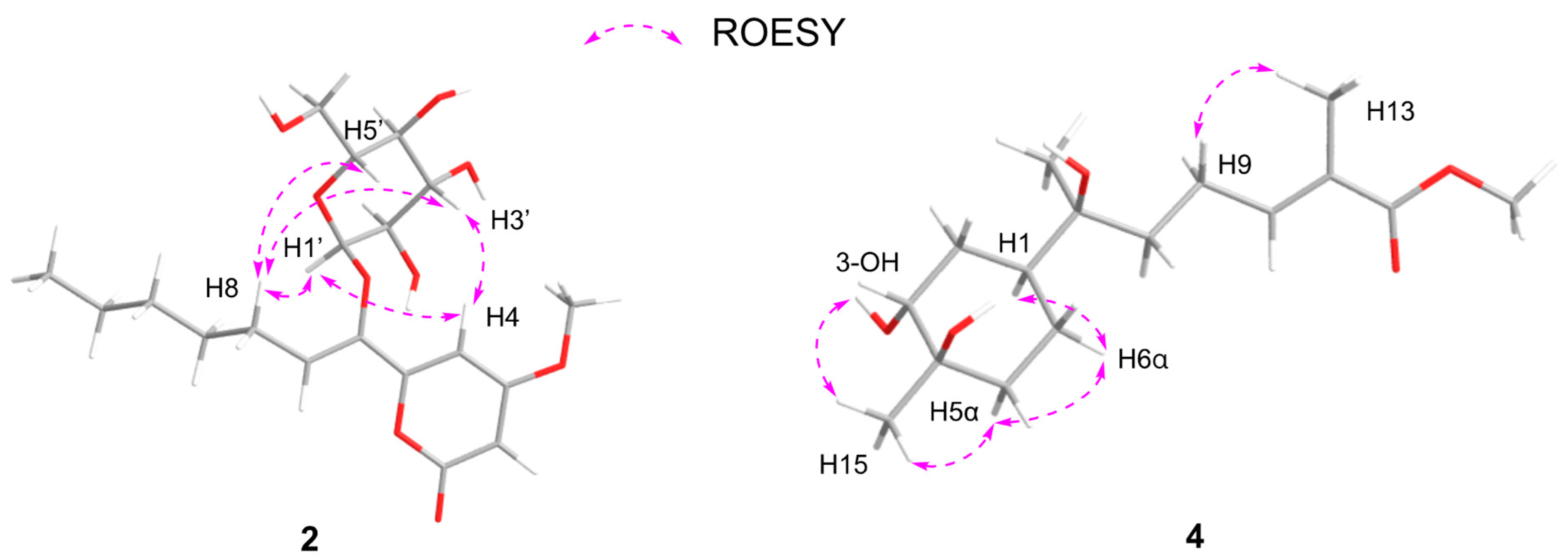
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
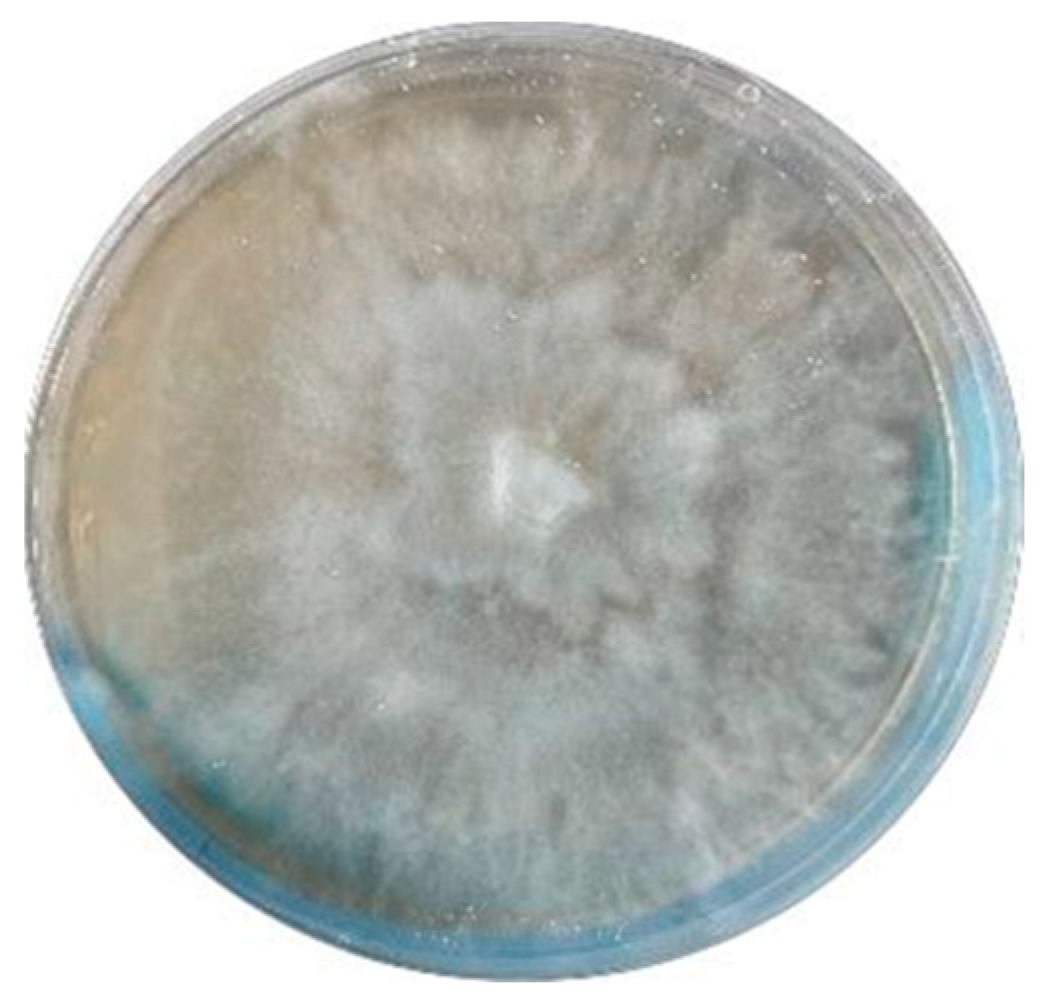
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. Spectral and Physical Data of Compounds 1–5
3.5. Computational Details (TDDFT-ECD) of 1
3.6. Biological Assays
3.6.1. Antimicrobial Activity
3.6.2. Anti-Inflammatory Activity
3.6.3. Alpha-Glucosidase-Inhibition Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tan, H.; Chen, Y.; Guo, X.; Wang, W.; Guo, H.; Liu, Z.; Zhang, W. Cytorhizins A-D, four highly structure-combined benzophenones from the endophytic fungus Cytospora rhizophorae. Org. Lett. 2019, 21, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Han, W.B.; Wang, G.Y.; Tang, J.J.; Wang, W.J.; Liu, H.; Gil, R.R.; Navarro-Vázquez, A.; Lei, X.X.; Gao, J.M. Herpotrichones A and B, two intermolecular [4+2] adducts with anti-neuroinflammatory activity from a Herpotrichia Species. Org. Lett. 2020, 22, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Yoiprommarat, S.; Unagul, P.; Suvannakad, R.; Klaysuban, A.; Suetrong, S.; Bunyapaiboonsri, T. Eremophilane sesquiterpenes from the mangrove fungus BCC 60405. Phytochem. Lett. 2019, 34, 84–90. [Google Scholar] [CrossRef]
- Wu, W.; Dai, H.; Bao, L.; Ren, B.; Lu, J.; Luo, Y.; Guo, L.; Zhang, L.; Liu, H. Isolation and structural elucidation of proline-containing cyclopentapeptides from an endolichenic Xylaria sp. J. Nat. Prod. 2011, 74, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Li, R.; Zhu, R.; Li, X.; Xu, Y.; He, Q.; Xie, F.; Qiao, Y.; Luan, X.; Lou, H. Xylarins A–D, two pairs of diastereoisomeric isoindoline alkaloids from the endolichenic fungus Xylaria sp. Org. Lett. 2021, 23, 7751–7754. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yu, M.; Chen, S.; Gong, T.; Xie, L.; Liu, J.; Bian, C.; Huang, G.; Zheng, C. Structures and biological activities of secondary metabolites from Xylaria spp. J. Fungi 2024, 10, 190. [Google Scholar] [CrossRef]
- Han, W.B.; Zhai, Y.J.; Gao, Y.; Zhou, H.Y.; Xiao, J.; Pescitelli, G.; Gao, J.M. Cytochalasins and an abietane-type diterpenoid with allelopathic activities from the endophytic fungus Xylaria species. J. Agric. Food. Chem. 2019, 67, 3643–3650. [Google Scholar] [CrossRef]
