
New Transferrin Receptor-Targeted Peptide–Doxorubicin Conjugates: Synthesis and In Vitro Antitumor Activity


Abstract
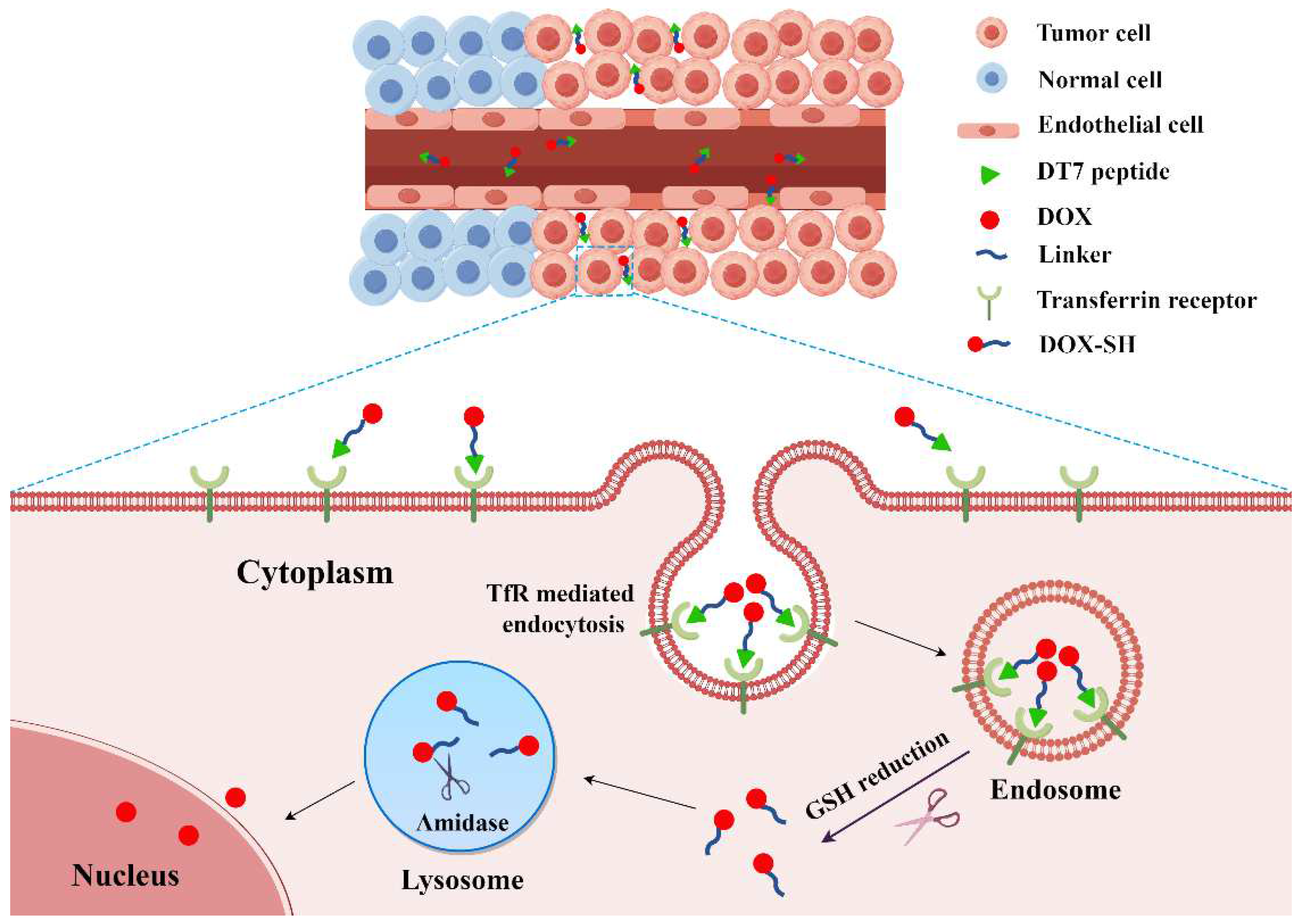
1. Introduction
2. Results and Discussion
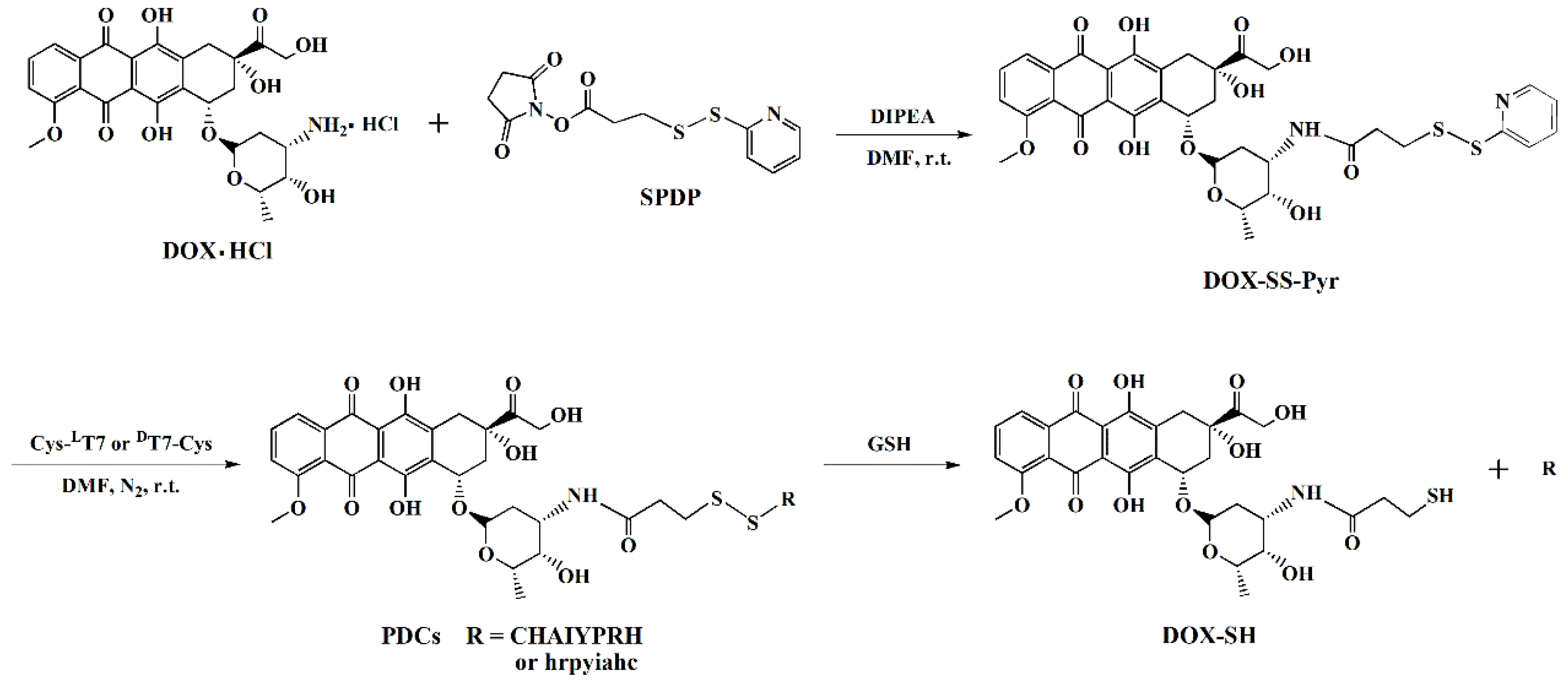
2.1. Synthesis of PDCs
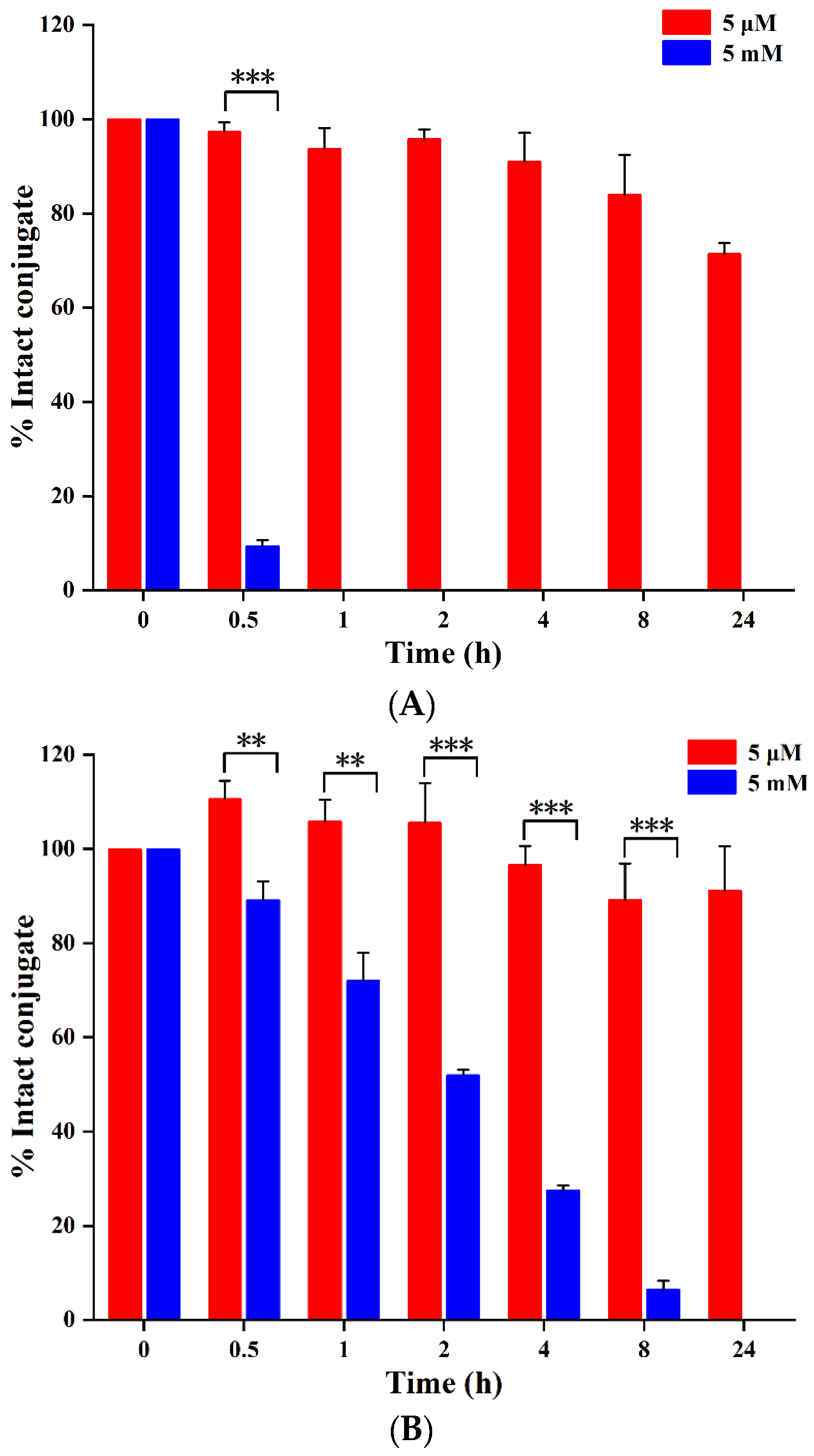
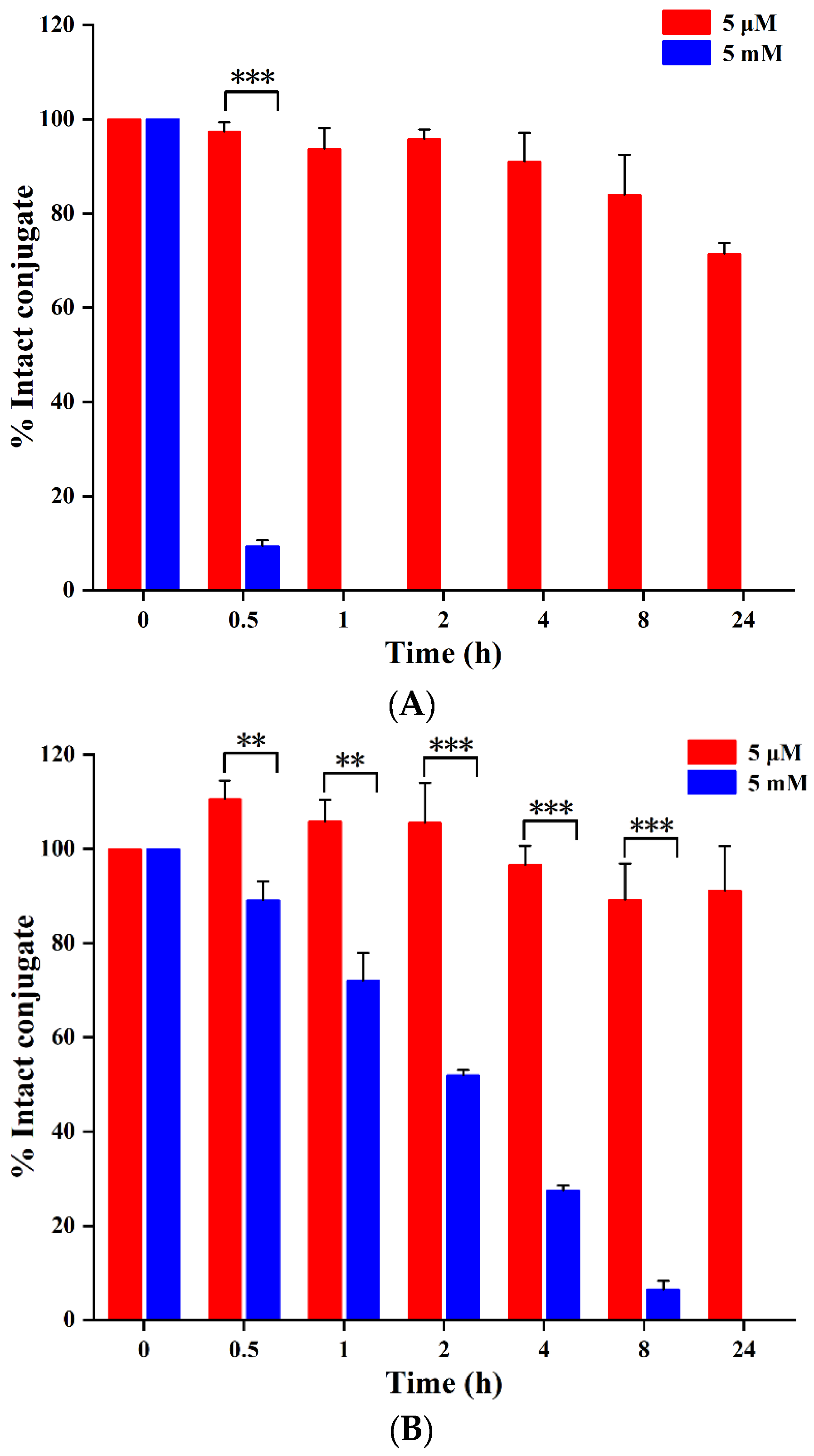
2.2. Serum Stability and Drug Release of PDCs
2.2.1. Serum Stability
2.2.2. Reduction-Triggered Drug Release
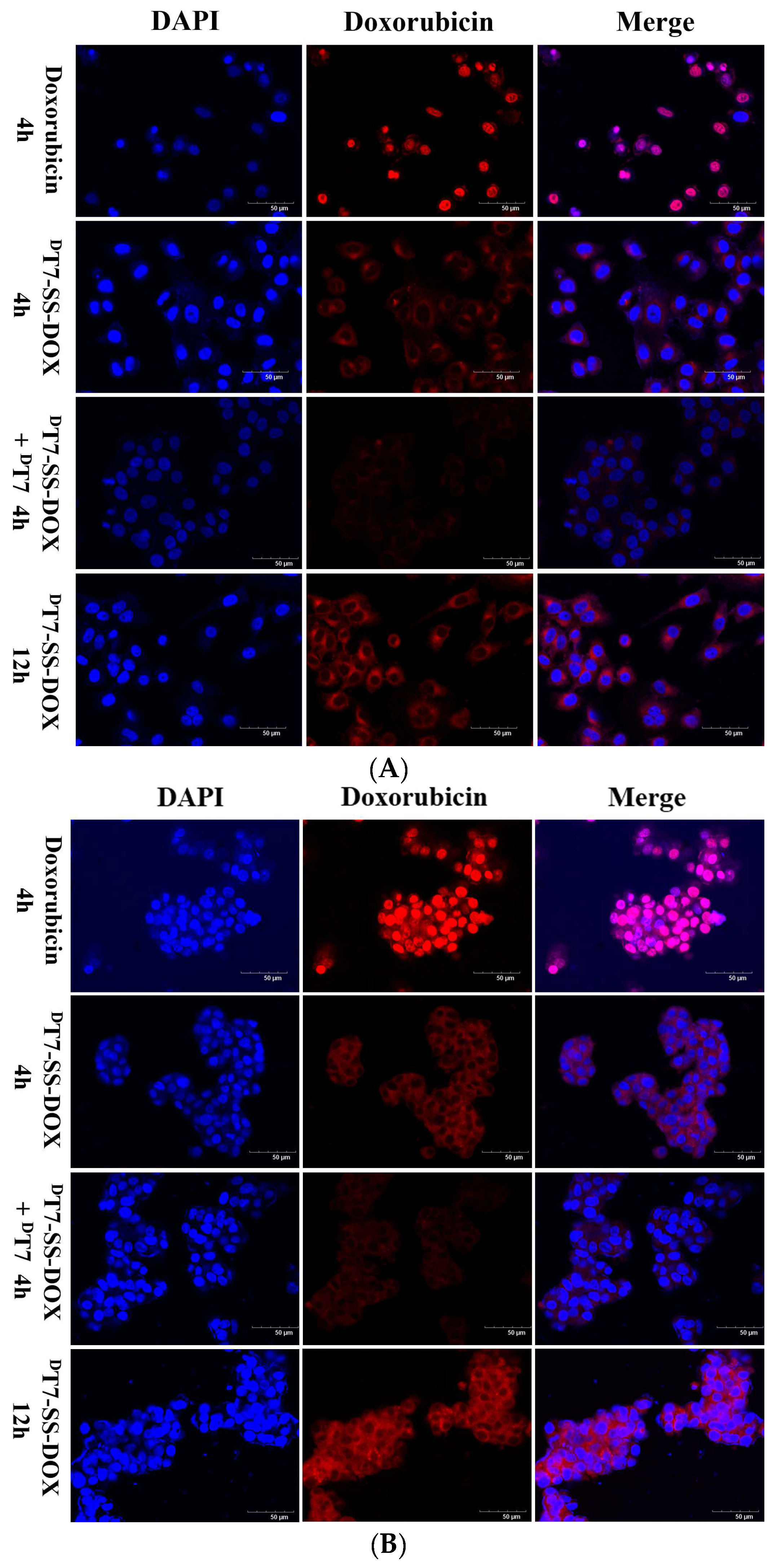
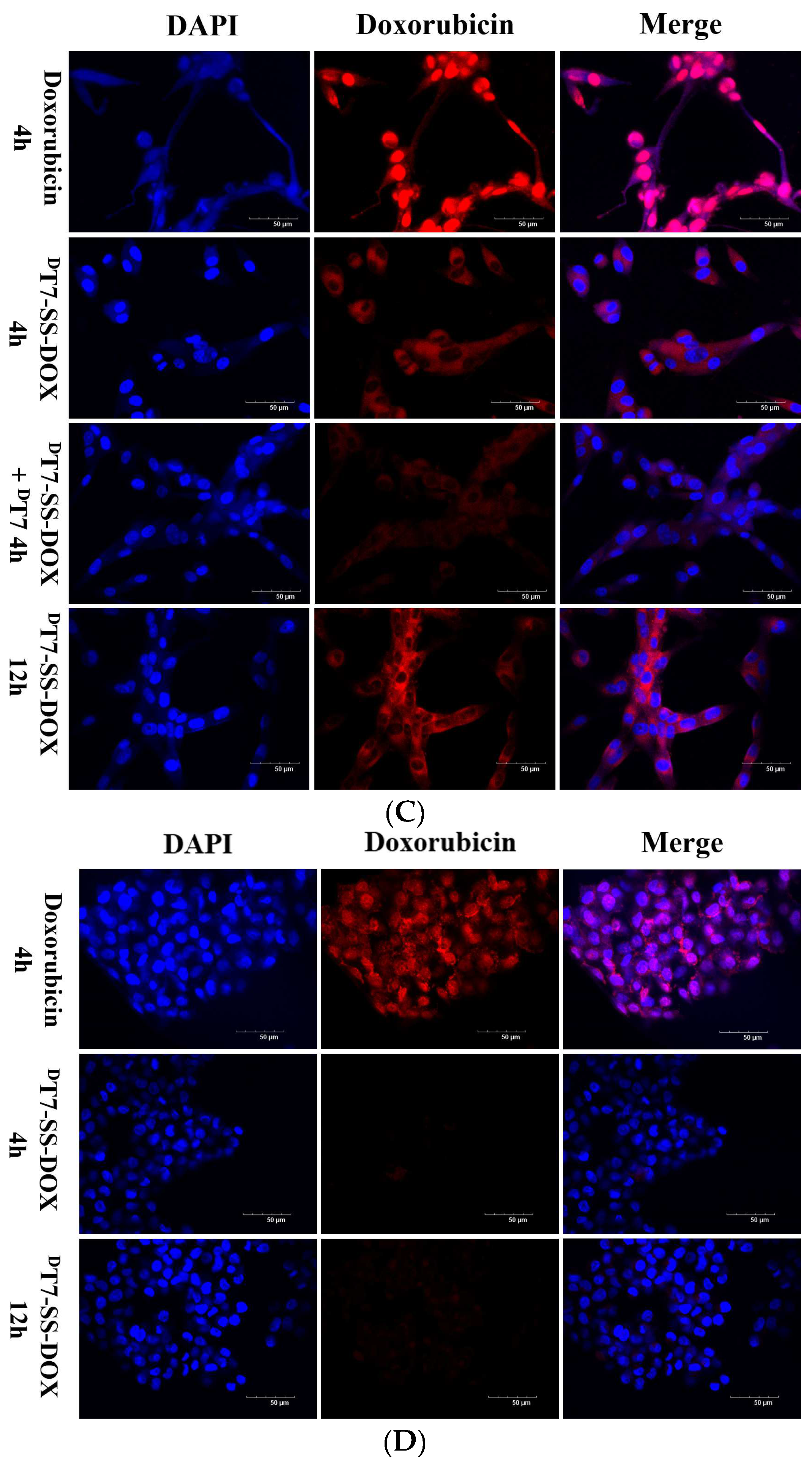
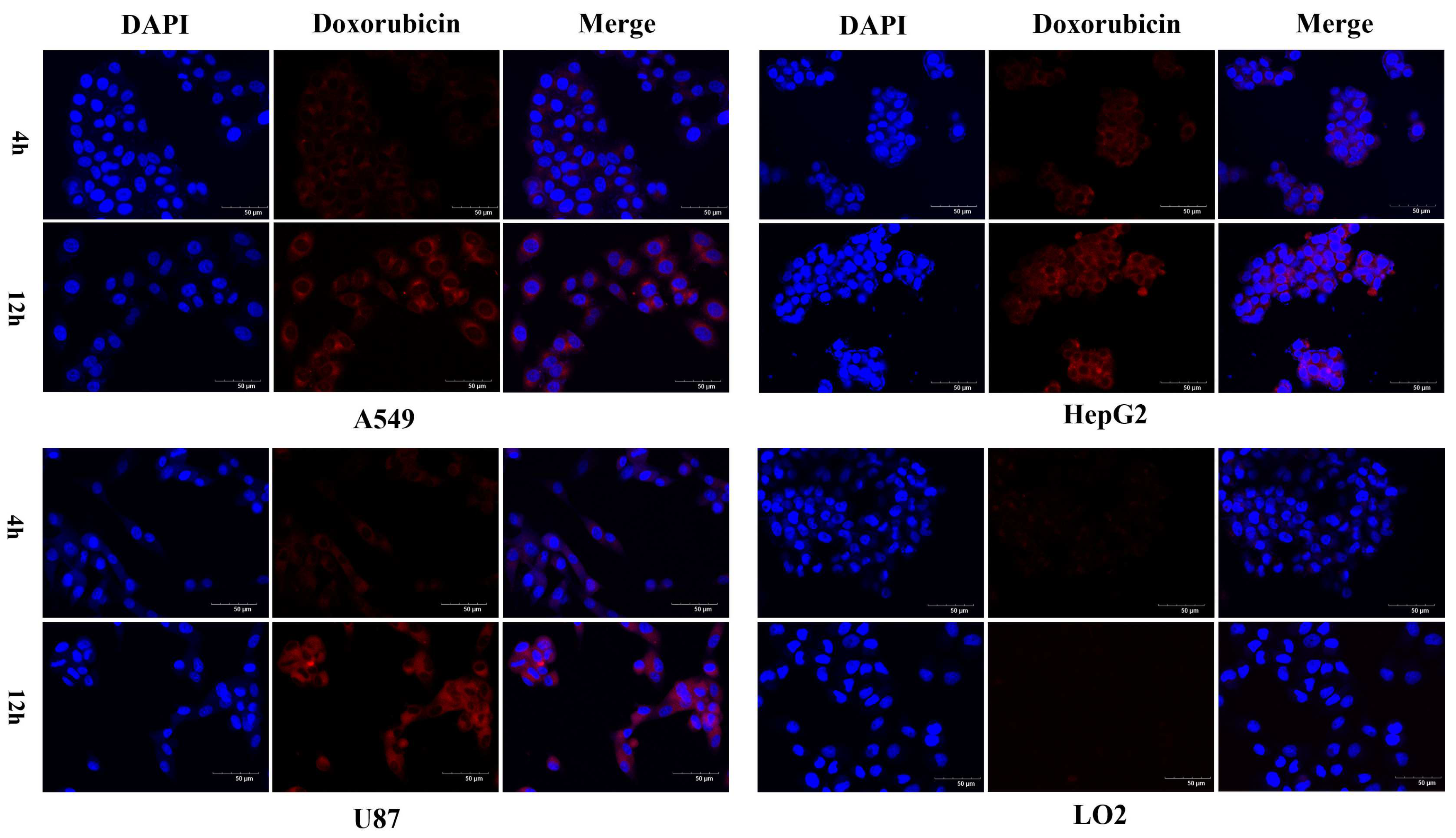
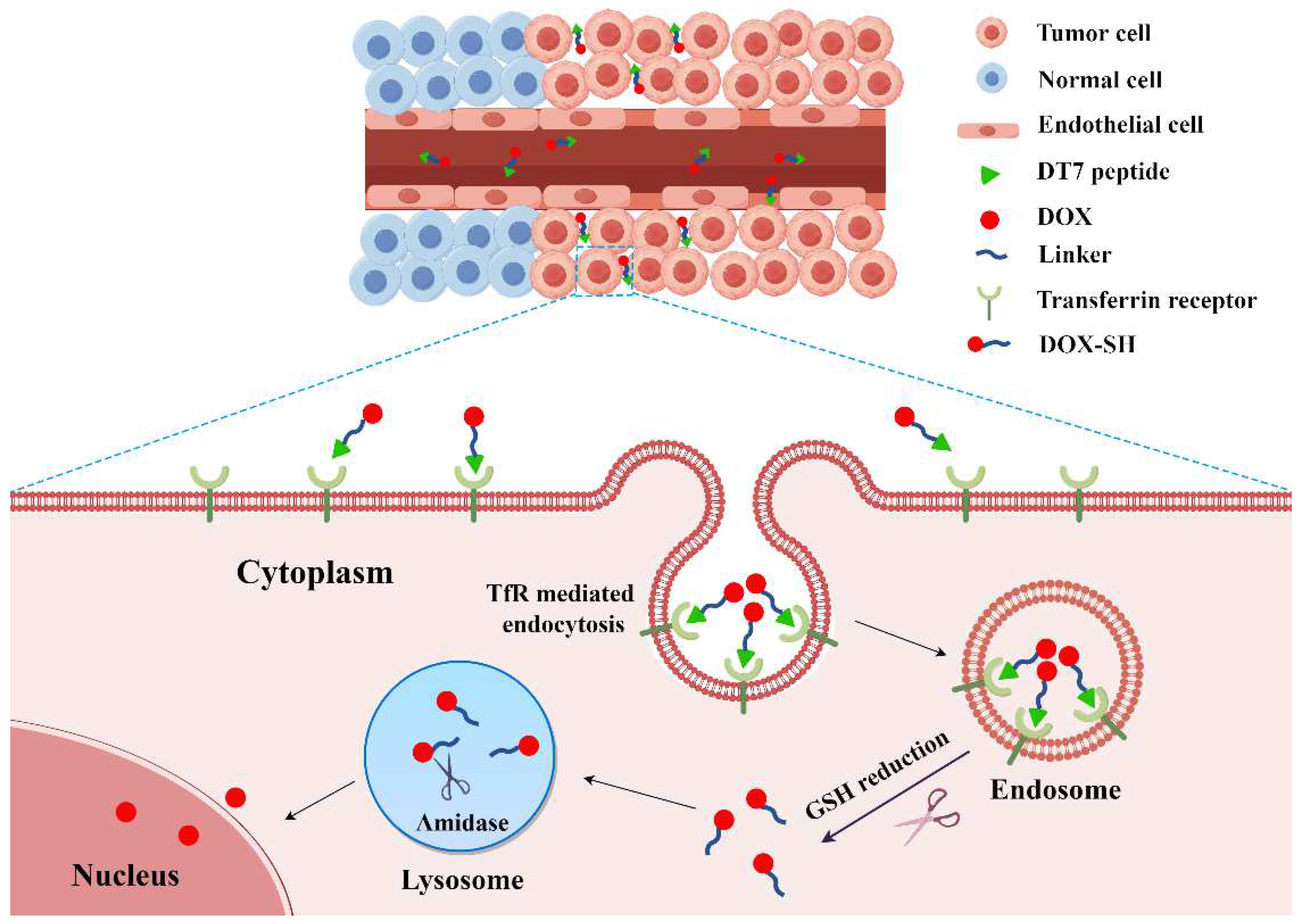
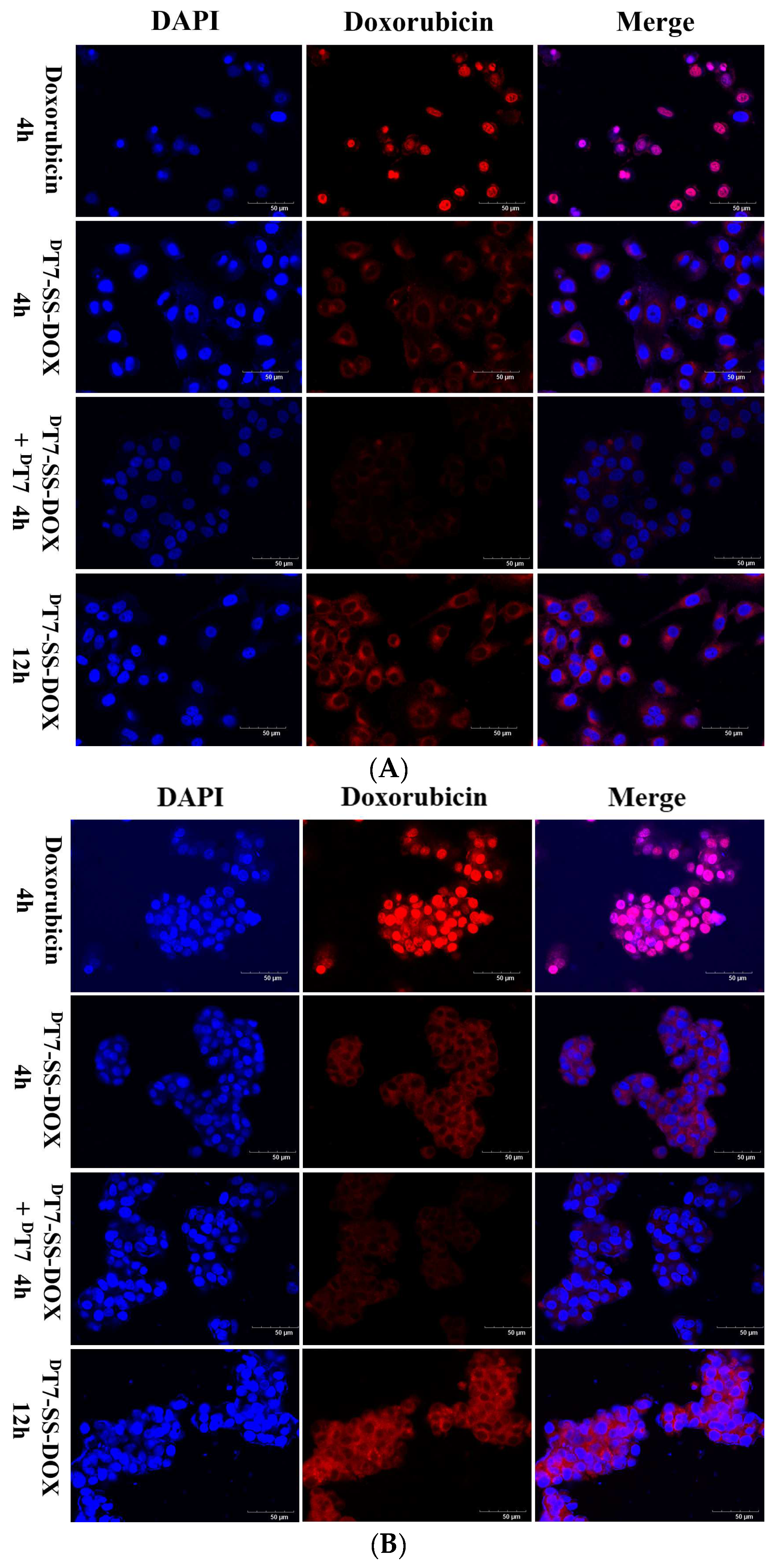
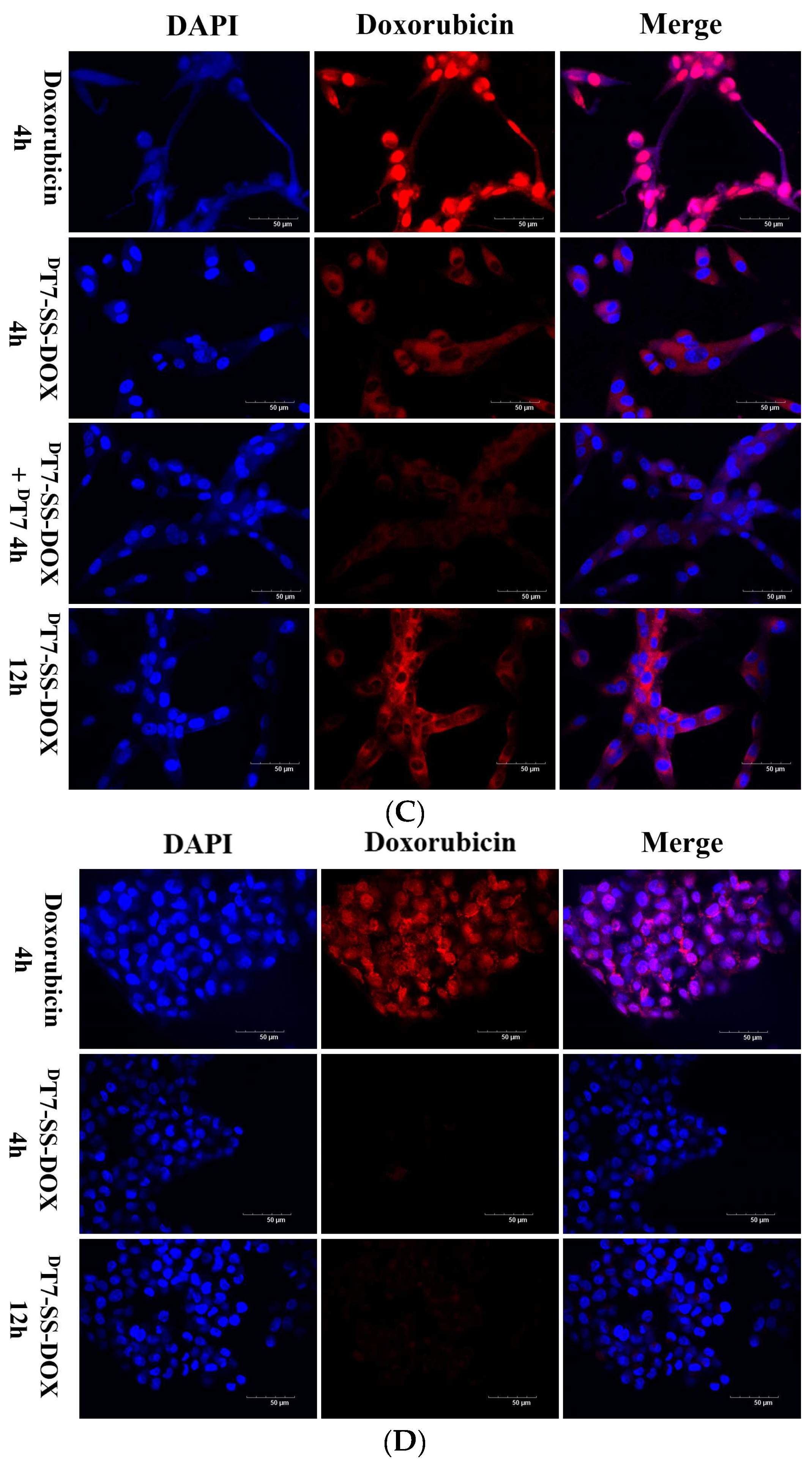
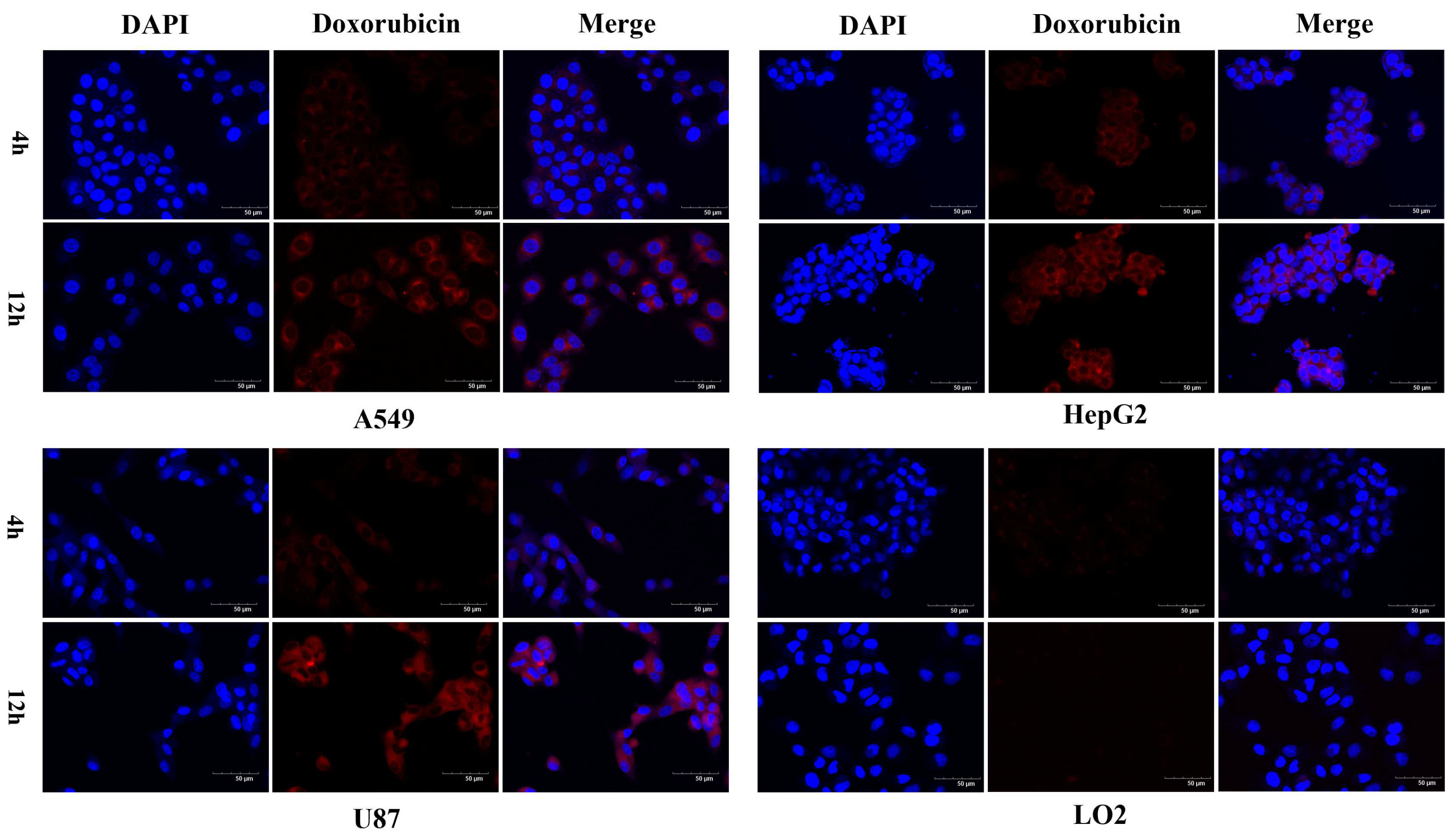
2.3. Confocal Microscopy Analysis of Cellular Uptake
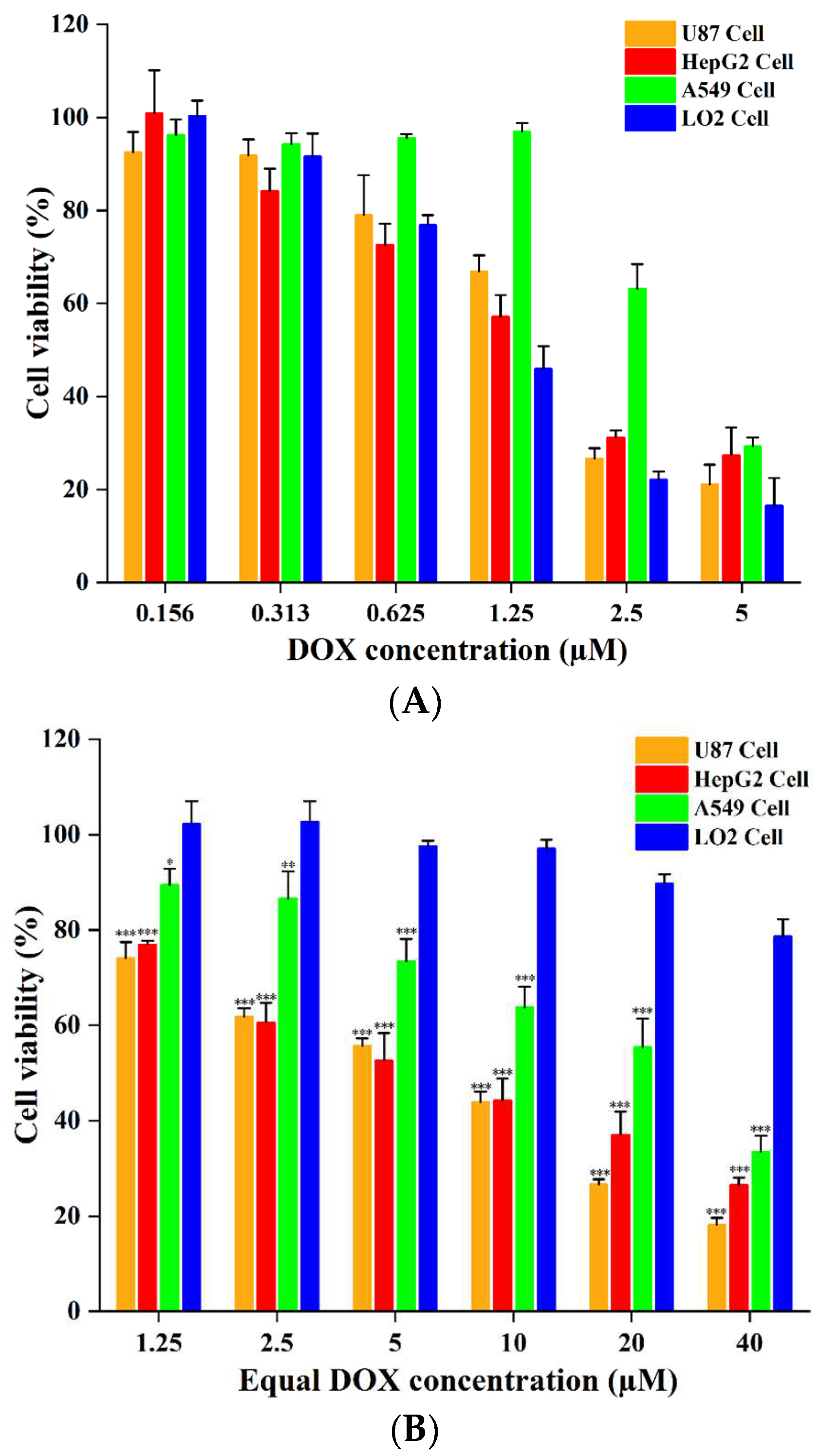
2.4. In Vitro Cytotoxicity
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Peptide–DOX Conjugates
3.2.1. Synthesis of DOX-SS-Pyr
3.2.2. Synthesis of Peptide–DOX Conjugates
3.3. In Vitro Stability of Conjugates
3.4. In Vitro Drug Release of Conjugates
3.5. Confocal Microscopy Studies
3.6. In Vitro Cytotoxicity Assay
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, R.H.; Gao, W.; Zhang, L. Targeting drugs to tumours using cell membrane-coated nanoparticles. Nat. Rev. Clin. Oncol. 