
Substitution Effects in Aryl Halides and Amides into the Reaction Mechanism of Ullmann-Type Coupling Reactions

 , ,
, ,  , , and
, , and
Abstract
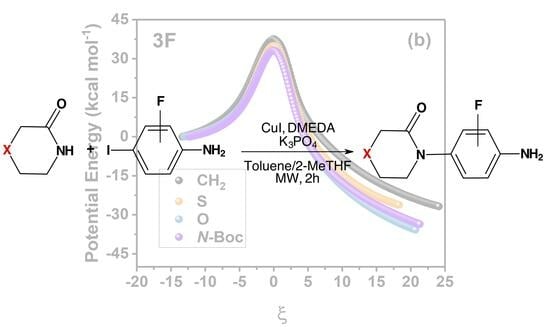
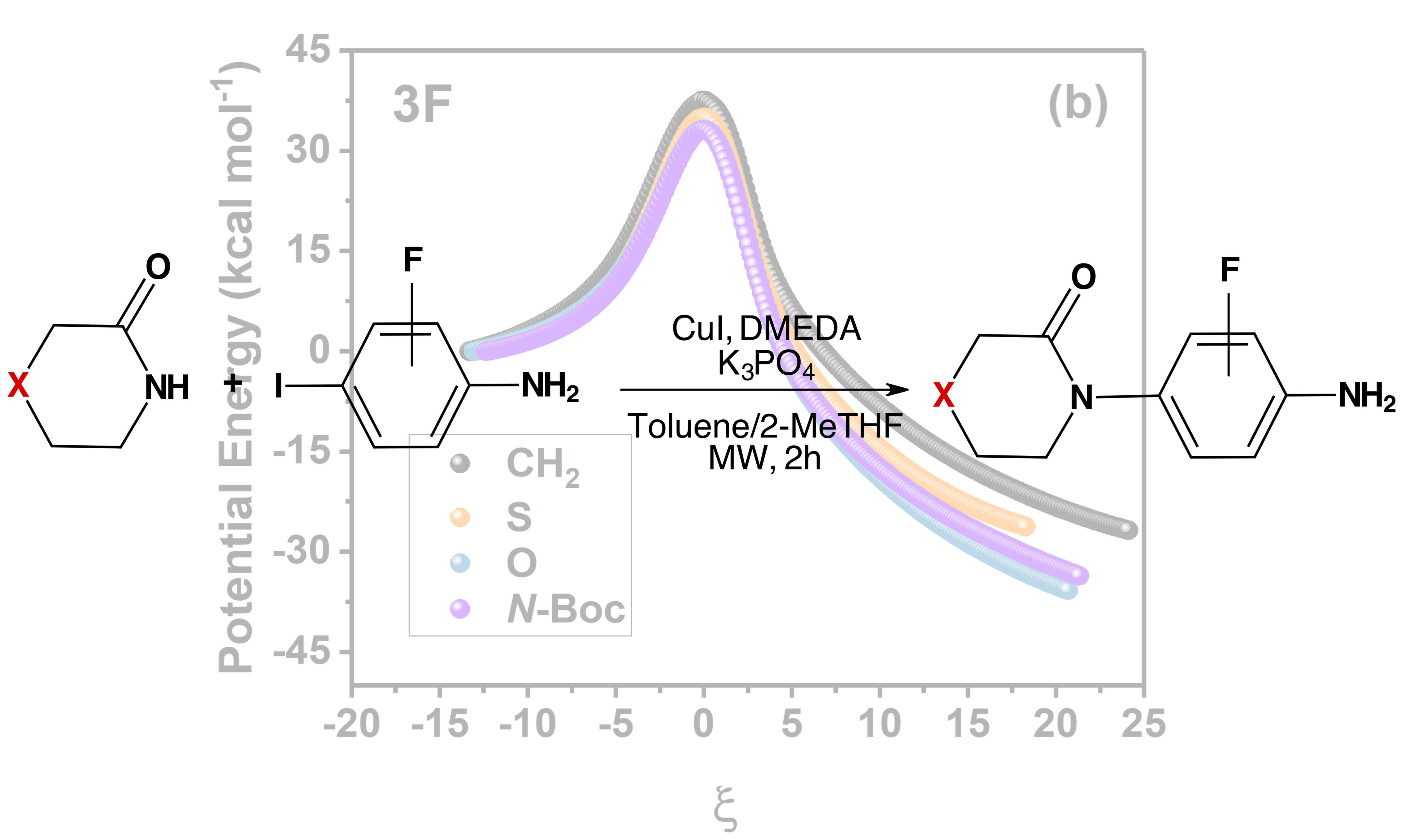
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
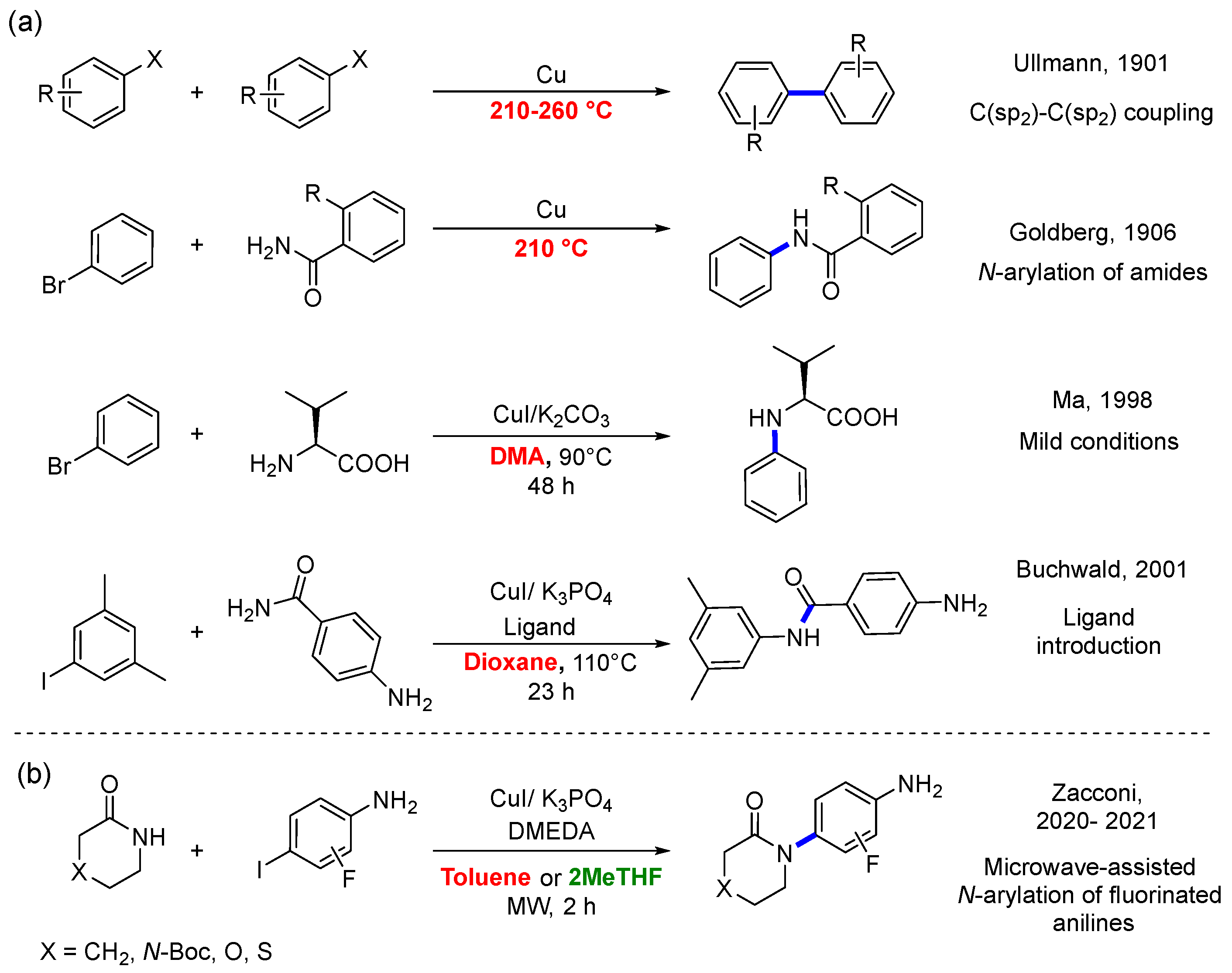
1. Introduction
1.1. Theoretical Background
1.1.1. Conceptual Density Functional Theory (cDFT)
1.1.2. Activation Strain Model (ASM)
1.1.3. Non-Covalent Interactions Index (NCI)
2. Results
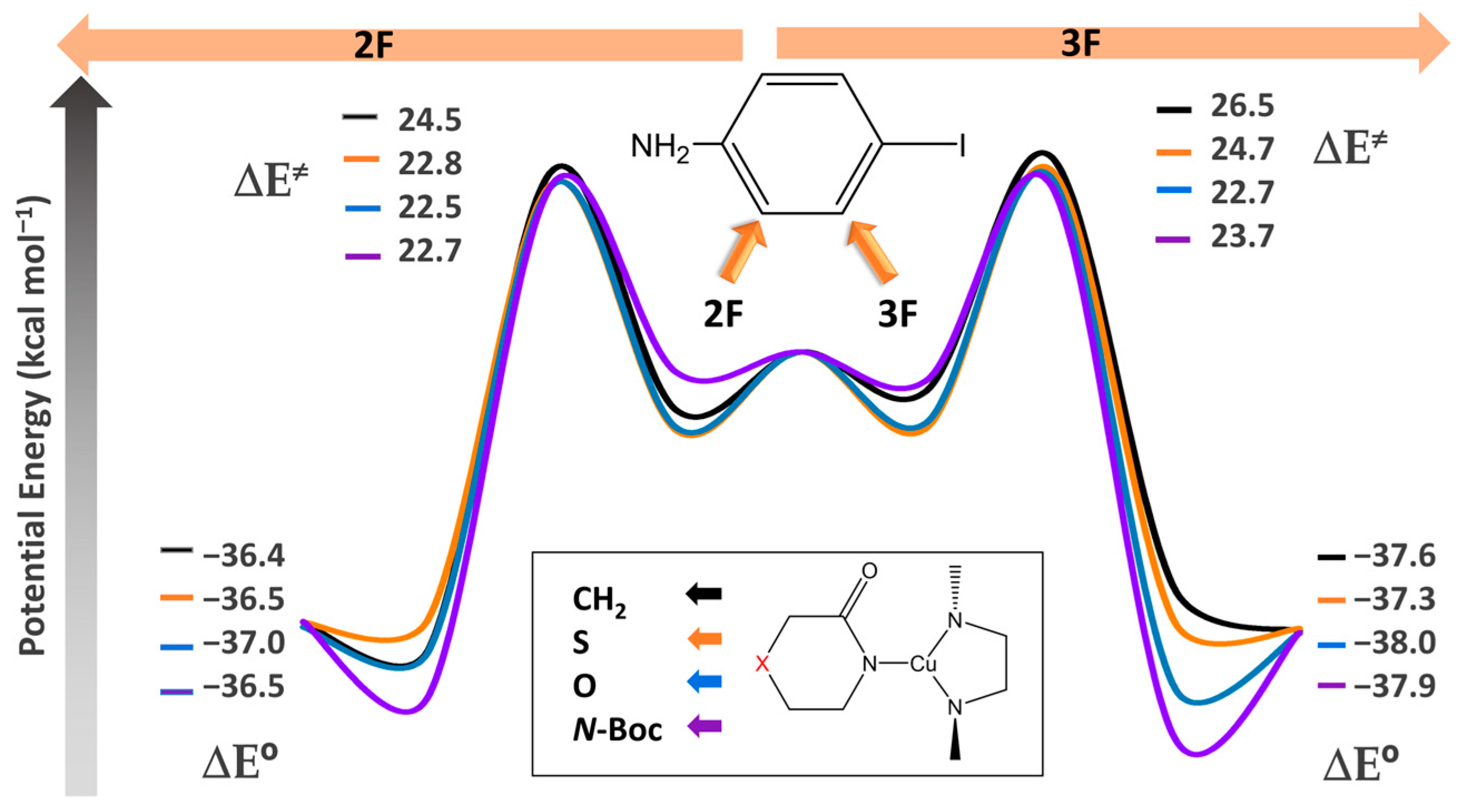
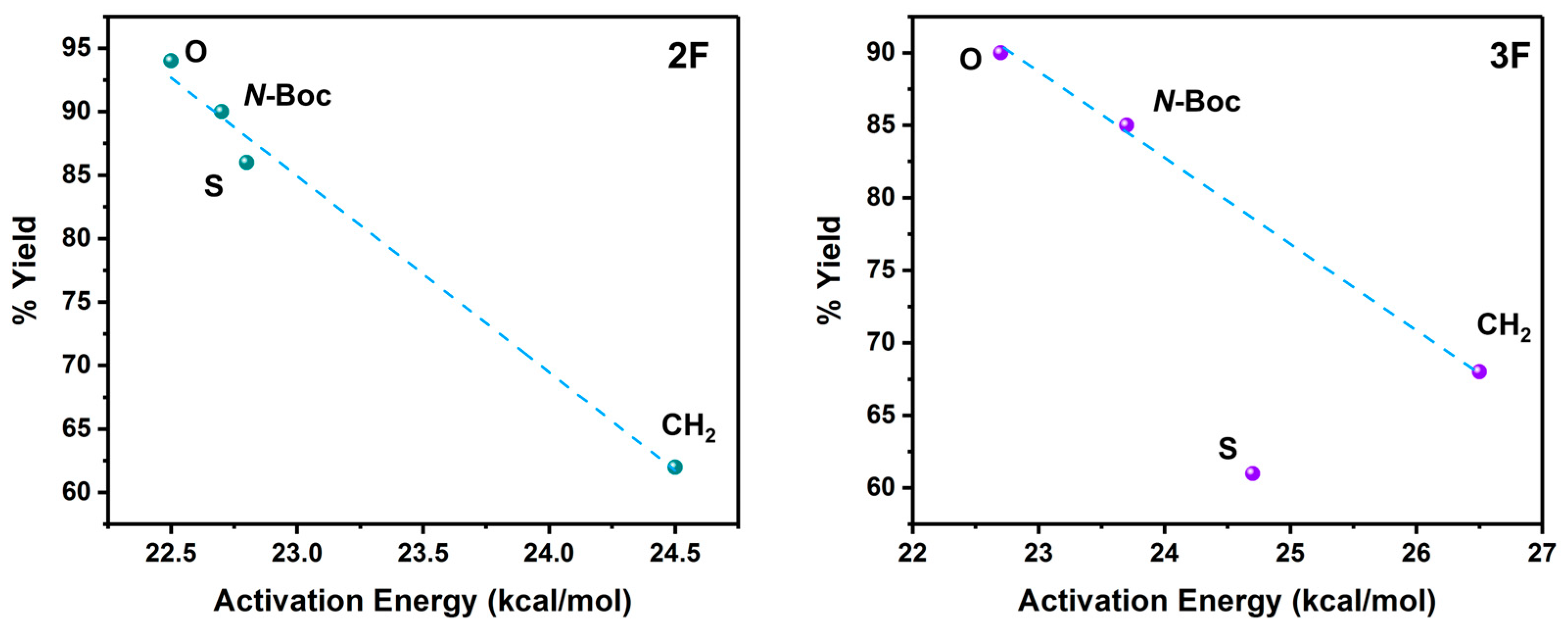
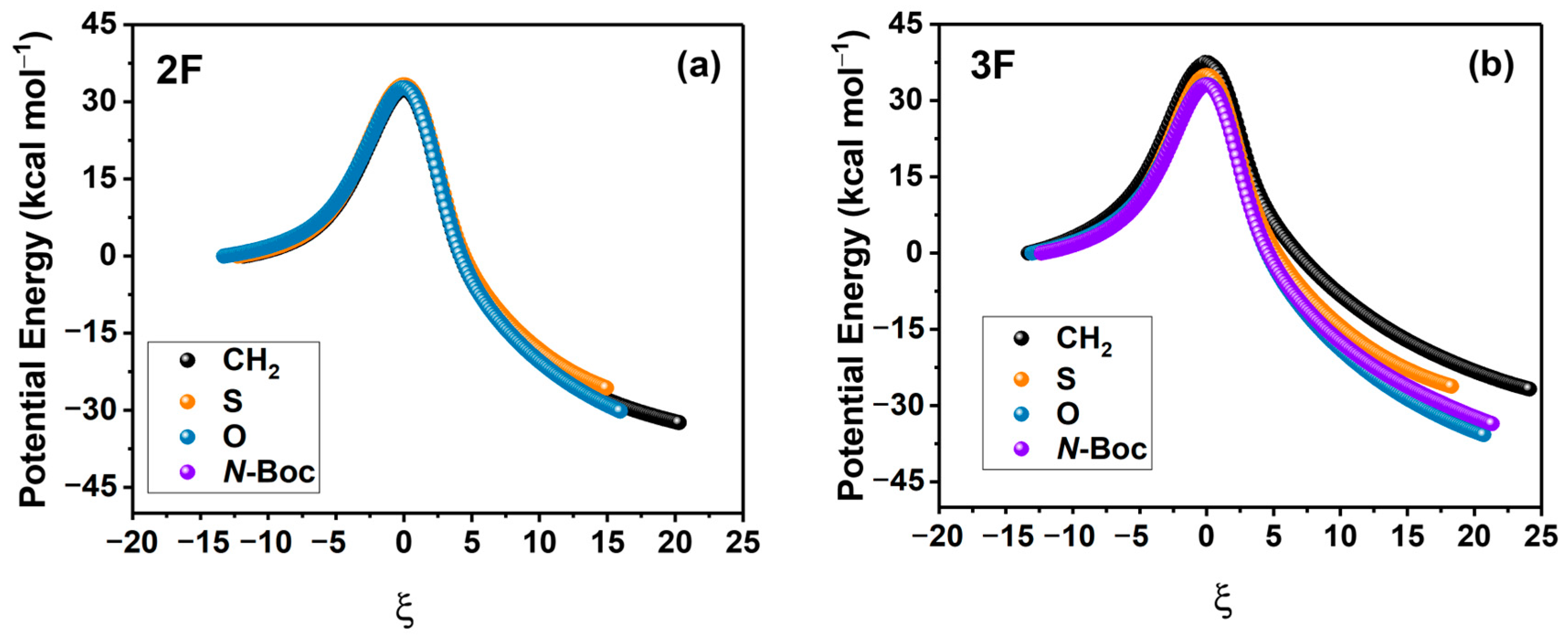
2.1. Energies and Reaction Profiles
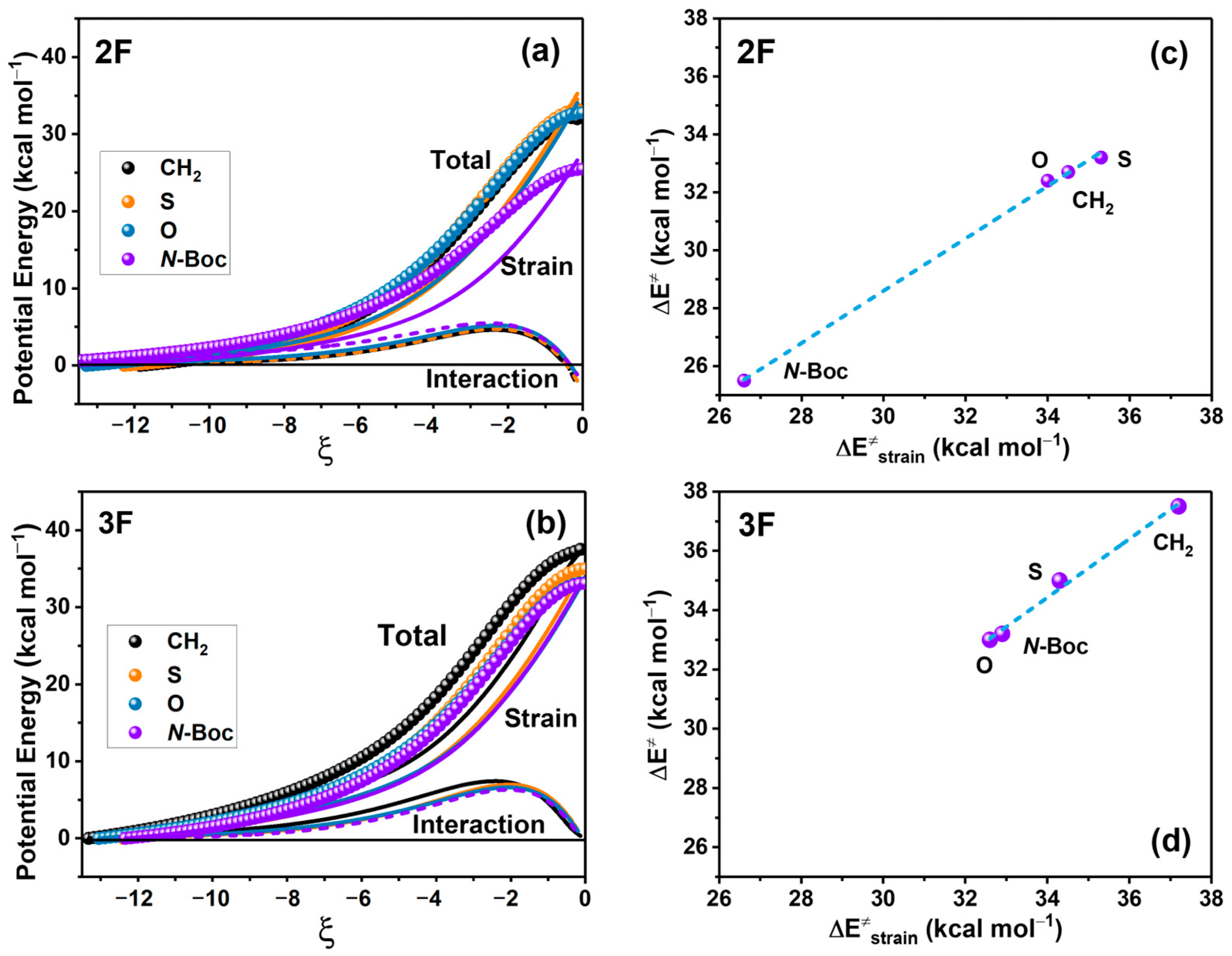
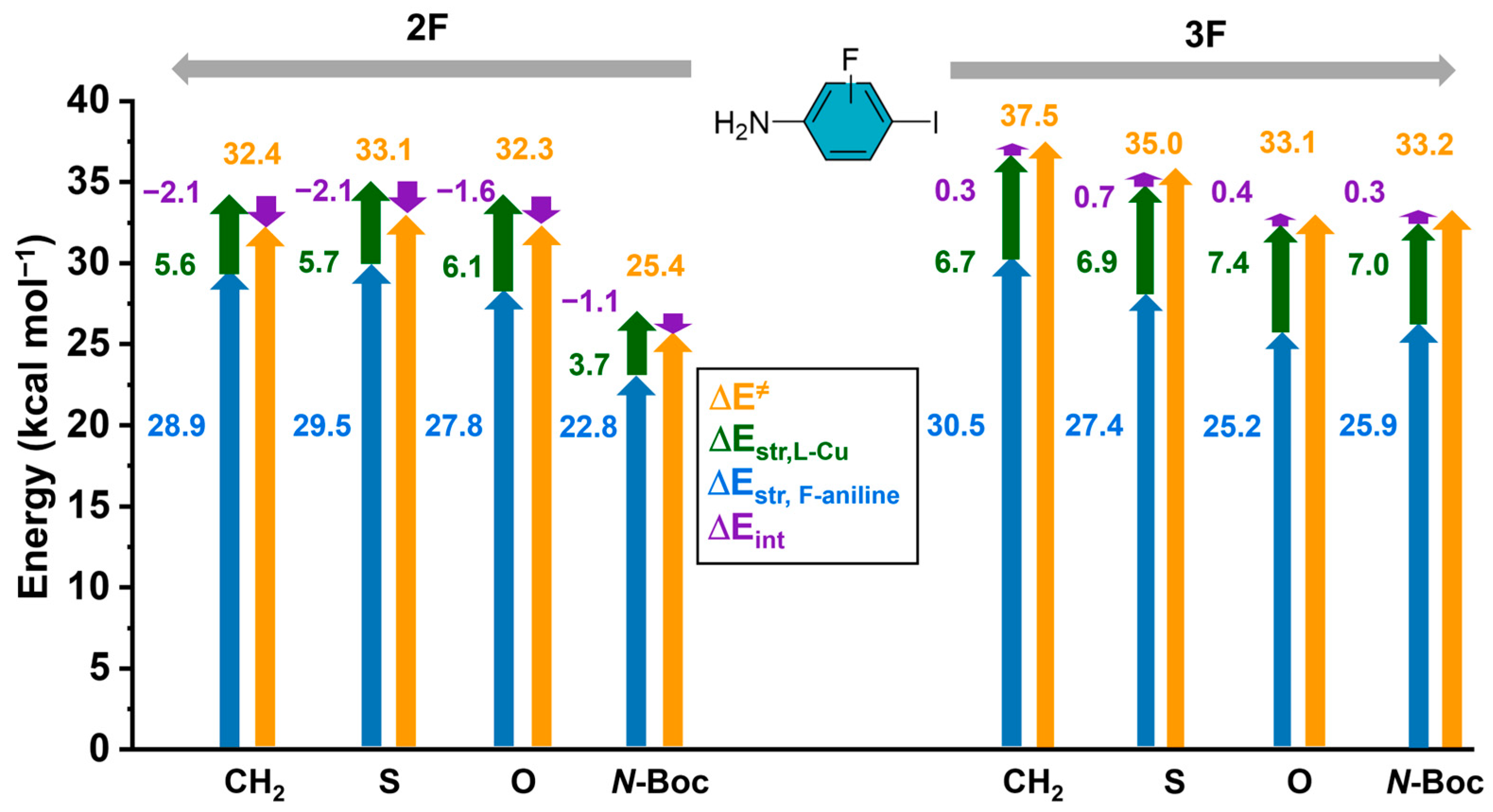
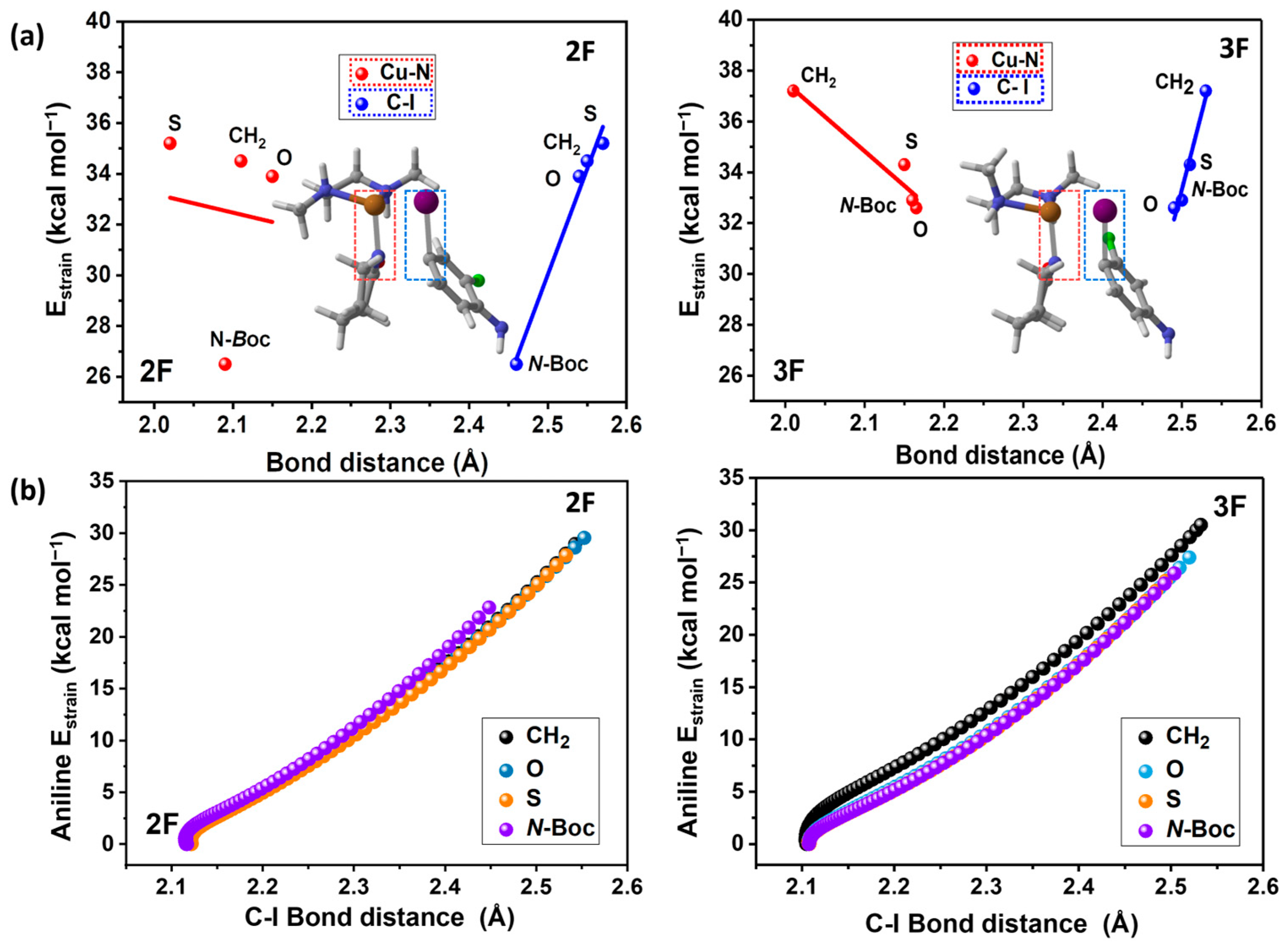
2.2. EDA and Strain Energy Analysis
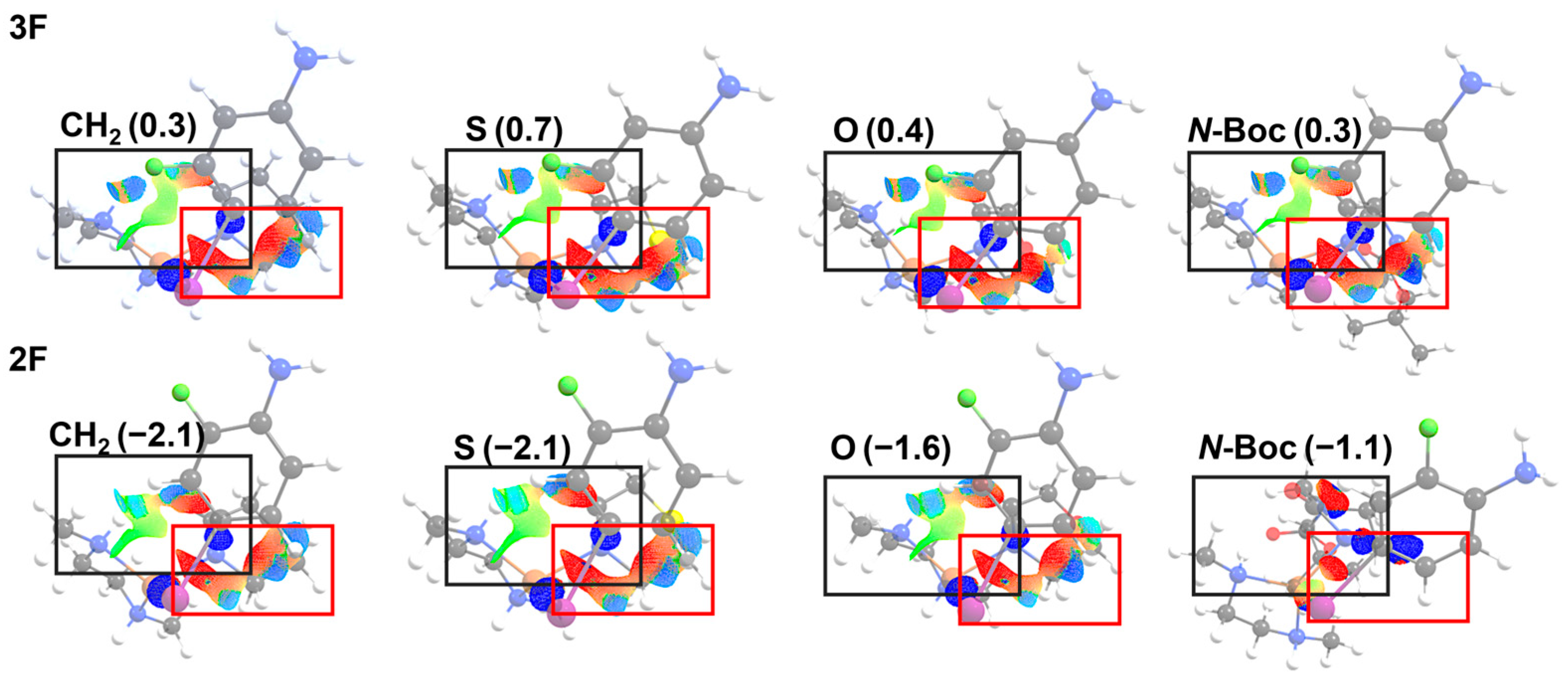
2.3. NCI Analysis
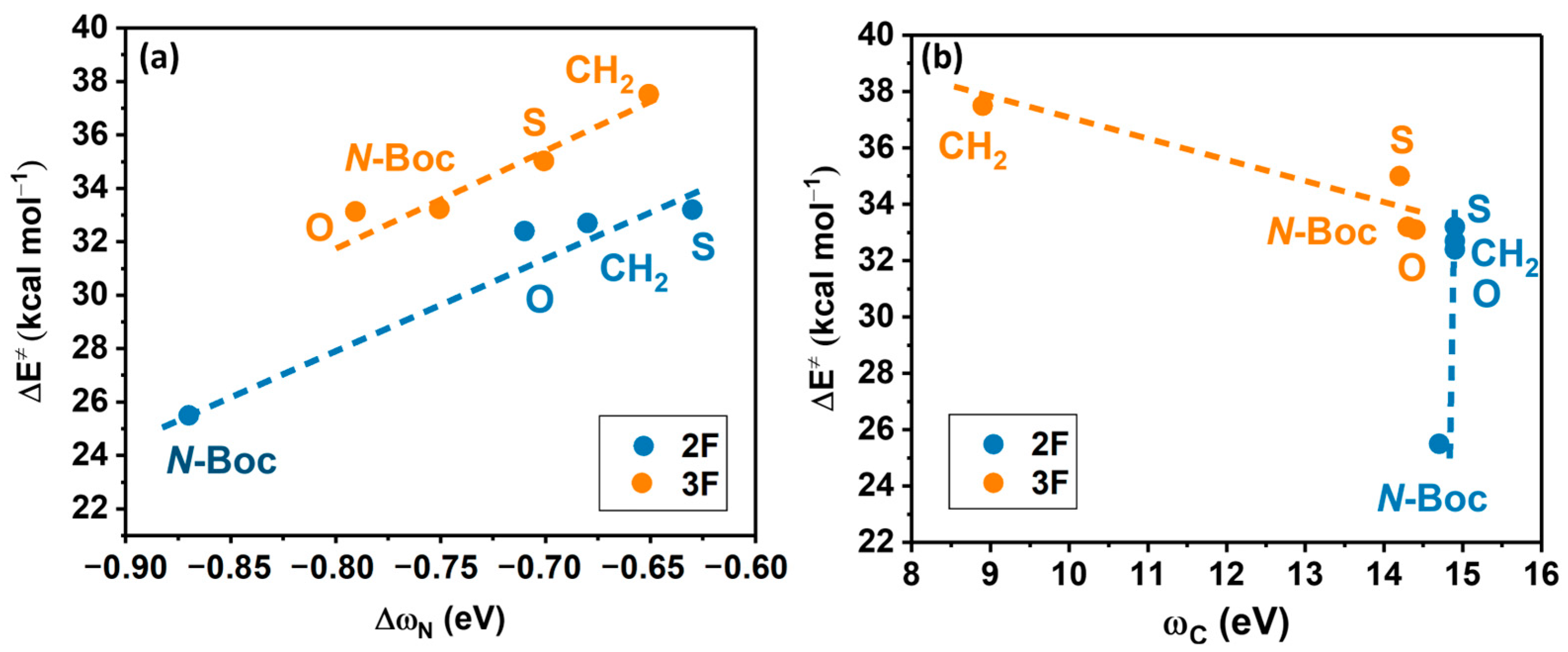
2.4. Local Electrophilicity
3. Discussion
3.1. Energies and Reaction Profiles
3.2. EDA and Strain Energy Analysis
3.3. NCI Analysis
3.4. Local Electrophilicity
4. Materials and Methods
Computational Details
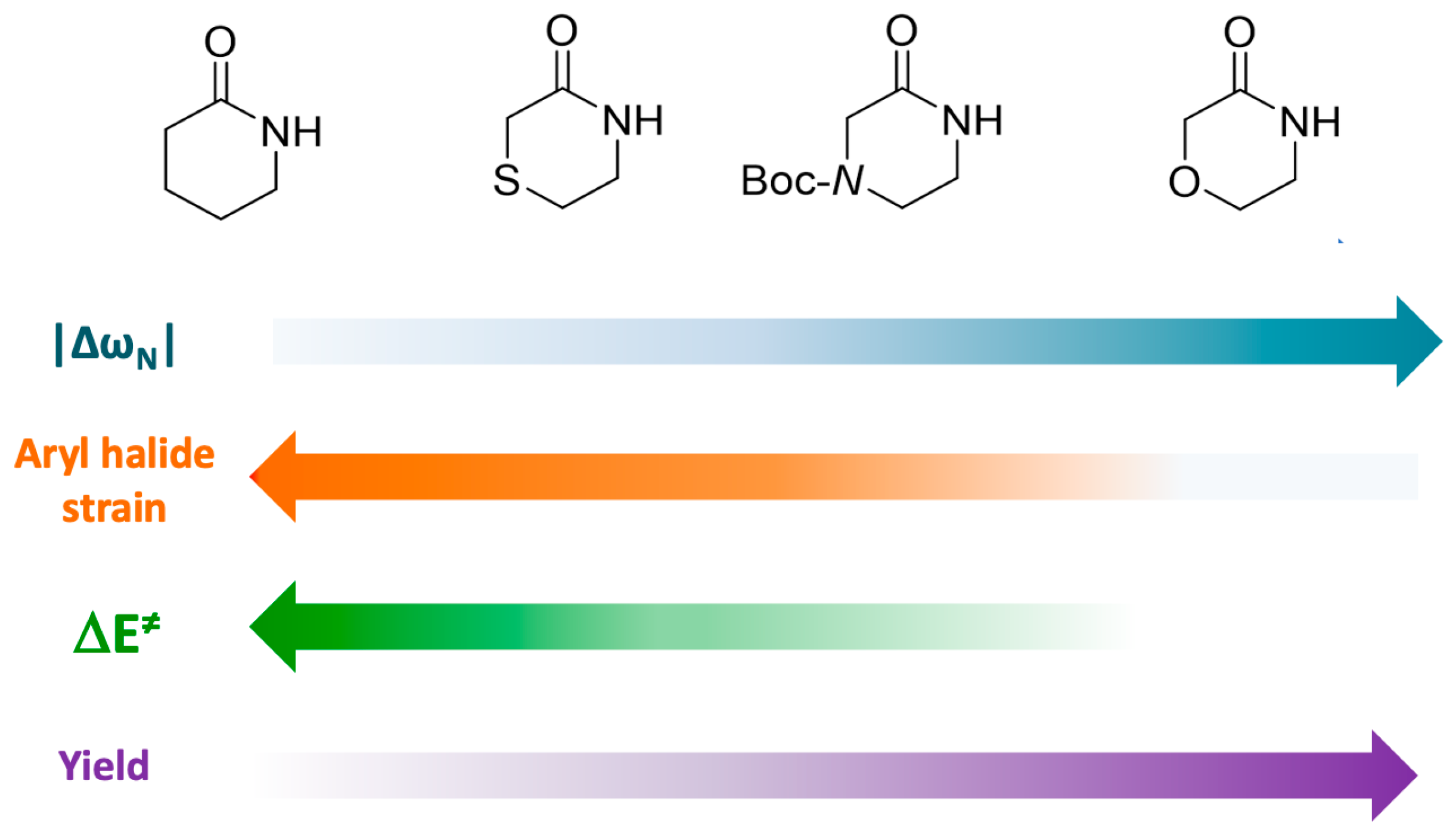
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ullmann, F.; Bielecki, J. Ueber Synthesen in Der Biphenylreihe. Berichte Dtsch. Chem. Ges. 1901, 34, 2174–2185. [Google Scholar] [CrossRef]
- Goldberg, I. Ueber Phenylirungen Bei Gegenwart von Kupfer Als Katalysator. Berichte Dtsch. Chem. Ges. 1906, 39, 1691–1692. [Google Scholar] [CrossRef]