- Jia, S.; Li, J.; Li, J.; Su, X.; Li, X.N.; Yao, Y.; Xue, Y. Anti-neuroinflammatory activity of new naturally occurring benzylated hydroxyacetophenone analogs from the endophytic fungus Alternaria sp. J030. Chem. Biodivers. 2022, 19, e202200751. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Su, X.; Yan, W.; Wu, M.; Wu, Y.; Lu, J.; He, X.; Ding, X.; Xue, Y. Acorenone C: A new spiro-sesquiterpene from a mangrove-associated fungus, Pseudofusicoccum sp. J003. Front. Chem. 2021, 9, 780304. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Su, X.; Lu, J.; Wu, M.; Yang, S.Y.; Mai, Y.; Deng, W.; Xue, Y. In Vitro and in silico analysis of phytochemicals from Fallopia dentatoalata as dual functional cholinesterase inhibitors for the treatment of Alzheimer’s disease. Front. Pharmacol. 2022, 13, 905708. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.W.; Lu, L.W.; Zhang, X.Q.; Bao, S.S.; Cao, F.; Guo, Z.Y.; Deng, Z.S.; Proksch, P. Xylariaopyrones E-I, five new α-pyrone derivatives from the endophytic fungus Xylariales sp. (HM-1). Nat. Prod. Res. 2022, 36, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Tai, A.; Fujinami, Y.; Sasaki, K.; Okazaki, S. Synthesis and characterization of a series of novel monoacylated ascorbic acid derivatives, 6-O-Acyl-2-O-α-D-glucopyranosyl-L-ascorbic acids, as skin antioxidants. J. Med. Chem. 2002, 45, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, C.X.; Yu, Y.; Wang, G.Q.; Zheng, Q.C.; Chen, G.D.; Lian, Y.Y.; Lin, F.; Guo, L.D.; Gao, H. Nodulisporipyrones A-D, new bioactive α-pyrone derivatives from Nodulisporium sp. J. Asian Nat. Prod. Res. 2015, 17, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, F.W.; Martin, D.; Weber, T.; Schiweck, H. Studies on Ketoses, 7–5-(α-D-Glucosyloxymethyl)furfural: Preparation from isomaltulose and exploration of its ensuing chemistry. Eur. J. Org. Chem. 1993, 9, 967–974. [Google Scholar] [CrossRef]
- Miyazawa, M.; Nankai, H.; Kameoka, H. Biotransformation of (–)-α-bisabolol by plant pathogenic fungus, Glomerella cingulata. Phytochemistry 1995, 40, 69–72. [Google Scholar] [CrossRef]
- Xu, J. The Preparation Method and Application of a Pyranoid Compound with Immunosuppressive Activity. CN 108913731 16 November 2021. [Google Scholar]