2023, 20, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tang, K.; Lv, J. Peptide-drug conjugate-based novel molecular drug delivery system in cancer. Trends Pharmacol. Sci. 2021, 42, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, J.; Xia, M.; Yin, L.; Zhang, L.; Liu, X.; Cheng, Y. Peptide-drug conjugates: A new paradigm for targeted cancer therapy. Eur. J. Med. Chem. 2024, 265, 116119. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Huang, W.; Yang, N.; Liu, Y. Learn from antibody-drug conjugates, consideration in the future construction of peptide-drug conjugates for cancer therapy. Exp. Hematol. Oncol. 2022, 11, 93. [Google Scholar] [CrossRef]
- Mojarad-Jabali, S.; Mahdinloo, S.; Farshbaf, M.; Sarfraz, M.; Fatahi, Y.; Atyabi, F.; Valizadeh, H. Transferrin receptor-mediated liposomal drug delivery: Recent trends in targeted therapy of cancer. Expert Opin. Drug Deliv. 2022, 19, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Zeng, C.; Tian, L.; Wang, T.; Yang, S.; Cheng, Q.; Zhang, J.; Meng, Q.; Zhang, C.; Meng, Z. Transferrin receptor targeting segment T7 containing peptide gene delivery vectors for efficient transfection of brain tumor cells. Drug Deliv. 2022, 29, 2375–2385. [Google Scholar] [CrossRef]