- Antilla, J.C.; Baskin, J.M.; Barder, T.E.; Buchwald, S.L. Copper−Diamine-Catalyzed N-Arylation of Pyrroles, Pyrazoles, Indazoles, Imidazoles, and Triazoles. J. Org. Chem. 2004, 69, 5578–5587. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, Y.; Yao, J.; Wu, S.; Tao, F. Accelerating Effect Induced by the Structure of α-Amino Acid in the Copper-Catalyzed Coupling Reaction of Aryl Halides with α-Amino Acids. Synthesis of Benzolactam-V8. J. Am. Chem. Soc. 1998, 120, 12459–12467. [Google Scholar] [CrossRef]
- Klapars, A.; Antilla, J.C.; Huang, X.; Buchwald, S.L. A General and Efficient Copper Catalyst for the Amidation of Aryl Halides and the N -Arylation of Nitrogen Heterocycles. J. Am. Chem. Soc. 2001, 123, 7727–7729. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.A.; Buchwald, S.L. Cu-Catalyzed Goldberg and Ullmann Reactions of Aryl Halides Using Chelating N- and O-Based Ligands. Nat. Protoc. 2007, 2, 2474–2479. [Google Scholar] [CrossRef] [PubMed]
- Al-Shboul, T.M.A.; Al-Tarawneh, S.S.; Ababneh, T.S.; Jazzazi, T.M.A. Post-Functionalization of Bromo-Substituted Ether-Linked Polymers via Ullman Coupling Reaction: Synthesis, Characterization and Their Role toward Carbon Dioxide Capture. Separations 2022, 9, 55. [Google Scholar] [CrossRef]
- Manna, K.; Ji, P.; Lin, Z.; Greene, F.X.; Urban, A.; Thacker, N.C.; Lin, W. Chemoselective Single-Site Earth-Abundant Metal Catalysts at Metal–Organic Framework Nodes. Nat. Commun. 2016, 7, 12610. [Google Scholar] [CrossRef] [PubMed]
- Hayler, J.D.; Leahy, D.K.; Simmons, E.M. A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics 2019, 38, 36–46. [Google Scholar] [CrossRef]