- Cook, L.; Ternai, B.; Ghosh, P. Inhibition of human sputum elastase by substituted 2-pyrones. J. Med. Chem. 1987, 30, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.Y.; Lu, L.W.; Bao, S.S.; Liu, C.X.; Deng, Z.S.; Cao, F.; Liu, S.P.; Zou, K.; Proksch, P. Xylariaopyrones A-D, four new antimicrobial α-pyrone derivatives from endophytic fungus Xylariales sp. Phytochem. Lett. 2018, 28, 98–103. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, Z.; Guo, Z.; Peng, Y.; Huang, N.; He, H.; Tu, X.; Zou, K. Effect of culture conditions on metabolite production of Xylaria sp. Molecules 2015, 20, 7940–7950. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.F.; Yu, R.J.; Li, X.X.; Gao, H.; Guo, L.D.; Tang, J.S.; Yao, X.S. 1H and 13C NMR spectral assignments of 2-pyrone derivatives from an endophytic fungus of sarcosomataceae. Magn. Reson. Chem. 2015, 53, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Castorena, A.L.; Arciniegas, A.; Guzmán, S.L.; Villaseñor, J.L.; Vivar, A.R. Eremophilanes from Senecio mairetianus and some reaction products. J. Nat. Prod. 2006, 69, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.; Sharma, N.; Singamaneni, V.; Sharma, V.; Kushwaha, M.; Abrol, V.; Guru, S.; Sharma, S.; Gupta, A.P.; Bhushan, S.; et al. Isolation and characterization of bioactive metabolites from Xylaria psidii, an endophytic fungus of the medicinal plant Aegle marmelos and their role in mitochondrial dependent apoptosis against pancreatic cancer cells. Phytomedicine 2016, 23, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Hallock, Y.F.; Clardy, J.; Kenfield, D.S.; Strobel, G. De-O-methyldiaporthin, a phytotoxin from Drechslera siccans. Phytochemistry 1988, 27, 3123–3125. [Google Scholar] [CrossRef]
- Feng, C.C.; Chen, G.D.; Zhao, Y.Q.; Xin, S.C.; Li, S.; Tang, J.S.; Li, X.X.; Hu, D.; Liu, X.Z.; Gao, H. New isocoumarins from a cold-adapted fungal strain Mucor sp. and their developmental toxicity to zebrafish embryos. Chem. Biodivers. 2014, 11, 1099–1108. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, X.; Feng, S.; Jiang, G.; Zhou, S.; Vrijmoed, L.L.P.; Jones, E.B.G. A novel N-cinnamoylcyclopeptide containing an allenic ether from the fungus Xylaria sp. (strain #2508) from the South China Sea. Tetrahedron Lett. 2001, 42, 449–451. [Google Scholar]