- Falvo, E.; Damiani, V.; Conti, G.; Boschi, F.; Messana, K.; Giacomini, P.; Milella, M.; De Laurenzi, V.; Morea, V.; Sala, G.; et al. High activity and low toxicity of a novel CD71-targeting nanotherapeutic named The-0504 on preclinical models of several human aggressive tumors. J. Exp. Clin. Cancer Res. 2021, 40, 63. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, Q.; Luan, W.; Li, T.; Xiao, Y.; Zhang, L.; Li, Y.; Rong, R.; Ren, C. LWJ-M30, a conjugate of DM1 and B6, for the targeted therapy of colorectal cancer with improved therapeutic effects. RSC Adv. 2023, 13, 10840–10846. [Google Scholar] [CrossRef]
- Mojarad-Jabali, S.; Farshbaf, M.; Hemmati, S.; Sarfraz, M.; Motasadizadeh, H.; Mojarrad, J.S.; Atyabi, F.; Zakeri-Milani, P.; Valizadeh, H. Comparison of three synthetic transferrin mimetic small peptides to promote the blood-brain barrier penetration of vincristine liposomes for improved glioma targeted therapy. Int. J. Pharm. 2022, 613, 121395. [Google Scholar] [CrossRef]
- Sun, P.; Xiao, Y.; Di, Q.; Ma, W.; Ma, X.; Wang, Q.; Chen, W. Transferrin Receptor-Targeted PEG-PLA Polymeric Micelles for Chemotherapy Against Glioblastoma Multiforme. Int. J. Nanomed. 2020, 15, 6673–6688. [Google Scholar] [CrossRef] [PubMed]
- Elfadadny, A.; Ragab, R.F.; Hamada, R.; Al Jaouni, S.K.; Fu, J.; Mousa, S.A.; El-Far, A.H. Natural bioactive compounds-doxorubicin combinations targeting topoisomerase II-alpha: Anticancer efficacy and safety. Toxicol. Appl. Pharmacol. 2023, 461, 116405. [Google Scholar] [CrossRef] [PubMed]