- Rana, S.; Biswas, J.P.; Paul, S.; Paik, A.; Maiti, D. Organic Synthesis with the Most Abundant Transition Metal–Iron: From Rust to Multitasking Catalysts. Chem. Soc. Rev. 2021, 50, 243–472. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, Y.; Ma, D. Cu-Mediated Ullmann-Type Cross-Coupling and Industrial Applications in Route Design, Process Development, and Scale-up of Pharmaceutical and Agrochemical Processes. Org. Process Res. Dev. 2022, 26, 1690–1750. [Google Scholar] [CrossRef]
- Koenig, S.G.; Dankwardt, J.W.; Liu, Y.; Zhao, H.; Singh, S.P. Copper-Catalyzed Synthesis of Indoles and Related Heterocycles in Renewable Solvents. ACS Sustain. Chem. Eng. 2014, 2, 1359–1363. [Google Scholar] [CrossRef]
- Santana-Romo, F.; Lagos, C.F.; Duarte, Y.; Castillo, F.; Moglie, Y.; Maestro, M.A.; Charbe, N.; Zacconi, F.C. Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXa) Inhibitors: Drug Design, Synthesis, and Biological Evaluation. Molecules 2020, 25, 491. [Google Scholar] [CrossRef] [PubMed]
- Sambiagio, C.; Munday, R.H.; John Blacker, A.; Marsden, S.P.; McGowan, P.C. Green Alternative Solvents for the Copper-Catalysed Arylation of Phenols and Amides. RSC Adv. 2016, 6, 70025–70032. [Google Scholar] [CrossRef]
- Rodríguez, D.F.; Durán-Osorio, F.; Duarte, Y.; Olivares, P.; Moglie, Y.; Dua, K.; Zacconi, F.C. Green by Design: Convergent Synthesis, Computational Analyses, and Activity Evaluation of New FXa Inhibitors Bearing Peptide Triazole Linking