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Shi, Z.; Guo, Y.; Xie, S.; Qiao, Y.; Li, X.N.; Xue, Y.; Luo, Z.; Zhu, H.; Chen, C.; et al. The absolute configurations of hyperilongenols A–C: Rare 12,13-seco-spirocyclic polycyclic polyprenylated acylphloroglucinols with enolizable β, β′-tricarbonyl systems from Hypericum longistylum Oliv. Org. Chem. Front. 2019, 6, 1491–1502. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, S.; Hu, Z.; Wu, Z.; Guo, Y.; Zhang, J.; Wang, J.; Xue, Y.; Zhang, Y. Triterpenoids from whole plants of Phyllanthus urinaria. Chin. Herb. Med. 2017, 9, 193–196. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Zhao, D.G.; Zhou, A.Y.; Zhang, Y.; Du, Z.; Zhang, K. α-Glucosidase inhibition and antihyperglycemic activity of phenolics from the flowers of Edgeworthia gardneri. J. Agric. Food. Chem. 2015, 63, 8162–8169. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | δH, Mult (J Hz) | δC, Type |
|---|---|---|
| 1 | 167.0, C | |
| 2 | 5.56, d (2.1) | 88.6, CH |
| 3 | 173.8, C | |
| 4 | 6.22, d (2.1) | 99.9, CH |
| 5 | 168.7, C | |
| 6 | 4.35, dd (7.2, 4.8) | 70.7, CH |
| 7 | 1.83, m | 35.2, CH2 |
| 1.68, m, overlap | ||
| 8 | 1.76, m | 21.7, CH2 |
| 1.69, m, overlap | ||
| 9 | 2.38, t (6.7) | 34.4, CH2 |
| 10 | 175.6, C | |
| 11 | 3.66, s | 52.0, CH3 |
| 12 | 3.87, s | 57.0, CH3 |
| No. | δH, Mult (J Hz) a | δC, Type a | δH, Mult (J Hz) b | δC, Type b | δH, Mult (J Hz) c | δC, Type c |
|---|---|---|---|---|---|---|
| 1 | 166.7, C | 164.1, C | 162.7, C | |||
| 2 | 5.59, d (2.2) | 89.4, CH | 5.70, d (2.2) | 89.7, CH | 5.61, d (2.2) | 88.6, CH |
| 3 | 174.0, C | 172.3, C | 171.2, C | |||
| 4 | 6.95, d (2.2) | 100.2, CH | 7.64, d (2.2) | 99.7, CH | 6.98, d (2.2) | 98.3, CH |
| 5 | 157.9, C | 157.5 C | 155.8, C | |||
| 6 | 145.8, C | 146.1, CH | 144.3, C | |||
| 7 | 6.07, t (7.5) | 125.0, CH | 6.24, t (7.5) | 123.6, CH | 5.91, t (7.5) | 122.8, CH |
| 8 | 2.42, m | 27.1, CH2 | 2.61, q (7.5) | 26.9, CH2 | 2.33, dd (15.0, 7.6) | 25.3, CH2 |
| 9 | 1.48, m | 30.0, CH2 | 1.33, m | 29.7, CH2 | 1.39, m | 28.4, CH2 |
| 10 | 1.36, m, overlap | 32.8, CH2 | 1.19, m, overlap | 32.3, CH2 | 1.28, m, overlap | 31.1, CH2 |
| 11 | 1.37, m, overlap | 23.6, CH2 | 1.18, m, overlap | 23.2, CH2 | 1.29, m, overlap | 22.0, CH2 |
| 12 | 0.92, t (6.9) | 14.4, CH3 | 0.75, t (7.0) | 14.6, CH3 | 0.87, t (6.9) | 13.9, CH3 |
| 13 | 3.87, s | 57.0, CH3 | 3.60, s | 56.5, CH3 | 3.81, s | 56.4, CH3 |
| 1′ | 5.12, d (3.7) | 102.8, CH | 5.68, d (3.7) | 103.4, CH | 4.96, d (3.7) | 101.3, CH |
| 2′ | 3.53, dd (10.0, 3.7) | 73.4, CH | 4.27, dd (10.5, 4.1) | 74.0, CH | 3.32, m, overlap | 71.7, CH |