- Paskeh, M.D.A.; Saebfar, H.; Mahabady, M.K.; Orouei, S.; Hushmandi, K.; Entezari, M.; Hashemi, M.; Aref, A.R.; Hamblin, M.R.; Ang, H.L.; et al. Overcoming doxorubicin resistance in cancer: siRNA-loaded nanoarchitectures for cancer gene therapy. Life Sci. 2022, 298, 120463. [Google Scholar] [CrossRef] [PubMed]
- Radu, E.R.; Semenescu, A.; Voicu, S.I. Recent Advances in Stimuli-Responsive Doxorubicin Delivery Systems for Liver Cancer Therapy. Polymers 2022, 14, 5249. [Google Scholar] [CrossRef] [PubMed]
- Almajidi, Y.Q.; Kadhim, M.M.; Alsaikhan, F.; Jalil, A.T.; Sayyid, N.H.; Ramírez-Coronel, A.A.; Jawhar, Z.H.; Gupta, J.; Nabavi, N.; Yu, W.; et al. Doxorubicin-loaded micelles in tumor cell-specific chemotherapy. Environ. Res. 2023, 227, 115722. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Luo, Y.; Gan, D.; Zhang, Y.; Deng, H.; Liu, G. Advances in Doxorubicin-based nano-drug delivery system in triple negative breast cancer. Front. Bioeng. Biotechnol. 2023, 11, 1271420. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Engler, J.A.; Collawn, J.F.; Moore, B.A. Receptor mediated uptake of peptides that bind the human transferrin receptor. Eur. J. Biochem. 2001, 268, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Lin, W. Peptide-Functionalized Nanoparticles-Encapsulated Cyclin-Dependent Kinases Inhibitor Seliciclib in Transferrin Receptor Overexpressed Cancer Cells. Nanomaterials 2021, 11, 772. [Google Scholar] [CrossRef]