Units. Pharmaceutics 2021, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Andrada, D.; Soria-Castro, S.; Caminos, D.; Argüello, J.; Peñéñory, A. Understanding the Heteroatom Effect on the Ullmann Copper-Catalyzed Cross-Coupling of X-Arylation (X = NH, O, S) Mechanism. Catalysts 2017, 7, 388. [Google Scholar] [CrossRef]
- Gurjar, K.K.; Sharma, R.K. Synthetic and Computational Studies on CuI/Ligand Pair Promoted Activation of C(Aryl)-Cl Bond in C–N Coupling Reactions. Heliyon 2020, 6, e03233. [Google Scholar] [CrossRef] [PubMed]
- Huffman, L.M.; Stahl, S.S. Carbon-Nitrogen Bond Formation Involving Well-Defined Aryl-Copper(III) Complexes. J. Am. Chem. Soc. 2008, 130, 9196–9197. [Google Scholar] [CrossRef] [PubMed]
- Clavé, G.; Garel, C.; Poullain, C.; Renard, B.-L.; Olszewski, T.K.; Lange, B.; Shutcha, M.; Faucon, M.-P.; Grison, C. Ullmann Reaction through Ecocatalysis: Insights from Bioresource and Synthetic Potential. RSC Adv. 2016, 6, 59550–59564. [Google Scholar] [CrossRef]
- Sambiagio, C.; Marsden, S.P.; Blacker, A.J.; McGowan, P.C. Copper Catalysed Ullmann Type Chemistry: From Mechanistic Aspects to Modern Development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar] [CrossRef]
- Lewis, E.A.; Marcinkowski, M.D.; Murphy, C.J.; Liriano, M.L.; Therrien, A.J.; Pronschinske, A.; Sykes, E.C.H. Controlling Selectivity in the Ullmann Reaction on Cu(111). Chem. Commun. 2017, 53, 7816–7819. [Google Scholar] [CrossRef] [PubMed]