| 3′ | 3.78, t (9.3) | 74.4, CH | 4.71, t (9.3) | 75.2, CH | 3.70, m | 72.5, CH |
| 4′ | 3.45, t (9.3) | 71.0, CH | 4.35, t (10.0) | 71.9, CH | 3.22, m | 69.4, CH |
| 5′ | 3.90, m | 75.5, CH | 4.66, dt (10.0, 3.5) | 76.8, CH | 3.54, m, overlap | 74.7, CH |
| 6′ | 3.76, m | 62.1, CH2 | 4.48, br s | 62.9, CH2 | 3.55, m, overlap | 60.3, CH2 |
| 2′-OH | 5.52, d (4.9) | |||||
| 3′-OH | 5.10, m | |||||
| 4′-OH | 5.10, m | |||||
| 6′-OH | 4.53, t (5.8) |
| No. | δH, Mult (J Hz) a | δC, Type a | δH, Mult (J Hz) b | δC, Type b | δH, Mult (J Hz) c | δC, Type c |
|---|---|---|---|---|---|---|
| 1 | 167.1, C | 164.5, C | 164.2, C | |||
| 2 | 5.57, d (2.2) | 89.1, CH | 5.68, d (2.3) | 89.1, CH | 5.57, d (2.2) | 88.0, CH |
| 3 | 173.6, C | 171.9, C | 170.9, C | |||
| 4 | 6.55, d (2.2) | 101.9, CH | 6.99, d (2.3) | 100.5, CH | 6.50, d (2.2) | 99.4, CH |
| 5 | 165.5, C | 165.8, C | 163.3, C | |||
| 6 | 4.52, t (6.2) | 75.3, CH | 4.82, dd (7.8, 4.4) | 74.8, CH | 4.38, dd (6.8, 4.8) | 72.9, CH |
| 7 | 1.85, dd (14.0, 7.6) | 35.1, CH2 | 1.89, m | 35.2, CH2 | 1.70, m | 33.5, CH2 |
| 1.84, m | ||||||
| 8 | 1.43, m | 26.2, CH2 | 1.51, m | 26.2, CH2 | 1.32, m | 24.4, CH2 |
| 9 | 1.35, m | 30.1, CH2 | 1.18, m, overlap | 29.7, CH2 | 1.27, m, overlap | 28.4, CH2 |
| 1.33, m | 1.10, m, overlap | |||||
| 10 | 1.31, m, overlap | 32.8, CH2 | 1.08, m, overlap | 32.2, CH2 | 1.23, m, overlap | 31.1, CH2 |
| 11 | 1.32, m, overlap | 23.7, CH2 | 1.16, m, overlap | 23.3, CH2 | 1.25, m, overlap | 22.0, CH2 |
| 12 | 0.90, t (6.8) | 14.4, CH3 | 0.78, t (7.3) | 14.7, CH3 | 0.85, t (6.8) | 14.0, CH3 |
| 13 | 3.87, s | 57.0, CH3 | 3.63, s | 56.4, CH3 | 3.81, s | 56.4, CH3 |
| 1′ | 4.80, d (3.8) | 98.5, CH | 5.42, d (3.8) | 99.3, CH | 4.66, d (3.8) | 97.2, CH |
| 2′ | 3.41, dd (9.8, 3.8) | 73.2, CH | 4.22, dd (9.6, 3.8) | 74.0, CH | 3.22, m | 71.5, CH |
| 3′ | 3.68, m, overlap | 74.8, CH | 4.67, t (9.6) | 75.6, CH | 3.45, m, overlap | 73.7, CH |
| 4′ | 3.29, m | 71.7, CH | 4.24, t (9.6) | 72.6, CH | 3.07, m | 70.1, CH |
| 5′ | 3.68, m, overlap | 74.6, CH | 4.44, t (9.6) | 75.8, CH | 3.45, m, overlap | 73.0, CH |
| 6′ | 3.68, m, overlap | 62.7, CH2 | 4.57, dd (9.6, 5.6) | 63.3, CH2 | 3.61, m | 60.9, CH2 |
| 3.83, m | 4.43, m | 3.45, m, overlap | ||||
| 2′-OH | 5.04, d (5.9) | |||||
| 3′-OH | 4.92, d (4.1) | |||||