- Piantino, M.; Louis, F.; Shigemoto-Mogami, Y.; Kitamura, K.; Sato, K.; Yamaguchi, T.; Kawabata, K.; Yamamoto, S.; Iwasaki, S.; Hirabayashi, H.; et al. Brain microvascular endothelial cells derived from human induced pluripotent stem cells as in vitro model for assessing blood-brain barrier transferrin receptor-mediated transcytosis. Mater. Today Bio 2022, 14, 100232. [Google Scholar] [CrossRef]
- Riaz, M.K.; Zhang, X.; Wong, K.; Chen, H.; Liu, Q.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Pulmonary delivery of transferrin receptors targeting peptide surface-functionalized liposomes augments the chemotherapeutic effect of quercetin in lung cancer therapy. Int. J. Nanomed. 2019, 14, 2879–2902. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhai, M.; Chen, Z.; Han, X.; Yu, F.; Li, Z.; Xie, X.; Han, C.; Yu, L.; Yang, Y.; et al. Dual-modified liposome codelivery of doxorubicin and vincristine improve targeting and therapeutic efficacy of glioma. Drug Deliv. 2017, 24, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, Q.; Yu, Q.; Qiu, Y.; Mei, L.; Wan, D.; Wang, X.; Li, M.; He, Q. A stabilized retro-inverso peptide ligand of transferrin receptor for enhanced liposome-based hepatocellular carcinoma-targeted drug delivery. Acta Biomater. 