- Strieter, E.R.; Blackmond, D.G.; Buchwald, S.L. The Role of Chelating Diamine Ligands in the Goldberg Reaction: A Kinetic Study on the Copper-Catalyzed Amidation of Aryl Iodides. J. Am. Chem. Soc. 2005, 127, 4120–4121. [Google Scholar] [CrossRef] [PubMed]
- Sperotto, E.; Van Klink, G.P.M.; Van Koten, G.; De Vries, J.G. The Mechanism of the Modified Ullmann Reaction. Dalton Trans. 2010, 39, 10338. [Google Scholar] [CrossRef]
- Xie, C.; Guo, Q.; Yang, Z.; Zi, G.; Huang, Y.; Hou, G. Enantioselective Synthesis of Chiral 2,2,2-Trifluoroethyl Lactams via Asymmetric Hydrogenation. Org. Chem. Front. 2023, 10, 2498–2504. [Google Scholar] [CrossRef]
- Halder, M.; Islam, M.M.; Ansari, Z.; Ahammed, S.; Sen, K.; Islam, S.M. Biogenic Nano-CuO-Catalyzed Facile C–N Cross-Coupling Reactions: Scope and Mechanism. ACS Sustain. Chem. Eng. 2017, 5, 648–657. [Google Scholar] [CrossRef]
- Durán, R.; Núñez-Navarro, N.; Zacconi, F.C.; Herrera, B. Theoretical Study of C-Arylations with Aryl Halides to Determine the Reaction Mechanism, the Effect of Substituents and Heteroatoms. Phys. Chem. Chem. Phys. 2019, 21, 10163–10170. [Google Scholar] [CrossRef] [PubMed]
- van Zeist, W.-J.; Bickelhaupt, F.M. The Activation Strain Model of Chemical Reactivity. Org. Biomol. Chem. 2010, 8, 3118. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.; Bickelhaupt, F.M. The Activation Strain Model and Molecular Orbital Theory: Understanding and Designing Chemical Reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Boto, R.A.; Izquierdo-Ruiz, F.; Reva, I.; Woller, T.; Alonso, M. A Benchmark for the Non-Covalent Interaction (NCI) Index Or… Is It Really All in the Geometry? Theor. Chem. Acc. 2016, 135, 242. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Non-Covalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R.; Contreras-García, J. Revealing Non-Covalent Interactions in Solids: NCI Plots Revisited. Phys. Chem. Chem. Phys. 2012, 14, 12165. [Google Scholar] [CrossRef] [PubMed]
- Parr, R. Some Functional Relations in the Density Functional Theory of Finite Interacting Electronic Systems. Chem. Phys. Lett. 1997, 276, 164–166. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of the Electronic Structure of Molecules. Annu. Rev. Phys. Chem. 1995, 46, 701–728. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules, 1st ed.; International Series Monographs in Chemistry; Oxford University Press: Oxford, UK, 1989; Volume 1, ISBN 978-0-19-509276-9. [Google Scholar]
- Parr, R. Chemical Reactivity Theory; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar] [CrossRef]
- Parr, R.G. Aspects of Density Functional Theory. Philosophical. Mag. B 1994, 69, 737–743. [Google Scholar] [CrossRef]
- Geerlings, P.; Proft, F.D.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; Proft, F.D.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual Density Functional Theory: Status, Prospects, Issues. Theor. Chem. Acc. 2020, 139, 36. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1987; Volume 1, ISBN 978-3-527-29482-4. [Google Scholar]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Parthasarathi, R.; Elango, M.; Subramanian, V.; Krishnamoorthy, B.S.; Gutierrez-Oliva, S.; Toro-Labbé, A.; Roy, D.R.; Chattaraj, P.K. Multiphilic Descriptor for Chemical Reactivity and Selectivity. J. Phys. Chem. A 2007, 111, 9130–9138. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Duley, S.; Domingo, L.R. Understanding Local Electrophilicity/Nucleophilicity Activation through a Single Reactivity Difference Index. Org. Biomol. Chem. 2012, 10, 2855. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Chiodo, S.; Russo, N.; Sicilia, E. LANL2DZ Basis Sets Recontracted in the Framework of Density Functional Theory. J. Chem. Phys. 2006, 125, 104107. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Scott, A.P.; Radom, L. Harmonic Vibrational Frequencies: An Evaluation of Hartree−Fock, Møller−Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors. J. Phys. Chem. 1996, 100, 16502–16513. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction Path Following in Mass-Weighted Internal Coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Fukui, K. The Path of Chemical Reactions—The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- van Zeist, W.-J.; Koers, A.H.; Wolters, L.P.; Bickelhaupt, F.M. Reaction Coordinates and the Transition-Vector Approximation to the IRC. J. Chem. Theory Comput. 2008, 4, 920–928. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16, Revision B.01, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Zhurko, G.; Zhurko, D. ChemCraft. Available online: http://www.chemcraftprog.com (accessed on 1 January 2019).
- Legault, C. CYLVIEW 10.b. Available online: http://www.cylview.org (accessed on 1 January 2021).
- Lin, Y.-S.; Li, G.-D.; Mao, S.-P.; Chai, J.-D. Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections. J. Chem. Theory Comput. 2013, 9, 263–272. [Google Scholar] [CrossRef]
- Boto, R.A.; Contreras-García, J.; Tierny, J.; Piquemal, J.-P. Interpretation of the reduced density gradient. Mol. Phys. 2016, 114, 1406–1414. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Durán, R.; Barrales-Martínez, C.; Santana-Romo, F.; Rodríguez, D.F.; Zacconi, F.C.; Herrera, B. Substitution Effects in Aryl Halides and Amides into the Reaction Mechanism of Ullmann-Type Coupling Reactions. Molecules 2024, 29, 1770. https://doi.org/10.3390/molecules29081770
Durán R, Barrales-Martínez C, Santana-Romo F, Rodríguez DF, Zacconi FC, Herrera B. Substitution Effects in Aryl Halides and Amides into the Reaction Mechanism of Ullmann-Type Coupling Reactions. Molecules. 2024; 29(8):1770. https://doi.org/10.3390/molecules29081770
Chicago/Turabian StyleDurán, Rocío, César Barrales-Martínez, Fabián Santana-Romo, Diego F. Rodríguez, Flavia C. Zacconi, and Barbara Herrera. 2024. "Substitution Effects in Aryl Halides and Amides into the Reaction Mechanism of Ullmann-Type Coupling Reactions" Molecules 29, no. 8: 1770. https://doi.org/10.3390/molecules29081770
APA StyleDurán, R., Barrales-Martínez, C., Santana-Romo, F., Rodríguez, D. F., Zacconi, F. C., & Herrera, B. (2024). Substitution Effects in Aryl Halides and Amides into the Reaction Mechanism of Ullmann-Type Coupling Reactions. Molecules, 29(8), 1770. https://doi.org/10.3390/molecules29081770