| 4′-OH | 4.99, d (5.2) | |||||
| 6′-OH | 4.52, t (5.4) |
| No. | δH, Mult (J Hz) a | δC, Type a | δH, Mult (J Hz) b | δC, Type b |
|---|---|---|---|---|
| 1 | 1.72, m | 39.1, CH | 1.59, m, overlap | 38.8, CH |
| 2 | 1.80, m | 29.3, CH2 | 1.59, m, overlap | 29.2, CH2 |
| 1.53 m | ||||
| 3 | 3.66, br s | 74.0, CH | 3.36, m, overlap | 72.6, CH |
| 4 | 70.9, C | 69.5, C | ||
| 5 | 1.74, m | 33.6, CH2 | 1.49, m | 33.5, CH2 |
| 1.55, m | 1.29, m, overlap | |||
| 6 | 1.49, m | 22.1, CH2 | 1.29, m, overlap | 21.7, CH2 |
| 1.40, m | ||||
| 7 | 74.0, C | 72.0, C | ||
| 8 | 1.60, m | 38.8, CH2 | 1.41, m | 38.1, CH2 |
| 9 | 2.26, m | 22.9, CH2 | 2.18, q (8.0) | 22.7, CH2 |
| 10 | 6.77, td (7.5, 1.2) | 142.6, CH | 6.71, td (7.5, 0.9) | 143.5, CH |
| 11 | 127.6, C | 126.4, C | ||
| 12 | 168.8, C | 0.90, t (6.8) | 167.7, C | |
| 13 | 1.84, s | 12.4, CH3 | 1.77, s | 12.2, CH3 |
| 14 | 1.14, s | 23.7, CH3 | 0.96, s | 23.8, CH3 |
| 15 | 1.26, s | 27.6, CH3 | 1.04, s | 27.9, CH3 |
| 16 | 3.73, s | 51.8, CH3 | 3.64, s | 51.6, CH3 |
| 3-OH | 4.36, d (4.0) | |||
| 4-OH | 3.96, s | |||
| 7-OH | 3.88, s |
| Compounds | Concentration | NO Production Inhibition (%) a |
|---|---|---|
| 2 | 50 μM | 4.51 ± 0.35 |
| 3 | 50 μM | –3.93 ± 2.43 |
| 4 | 50 μM | 1.85 ± 3.18 |
| 5 | 50 μM | –1.61 ± 0.53 |
| 12 | 50 μM | 0.92 ± 2.97 |
| 13 | 50 μM | 4.67 ± 2.36 |
| 14 | 50 μM | 3.54 ± 1.26 |
| 18 | 50 μM | –0.92 ± 2.21 |
| Crude extract | 50 μg/mL | 77.28 ± 0.82 |
| 6.25 μg/mL | 7.78 ± 3.29 | |
| L-NMMA b | 50 μM | 53.75 ± 1.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Jin, Y.; Yan, W.; Gu, P.; Zeng, Z.; Li, Z.; Zhang, G.; Wei, M.; Xue, Y. New Pyranone Derivatives and Sesquiterpenoid Isolated from the Endophytic Fungus Xylaria sp. Z184. Molecules 2024, 29, 1728. https://doi.org/10.3390/molecules29081728
Zhang Y, Jin Y, Yan W, Gu P, Zeng Z, Li Z, Zhang G, Wei M, Xue Y. New Pyranone Derivatives and Sesquiterpenoid Isolated from the Endophytic Fungus Xylaria sp. Z184. Molecules. 2024; 29(8):1728. https://doi.org/10.3390/molecules29081728
Chicago/Turabian StyleZhang, Yan, Yang Jin, Wensi Yan, Peishan Gu, Ziqian Zeng, Ziying Li, Guangtao Zhang, Mi Wei, and Yongbo Xue. 2024. "New Pyranone Derivatives and Sesquiterpenoid Isolated from the Endophytic Fungus Xylaria sp. Z184" Molecules 29, no. 8: 1728. https://doi.org/10.3390/molecules29081728