2019, 83, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Su, D.; Yang, Y.; Qin, L.; Hu, C.; Liu, R.; Zhou, Y.; Yang, C.; Yang, X.; Wang, G.; et al. D-T7 Peptide-Modified PEGylated Bilirubin Nanoparticles Loaded with Cediranib and Paclitaxel for Antiangiogenesis and Chemotherapy of Glioma. ACS Appl. Mater. Interfaces 2019, 11, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Zhu, L.; Wang, L.; Liu, X.; Xiao, F.; Xie, Y.; Zheng, W.; Jiang, X. Screening on-chip fabricated nanoparticles for penetrating the blood-brain barrier. Nanoscale 2022, 14, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Guan, L.; Li, W.; Jia, R.; Jia, L.; Zhang, Y.; Wen, X.; Meng, S.; Ma, D.; Zhang, N.; et al. DT7 peptide-modified lecithin nanoparticles co-loaded with γ-secretase inhibitor and dexamethasone efficiently inhibit T-cell acute lymphoblastic leukemia and reduce gastrointestinal toxicity. Cancer Lett. 2022, 533, 215608. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Chuan, X.; Chen, B.; He, B.; Zhang, H.; Dai, W.; Wang, X.; Zhang, Q. A smart tumor targeting peptide-drug conjugate, pHLIP-SS-DOX, synthesis and cellular uptake on MCF-7 and MCF-7/Adr cells. Drug Deliv. 2016, 23, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, Y.; Zhuang, X.; Li, D.; Cheng, B.; Tan, S.; Zhang, Z. D-α-tocopherol polyethylene glycol succinate-based redox-sensitive paclitaxel prodrug for overcoming multidrug resistance in cancer cells. Mol. Pharm. 2014, 11, 3196–3209. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Guo, X.; Yang, T.; Yu, M.; Chen, D.; Wang, J. Brain tumor-targeted therapy by systemic delivery of siRNA with Transferrin receptor-mediated core-shell nanoparticles. Int. J. Pharm. 2016, 510, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Xiong, Y.; Su, Z.; Zhang, Q.; Dai, X.; Li, L.; Xiao, X.; Huang, Y. Expression of curcin-transferrin receptor binding peptide fusion protein and its anti-tumor activity. Protein Expr. Purif. 2013, 89, 181–188. [Google Scholar] [CrossRef]
- Liang, Y.; Li, S.; Wang, X.; Zhang, Y.; Sun, Y.; Wang, Y.; Wang, X.; He, B.; Dai, W.; Zhang, H.; et al. A comparative study of the antitumor efficacy of peptide-doxorubicin conjugates with different linkers. J. Control Release 2018, 275, 129–141. [Google Scholar] [CrossRef]
- Yu, J.; Li, S.; Zhao, G. Preparation of transferrin receptor binding peptide derivative. Chem. Bioeng. 2024, 41, 33–35. [Google Scholar]
- Feng, C.; Rui, M.; Shen, H.; Xin, Y.; Zhang, J.; Li, J.; Yue, L.; Lai, W.; Xu, X. Tumor-specific delivery of doxorubicin through conjugation of pH-responsive peptide for overcoming drug resistance in cancer. Int. J. Pharm. 2017, 528, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; El-Sayed, N.S.; Shamloo, K.; Lohan, S.; Kumar, S.; Sajid, M.I.; Tiwari, R.K. Targeted Delivery of Cabazitaxel Using Cyclic Cell-Penetrating Peptide and Biomarkers of Extracellular Matrix for Prostate and Breast Cancer Therapy. Bioconjug Chem. 2021, 32, 1898–1914. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | |||
|---|---|---|---|---|
| U87 | HepG2 | A549 | LO2 | |
| DT7-SS-DOX | 5.70 ± 0.22 | 7.01 ± 1.64 | 20.61 ± 4.81 | >100 |
| DOX | 1.65 ± 0.20 | 1.58 ± 0.19 | 3.55 ± 0.21 | 1.29 ± 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Mao, X.; Yang, X.; Zhao, G.; Li, S. New Transferrin Receptor-Targeted Peptide–Doxorubicin Conjugates: Synthesis and In Vitro Antitumor Activity. Molecules 2024, 29, 1758. https://doi.org/10.3390/molecules29081758
Yu J, Mao X, Yang X, Zhao G, Li S. New Transferrin Receptor-Targeted Peptide–Doxorubicin Conjugates: Synthesis and In Vitro Antitumor Activity. Molecules. 2024; 29(8):1758. https://doi.org/10.3390/molecules29081758
Chicago/Turabian StyleYu, Jiale, Xiaoxia Mao, Xue Yang, Guiqin Zhao, and Songtao Li. 2024. "New Transferrin Receptor-Targeted Peptide–Doxorubicin Conjugates: Synthesis and In Vitro Antitumor Activity" Molecules 29, no. 8: 1758. https://doi.org/10.3390/molecules29081758
APA StyleYu, J., Mao, X., Yang, X., Zhao, G., & Li, S. (2024). New Transferrin Receptor-Targeted Peptide–Doxorubicin Conjugates: Synthesis and In Vitro Antitumor Activity. Molecules, 29(8), 1758. https://doi.org/10.3390/molecules29081758